

Abstract
The prevalence of congenital heart disease in infants with Down syndrome is 40%, compared with 0.3% in children who have normal chromosomes. Atrioventricular and ventricular septal defects are often associated with chromosomal aberrations, such as in trisomy 21, whereas hypertrophic cardiomyopathy is chiefly thought to be secondary to specific gene mutations. We found only one reported case of congenital hypertrophic cardiomyopathy and atrioventricular septal defect in an infant with Down syndrome. Here, we report atrioventricular septal defect, hypertrophic cardiomyopathy, and pulmonary vein stenosis in a neonate with Down syndrome—an apparently unique combination. In addition, we discuss the relevant medical literature.
Keywords: Abnormalities, multiple/genetics; cardiomyopathy, hypertrophic/complications/congenital; Down syndrome/complications; fatal outcome; heart defects, congenital/diagnosis/epidemiology; heart septal defects, ventricular/embryology/physiopathology; hypertension, pulmonary/complications; infant, newborn; pulmonary veins/physiopathology
Congenital heart disease (CHD) is found in 40% of infants with Down syndrome but in only 0.3% of children who have normal chromosomes.1 In Down syndrome, ventricular septal defect and atrioventricular septal defect (AVSD) are typical malformations. Transient hypertrophic cardiomyopathy (HCM) in neonates has typically been attributed to maternal diabetes mellitus and has infrequently been reported in association with CHD. Atrioventricular septal defect is often associated with chromosomal aberration, whereas HCM is thought to be secondary to specific gene mutations. We found just one reported case of congenital HCM and AVSD in an infant with Down syndrome.2 Here, we report the case of a neonate who had Down syndrome with HCM, complete AVSD, and pulmonary vein (PV) stenosis, and we discuss the relevant medical literature.
Case Report
A male infant (twin B) from a dichorionic diamniotic pregnancy was born at our hospital. The 38-year-old mother underwent a cesarean section at 33 weeks of gestation because of uteroplacental insufficiency and intrauterine growth restriction. The infant's birth weight was 3 lb 6 oz, and his Apgar scores were 8 at 1 min and 9 at 5 min. Routine antenatal ultrasonographic results had raised suspicion of AVSD; other findings were the absence of a nasal bone and a poorly visible stomach in twin B. At 26 weeks of gestation, a prenatal echocardiogram had shown a common atrioventricular (AV) canal defect in twin B and no cardiac defects in twin A. No antenatal genetic tests were performed.
The initial physical examination revealed a neonate who was small for his gestational age and had Down facies, single palmar crease, and mild hypotonia. Cardiovascular examination revealed a normal precordium, no cyanosis, a normal S1, and a loud S2 (P2 component) with no murmur. The peripheral pulses were well palpable. The liver was palpable 2 cm below the costal margin. A chest radiograph showed an enlarged cardiac silhouette. A 12-lead electrocardiogram showed sinus rhythm, left-axis deviation, and possible biventricular hypertrophy. A transthoracic echocardiogram (TTE) showed a common AV canal defect (Rastelli type A), a thick ventricular septum, a large patent ductus arteriosus, and a patent foramen ovale. A postnatal chromosomal analysis revealed 47, XY, +21 (trisomy 21).
Serial TTEs were obtained during the infant's hospital stay. At 3 months, a TTE showed complete AVSD and severe asymmetric septal hypertrophy (Fig. 1), as well as left ventricular midcavitary obstruction (peak gradient, 25 mmHg). There was no systolic anterior motion of the mitral valve. Results of hemodynamic catheterization included pulmonary venous desaturation of 88% on the left due to chronic lung disease, and borderline elevation of pulmonary vascular resistance at 3.16 Wood units, normalized to 1.98 Wood units on 100% oxygen; the Qp/Qs ratio of 2.58:1 on 50% oxygen increased to 3.58:1 on 100% oxygen. Cardiac magnetic resonance was performed to better define the hypertrophy and evaluate myocardial viability. The findings confirmed severe (1.4-cm) asymmetric septal hypertrophy (Fig. 2), hypertrophied papillary muscles, a narrow left ventricular cavity with midcavitary obstruction, and hyperdynamic systolic function with low end-diastolic volumes. No evidence of myocardial delayed enhancement was found. The rest of the anatomy was consistent with a common AV canal defect. The infant's heart failure was medically managed with the use of diuretic agents. Propranolol was added to alleviate the progressive midcavitary obstruction and low end-diastolic volumes. The infant had oral aversion and was almost entirely fed first through a nasogastric tube and then through a gastrointestinal tube throughout the hospital course.
Fig. 1.
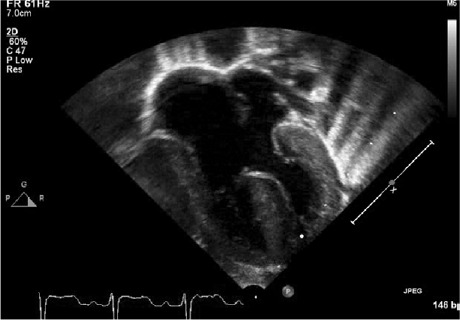
Transthoracic echocardiogram (4-chamber view) shows a complete atrioventricular septal defect and a hypertrophied septum.
Fig. 2.
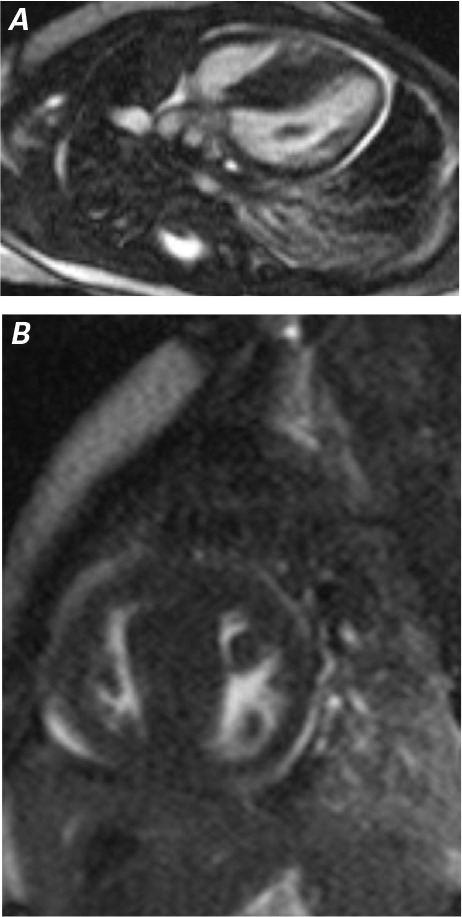
Cardiac magnetic resonance A) long-axis and B) short-axis steady-state free precession images show asymmetric hypertrophy of the ventricular septum.
Follow-up TTEs showed increased flow velocity in the right upper PV and subsequently in the left upper PV, indicating PV stenosis. The lower PVs had low-velocity laminar flow. When the infant was 6 months old, catheterization results revealed mild elevation of the pulmonary vascular resistance, severe stenosis in the proximal left lower PV, and severe diffuse hypoplasia of the right upper, left upper, and middle PVs (Fig. 3). Because of the diffuse hypoplasia, no intervention was attempted. After this catheterization, the infant had a profound hypoxic crisis and went into cardiac arrest. Despite aggressive medical therapy, maximal ventilator support, and inotropic support, his condition remained dismal. Further treatment was considered futile at this stage, and care was withdrawn after parental consent.
Fig. 3.
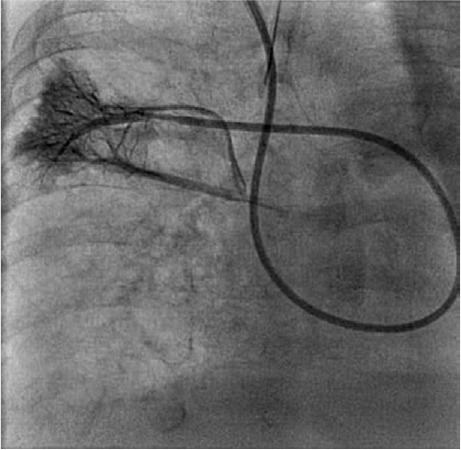
Angiogram of the right pulmonary artery shows hypoplastic right middle pulmonary veins in levophase.
Autopsy results confirmed the diagnosis of complete AVSD, Rastelli type A canal defect; asymmetric HCM; hypoplastic PVs with stenosis involving the left upper, right upper, and middle PVs with mild PV thickening; pulmonary arteriopathy; and lymphatic muscularization, along with chronic lung disease. There was no family history of HCM or sudden cardiac death, and the screening echocardiograms of the infant's siblings and both parents were normal. Genetic and metabolic findings in the infant were negative, including those of 18-gene HCM testing and tests for Pompe disease.
Discussion
To our knowledge, the combination of Down syndrome with common AV canal defect, HCM, and PV stenosis has not been described in the medical literature. In our patient, the results of genetic tests—notably an 18-gene panel for familial HCM—were negative, so this unusual constellation of conditions in Down syndrome was most likely a random event.
Garrod first described the association of CHD with mongolism.3 In Down syndrome, complete AVSD is often seen.4 The increased adhesiveness of trisomy 21 cells might keep the embryonal endocardial cushion from fusing, thereby causing persistent AVSD.5
Complete AVSD, which affects approximately 3% of all patients who have CHD,6 tends to be associated with chromosomal abnormalities—mainly Down syndrome, del (8p) syndrome, trisomy 9, and trisomy 18.7–9 This cardiac malformation is characterized by an ostium primum atrial septal defect, a common AV valve, and variably deficient ventricular septal inflow. In complete AVSD, substantial interatrial and interventricular systemic-to-pulmonary shunting contributes to an overload in right ventricular pressure and volume that leads to irreversible pulmonary hypertension.6 Among concomitant congenital conditions are tetralogy of Fallot, truncus arteriosus, aortic coarctation, and heterotaxy syndrome; acquired left ventricular outflow tract obstruction is a well-recognized complication.10 Although PV stenosis has rarely been documented in patients with Down syndrome, it has a high mortality rate,11 and balloon angioplasty and stenting for PV stenosis have yielded high rates of restenosis.12
In infants and children, HCM is typically isolated and has been reported chiefly in case reports or small case series.13 Infants with HCM have a worse prognosis than do older children with that condition. Infants can become severely symptomatic and die of progressive congestive heart failure in the first year of life.14 The onset of marked congestive heart failure during the first year appears to be a poor prognostic sign, and sudden death can occur secondary to heart failure or after surgical repair.
Substantial evidence indicates that HCM in children and adults is often genetically transmitted. Hypertrophic cardiomyopathy is associated with genetic syndromes such as Noonan syndrome and Costello syndrome, and with metabolic diseases such as long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and adenosine triphosphate synthase deficiency. Other reported origins of HCM are maternal diabetes mellitus and steroid use.15,16 In children, primary HCM can be caused by a mutation in one of several genes, and secondary HCM by an inborn error in metabolism such as glycogen storage disease or mucopolysaccharidosis.17 Primary HCM has occurred in association with other congenital conditions, such as ventricular septal defect and tetralogy of Fallot.18
The prognosis would appear to be poor in other patients like ours. The lone case report2 of the patient who had reached one year of age without any complications was encouraging; however, our patient had PV stenosis that further worsened his prognosis, and he died at the age of 6 months.
Acknowledgments
We thank Richard J. Martin, MD, and Michiko Watanabe, PhD, of Case Western Reserve University, for reviewing and editing the manuscript.
Footnotes
From: Division of Pediatric Cardiology, Rainbow Babies and Children's Hospital, Cleveland, Ohio 44106
References
- 1.Ferencz C, Rubin JD, McCarter RJ, Brenner JI, Neill CA, Perry LW et al. Congenital heart disease: prevalence at live-birth. The Baltimore-Washington Infant Study. Am J Epidemiol. 1985;121(1):31–6. doi: 10.1093/oxfordjournals.aje.a113979. [DOI] [PubMed] [Google Scholar]
- 2.Eidem BW, Jones C, Cetta F. Unusual association of hypertrophic cardiomyopathy with complete atrioventricular canal defect and Down syndrome. Tex Heart Inst J. 2000;27(3):289–91. [PMC free article] [PubMed] [Google Scholar]
- 3.Garrod AE. On the association of cardiac malformations with other congenital defects. St Barth Hosp Rep. 1894;30:53–61. Available from: http://hdl.handle.net/2027/nnc2.ark:/13960/t9q24mm0h?urlappend=%3Bseq=89 [cited 2015 Aug 7] [Google Scholar]
- 4.Spicer RL. Cardiovascular disease in Down syndrome. Pediatr Clin North Am. 1984;31(6):1331–43. doi: 10.1016/s0031-3955(16)34725-3. [DOI] [PubMed] [Google Scholar]
- 5.Kurnit DM, Aldridge JF, Matsuoka R, Matthysse S. Increased adhesiveness of trisomy 21 cells and atrioventricular canal malformations in Down syndrome: a stochastic model. Am J Med Genet. 1985;20(2):385–99. doi: 10.1002/ajmg.1320200222. [DOI] [PubMed] [Google Scholar]
- 6.Calabro R, Limongelli G. Complete atrioventricular canal. Orphanet J Rare Dis. 2006;1:8. doi: 10.1186/1750-1172-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmi R, Boughman JA, Ferencz C. Endocardial cushion defect: further studies of “isolated” versus “syndromic” occurrence. Am J Med Genet. 1992;43(3):569–75. doi: 10.1002/ajmg.1320430313. [DOI] [PubMed] [Google Scholar]
- 8.Marino B, Reale A, Giannotti A, Digilio MC, Dallapiccola B. Nonrandom association of atrioventricular canal and del (8p) syndrome. Am J Med Genet. 1992;42(4):424–7. doi: 10.1002/ajmg.1320420404. [DOI] [PubMed] [Google Scholar]
- 9.Francalanci P, Marino B, Boldrini R, Abella R, Iorio F, Bosman C. Morphology of the atrioventricular valve in asplenia syndrome: a peculiar type of atrioventricular canal defect. Cardiovasc Pathol. 1996;5(3):145–51. doi: 10.1016/1054-8807(95)00117-4. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Shacher G, Moller JH, Castaneda-Zuniga W, Edwards JE. Signs of membranous subaortic stenosis appearing after correction of persistent common atrioventricular canal. Am J Cardiol. 1981;48(2):340–4. doi: 10.1016/0002-9149(81)90617-2. [DOI] [PubMed] [Google Scholar]
- 11.Gowda S, Bhat D, Feng Z, Chang CH, Ross RD. Pulmonary vein stenosis with Down syndrome: a rare and frequently fatal cause of pulmonary hypertension in infants and children. Congenit Heart Dis. 2014;9(3):E90–7. doi: 10.1111/chd.12088. [DOI] [PubMed] [Google Scholar]
- 12.Latson LA, Prieto LR. Congenital and acquired pulmonary vein stenosis. Circulation. 2007;115(1):103–8. doi: 10.1161/CIRCULATIONAHA.106.646166. [DOI] [PubMed] [Google Scholar]
- 13.Maron BJ, Henry WL, Clark CE, Redwood DR, Roberts WC, Epstein SE. Asymmetric septal hypertrophy in childhood. Circulation. 1976;53(1):9–19. doi: 10.1161/01.cir.53.1.9. [DOI] [PubMed] [Google Scholar]
- 14.Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007;115(6):773–81. doi: 10.1161/CIRCULATIONAHA.106.621185. [DOI] [PubMed] [Google Scholar]
- 15.Clark CE, Henry WL, Epstein SE. Familial prevalence and genetic transmission of idiopathic hypertrophic subaortic stenosis. N Engl J Med. 1973;289(14):709–14. doi: 10.1056/NEJM197310042891402. [DOI] [PubMed] [Google Scholar]
- 16.Pare JA, Fraser RG, Pirozynski WJ, Shanks JA, Stubington D. Hereditary cardiovascular dysplasia. A form of familial cardiomyopathy. Am J Med. 1961;31:37–62. doi: 10.1016/0002-9343(61)90222-4. [DOI] [PubMed] [Google Scholar]
- 17.Berger S, Dhala A, Dearani JA. State-of-the-art management of hypertrophic cardiomyopathy in children. Cardiol Young. 2009;19(Suppl 2):66–73. doi: 10.1017/S1047951109991648. [DOI] [PubMed] [Google Scholar]
- 18.Somerville J, Becu L. Congenital heart disease associated with hypertrophic cardiomyopathy. Br Heart J. 1978;40(9):1034–9. doi: 10.1136/hrt.40.9.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]