

Abstract
The ErbB family (HER-1, HER-2, HER-3 and HER-4) of receptor tyrosine kinases has been the focus of cancer immunotherapeutic strategies while antiangiogenic therapies have focused on VEGF and its receptors VEGFR-1 and VEGFR-2. Agents targeting receptor tyrosine kinases in oncology include therapeutic antibodies to receptor tyrosine kinase ligands or the receptors themselves, and small-molecule inhibitors. Many of the US FDA-approved therapies targeting HER-2 and VEGF exhibit unacceptable toxicities, and show problems of efficacy, development of resistance and unacceptable safety profiles that continue to hamper their clinical progress. The combination of dif ferent peptide vaccines and peptidomimetics targeting specific molecular pathways that are dysregulated in tumors may potentiate anticancer immune responses, bypass immune tolerance and circumvent resistance mechanisms. The focus of this review is to discuss efforts in our laboratory spanning two decades of rationally developing peptide vaccines and therapeutics for breast cancer. This review highlights the prospective benefit of a new, untapped category of therapies biologically targeted to EGF receptor (HER-1), HER-2 and VEGF with potential peptide ‘blockbusters‘ that could lay the foundation of a new paradigm in cancer immunotherapy by creating clinical breakthroughs for safe and efficacious cancer cures.
Keywords: angiogenesis, cancer immunotherapy, chimeric peptide B-cell epitope vaccines, HER-2, VEGF
HER family receptors & antitumor strategies
The HER family of receptors plays a central role in the pathogenesis of several human cancers including breast, ovarian, renal, colon and lung carcinomas, and is associated with aggressive forms of several human cancers. They regulate cell growth, survival and differentiation via multiple signal transduction pathways [1], and participate in cellular proliferation and differentiation [2,3]. The family is made up of four main members: HER-1, HER-2, HER-3 and HER-4, also called ErbB1, ErbB2, ErbB3 and ErbB4, respectively [4]. All four HER receptors (HER-1 [EGF receptor (EGFR)], HER-2, HER-3 and HER-4) share structurally homologous extracellular domains (ECDs) [5]. HER-2 plays a major coordinating role in this network, since each receptor with a specific ligand seems to prefer HER-2 as its heterodimeric partner [6,7] owing to its constitutively ‘open’ conformation. HER-2-containing heterodimers potently amplify signaling because HER-2 reduces the rate of ligand dissociation, allowing strong activation of downstream signaling pathways [8,9], and those involving the activation of the PI3K–Akt and Ras–Raf–MEK–MAPK pathways [10].
The most important member of this family is HER-2 and its overexpression in several tumor types is regarded as the main cause of aggressive forms of cancer and poor clinical outcome [11,12]. The HER-2 protein has been shown to be an important therapeutic target in many cancers owing to the fact that it is the only member that does not have a ligand. Its oncogenic potential is likely due to being the preferred dimerization partner for the other family members, explaining why it is one of the most studied of this group of receptors [13]. Since the amount of HER-2 expressed on cancer cells is elevated when compared to normal adult tissues [14], it has the potential of reducing the toxicity of HER-2-targeting drugs. High expression of HER-2 in tumors is often characterized by homogeneous and intense immunohistochemical staining [15], signifying that HER-2-targeted therapy would be directed to killing tumor cells in a cancer patient. Additionally, HER-2 overexpression is found in both the primary and metastatic sites [16], suggesting that HER-2 targeted therapy may be effective in all disease sites. HER-2 overexpression and amplification has been characterized in a number of gastric, esophageal, endometrial, uterine, ovarian and lung cancers [17–22]. Several studies have shown that many cancers are caused by dysregulated signaling of the HER family receptors [4], and targeting more than one of the receptors may produce greater antitumor effects.
HER-2 therapeutic strategies
Cancer remains the second-most frequent cause of death in the USA according to the American Cancer Society [301]. An estimated 226,870 new cases of invasive breast cancer are expected to occur among women in the USA during 2012 with an estimated 39,510 breast cancer deaths. Breast cancer ranks as the second-leading cause of death after lung cancer. The US FDA approved the use of trastuzumab (Herceptin®, Genentech, Inc., CA, USA) for use in breast cancer patients. Trastuzumab is a recombinant humanized monoclonal antibody that binds domain IV of the ECD of HER-2 receptor [23] and inhibits intracellular signaling [24]. Another monoclonal antibody, referred to as pertuzumab (2C4 or Omnitarg®, Genentech), is being tested in Phase III clinical trials. This recombinant humanized HER-2 antibody inhibits dimerization of HER-2 with EGFR and HER-3 [25] by sterically binding to domain II of the HER-2 ECD and thus can block signaling from HER-2–HER-3 and HER-2–EGFR heterodimers (Figure 1). Members of this new class of antibody therapeutics are known as dimerization inhibitors; they prevent signaling by HER family receptors and interfere with signaling in cells that express normal levels of HER-2 [26]. When both trastuzumab and pertuzumab are used together the survival of the BT474 breast cancer cell line is greatly inhibited, demonstrating increased apoptosis. This synergistic inhibition demonstrated that combining HER-2-targeting agents may be more effective than using a single HER-2 therapeutic strategy [27–29]. Several Phase II trials in breast, non-small-cell lung [30], ovarian [31] and prostate [32] cancers show the importance of combination therapy.
Figure 1. HER and VEGF family pathways in cancer.

The signaling pathways of VEGF and HER family members and the current drugs that target these proteins in cancer are shown. HER-2 can heterodimerize with any of the ligand-activated HER receptors (HER-1, HER-3 or HER-4) and this association leads to intracellular signaling via two major pathways, the MAPK pathway and the PI3K pathway, leading to proliferation, cell survival, metastasis and angiogenesis. On the other hand, VEGF can bind to its main receptor, VEGFR-2 (KDR), and this binding causes intracellular phosphorylation of the receptor, thereby stimulating the PI3K pathway and stimulating angiogenesis. The signaling pathways can be targeted extracellularly using humanized monoclonal antibodies, such as trastuzumab and pertuzumab (HER-2), cetuximab (EGF receptor), as well as bevacizumab or VEGF Trap (VEGF), which can prevent ligand binding and activation of the receptors or can directly block binding of an activated receptor to another. At the intracellular level, small-molecule inhibitors, such as sunitinib (VEGF), lapatinib (HER-1 and HER-2) and erlotinib (HER-2), can disrupt the phosphorylation sites and directly prevent activation of the PI3K or MAPK pathways.
P: Phosphate; VEGFR: VEGF receptor.
Targeting HER-1
HER-1 or EGFR plays an important role in many types of epithelial tumors, such as lung, breast, pancreatic, colorectal, prostate, bladder and ovarian tumors, and a natural role in development. HER-1 is involved in cell growth and differentiation; its levels of expression are very low but receptor levels are significantly higher in malignant cells, mainly due to increased proliferative potential as a result of unregulated signaling [33]. The involvement of EGFR in numerous epithelial tumors has led to the development of many drugs that target the receptor. The drugs developed so far can be classified into two broad groups; the tyrosine kinase inhibitors (TKI) and the humanized monoclonal antibodies. Small-molecule TKIs (Figure 1) were the first group of drugs developed to target HER-1 and include erlotinib, gefitinib and the dual (HER-1/HER-2) inhibitor lapatanib, which are all approved by the FDA [34]. They are able to prolong patient survival for months but there are significant side effects and cause for concern when considering the quality of life of most patients during treatment [35]. Despite FDA approval of these small molecules, their use is hampered by the problems of non-specificity that results into unacceptable safety profiles. Humanized monoclonal antibodies that target HER-1 include panitumumab and cetuximab. Panitumumab gained FDA approval in 2006 for the treatment of HER-1-expressing metastatic colon cancer [34]. Cetuximab was the first therapeutic agent that was approved to target EGFR owing to its high overexpression in many epithelial tumors [36]. Both small-molecule TKIs and cetuximab have been associated with side effects such as skin rash, severe diarrhea and, in some cases, serious hypersensitivity reactions [37]. Most of the FDA-approved agents targeting HER-1 (cetuximab, erlotonib and gefitinib) and HER-2 (trastuzumab, pertuzumab and lapatanib) have significant toxicities (Table 1) with unacceptable safety profiles in most patients [38].
Table 1.
Toxicities associated with HER-1, HER-2 and VEGF therapies.
| Drug | Target(s) | Adverse side effects |
|---|---|---|
| Trastuzumab | HER-2 | Cardiotoxicity and congestive heart failure |
| Pertuzumab | HER-2 | Neutropenia, diarrhea and skin rash |
| Cetuximab | HER-1 | Neuropathy and venous thrombosis |
| Panitumumab | HER-1 | Conjunctivitis and severe infusion reaction |
| Lapatinib | HER-1 and HER-2 | Gastroesophageal reflux disease, skin rash and diarrhea |
| Bevacizumab | VEGF | Neutropenia, thrombocytopenia and pulmonary toxicity |
| Sunitinib | VEGFR-1 and VEGFR-2 | Pleural effusion, thrombocytopenia and hepatic steatosis |
VEGFR: VEGF receptor.
Targeting of HER-3 & HER-4
It is becoming increasingly clear that the EGFRs form a network of inter-receptor activation in cancer. Combined upregulation of HER-3 or HER-4 with other HER family members significantly increases the dysregulated signaling of the receptors and leads to more aggressive forms of cancer with decreased patient survival. HER-3 and HER-4 therefore play a substantial supporting role in HER-1- or HER-2-positive cancers [39]. HER-3 and HER-4 have been implicated in the development of resistance to anticancer agents that target endocrine tumors [40]. HER-1 can dimerize with HER-3, providing a platform for increased signaling and increased metastatic potential, and the expression of HER-3 has been shown to provide an alternative pathway when HER-1 and HER-2 dimers are blocked.
Unlike HER-2, HER-3 lacks the ability to form homodimers. As a result, HER-3 signaling is entirely dependent on heterodimerization [41,42]. Interestingly, the heterodimer of HER-2–HER-3 constitutes the most potent receptor with respect to strength of interaction, ligand-induced tyrosine phosphorylation, and downstream signaling [43,44]. It forms a high-affinity heregulin receptor with kinase activity [45]. In addition, the HER-2–HER-3 heterodimer was found to mediate the most mitogenic signal in vitro [42,46–48].
All this illustrates that there is extensive crosstalk among the HER family receptors, indicating that interrupting signaling from more than one member maybe beneficial in limiting drug resistance. A two-in-one antibody that targets both HER-1 and HER-3 has been shown to produce superior antitumor effects in cancer cells that have high levels of both receptors [39]. Inhibition of HER-2 activity is always accompanied by upregulation of HER-3, which indicates that targeting both HER-2 and HER-3 simultaneously will block any alternative pathway [49]. Furthermore, in HER-2-dependent cells, blockage of HER-3 signaling results in decreased signaling and low proliferation rates [50].
Rationale for targeting HER-3
Unfortunately, efforts at targeting HER-3 have lagged behind owing, in part, to its impaired kinase activity. However, several recent studies have shown that HER-3 is frequently upregulated in cancers with EGFR or HER-2 overexpression and that it synergistically increases the tumorigenic potency of HER-2 [46,48]. HER-3 also has the highest binding affinity for PI3K when compared with all other HER proteins. As a result, HER-3 serves as a key activator in downstream signaling of PI3K, which, ultimately, leads to apoptosis resistance in a wide range of cancers [51–54]. The combination of HER-2 and HER-3 receptors may be critical in breast cancer growth and progression, and HER-3 may be a necessary partner for the oncogenic activity of HER-2 [55]. HER-3 may also provide a route for resistance to agents that target EGFR or HER-2 [56–60]. Recent evidence suggests that HER-3 contributes to escape from therapeutic suppression by several TKIs in breast cancer [56,59,60]. In fact, HER-3 expression or signaling is associated with resistance to HER-2 inhibitors in HER-2-amplified breast cancers [60], EGFR inhibitors in lung cancers [61], pertuzumab in ovarian cancers [62], antiestrogen therapies in ER-positive breast cancers [62–65], EGFR inhibitors in head and neck cancer [66] and hormone therapies in prostate cancers [67].
HER-2 vaccines & recent advances
Peptide cancer vaccine strategies focus mainly on eliciting a cellular antigen-specific T-cell response [68–70]. Disis et al. showed that HER-2 breast cancer patients exhibit pre-existing T- and B-cell immunity against HER-2 [71,72]. CD4+ T-cell peptide epitopes elicited immunity to HER-2 [73] in a clinical trial in humans [74]. Several HER-2 cytotoxic T lymphocyte (CTL) epitopes have been identified to date [75] and CD4+ T-cell responses have been shown to be essential for an antitumor response [76,77] with several MHC class II-presented epitopes [71,78–82]. An exploratory Phase I/II clinical trial vaccinated breast cancer patients with E75, a HLA-A2/HLA-A3-restricted HER-2/neu (HER-2) peptide, and GM-CSF [83]. Several vaccines are currently undergoing clinical trials, most of which are CD8+ T-cell-eliciting vaccines. AE37 is a promising CD4+ T-cell-eliciting HER-2/neu breast cancer vaccine currently in clinical trials [84]. Preclinical studies [85] suggest that GP2 is a clinically relevant HER-2/neu-derived peptide epitope and a recent Phase I clinical trial [86] demonstrated that GP2-based vaccines were safe and effective in stimulating peptide-specific immunity. HER-2 vaccine strategies include whole-cell expressing tumor antigens [87–89], proteins [90], DNA [91–93], HER-2/neu phage-display libraries [94] and epitope mimics [95]. HER-2 epitopes from the ECD coupled to carrier proteins to tetanus toxoid (TT) or keyhole limpet hemocyanin were able to elicit antibodies that inhibited tumor cell growth [96]. The peptide mimotopes strategy [97] has been used to elucidate HER-2 epitopes by biopanning of trastuzumab. Riemer et al. [98] showed that it is feasible to identify complex HER-2 epitopes by high-throughput screening and phage-display technology that corresponds to trastuzumab. Similar strategies for generating peptide mimotopes specific for an anti-HER-2 monoclonal antibody [99] were selected from a constrained random 12-mer peptide phage library that elicited antipeptide antibodies when coupled with keyhole limpet hemocyanin, however, no antibodies were produced when the multiple antigen peptide strategy was used. A virosomal HER-2 multipeptide vaccines has been shown to induce HER-2-specific immunity, and to be safe and well-tolerated in a Phase I trial [100].
VEGF family receptors & antiangiogenesis strategies
The growth of new blood vessels from pre-existing vasculature has been termed angiogenesis. Tumors are able to induce uncontrolled expression of proangiogenic factors resulting in an increase in angiogenesis and abnormal tumor vasculature [101]. Although disorganized, the tumor vasculature is essential for tumor growth and metastasis, and the idea that tumor growth was angiogenesis-dependent was first suggested by Folkman in the 1970s [102]; a decade later, his group started reporting on the first angiogenesis inhibitors.
The tumor angiogenic microenvironment
Interactions between cancer cells and their microenvironment are critical for the development and progression of solid tumors [103–108]. Tumor growth and metastasis are critically dependent on the development and/or remodeling of the microvasculature [109]. The transition between dormancy and active growth in tumorigenesis appears to be triggered by an ‘angiogenic switch’ [109,110]. This angiogenic switch has recently been documented in several forms of cancer [111]. VEGF constitutes one of the most proangiogenic factors known today [112]. In many different types of cancer, the expression of VEGF is elevated and most tumor cells secrete VEGF [113]. It has been well established that VEGF is a key promoter of metastasis [114]. VEGF is one of the most studied angiogenesis factors and can activate both mechanisms via VEGF receptor (VEGFR)-1 and/or VEGFR-2 signaling [35,115,116]. Endothelial cells respond via VEGFR-2 activation, while infiltrating cells, such as macrophages, are activated via VEGFR-1 signaling, which is also involved in the recruitment of endothelial progenitor cells in neovascularization [117–120].
VEGF & VEGFR
VEGF is a homodimeric glycoprotein (34–42 kDa) that exhibits several isoforms arising from splice variants [112,113,121–123]. The VEGF protein family is made up of several different proteins including VEGF-A, VEGF-B, VEGF-C, VEGF-D and PlGF. The most important of these proteins is VEGF-A, which plays a major role in stimulating angiogenesis by binding to one of its receptors, VEGFR-2 [124]. The functions of the other proteins in this family are unrelated to angiogenesis and these proteins are able to bind only to VEGFR-1, while VEGF-A binds to both VEGFR-1 and VEGFR-2. There are many splice variants or isoforms of VEGF-A, which include VEGF-A [121], VEGF-A [125], and VEGF-A [126], which is the matrix-bound isoform expressed in pathological angiogenesis [127]. The VEGFR signaling mechanism comprises receptor dimerization, activation of the tyrosine kinase and creation of docking sites for signaling VEGFRs. The three known tyrosine kinase receptors that bind VEGF are Flt-1 (VEGFR-1), KDR (VEGFR-2, Flk-1) and Flt-4 (VEGFR-3). Although VEGFR-1 binds VEGF with higher affinity than VEGFR-2, the latter is considered the most biologically relevant receptor in solid tumor angiogenesis. VEGFR-2 is a tyrosine kinase receptor and is the most important VEGFR in angiogenesis activation. It can be overexpressed in breast, kidney, pancreas and GI tract cancer cells. This receptor is thus considered an important target for the development of kinase inhibitors aimed at targeting angiogenesis. Overexpression of VEGF also occurs in many different types of cancer and, thus, VEGF and its receptors, VEGFR-1 and VEGFR-2, are considered as prime targets for antiangiogenic intervention [128–130]. Many angiogenic inhibitors have been developed, mainly focused on the VEGF pathway, since its inhibition is correlated with suppression of tumor growth and angiogenesis [131].
Antiangiogenesis strategies
Some drugs have been successfully implemented in some specific areas of cancer treatment but their long-term side effects still remain a cause for concern, thus the need for better drugs [132]. Tumor angiogenesis is, today, one of the most attractive areas in clinical oncology [133], leading to the development of many antiangiogenic drugs (e.g., monoclonal antibodies [mAbs] and TKIs) that target VEGF and its receptors (see Figure 1).
Antibodies
Antibodies generally have neutralizing and inhibiting functions in addition to their ability to cause antibody-dependent cell-mediated cytotoxicity (ADCC) [134]. Antibodies against VEGF were the first to be explored and, after decades of studies, bevacizumab (Avastin®), a humanized monoclonal antibody developed by Genentech, was approved by the FDA as a drug to be used, as first- and second-line therapy in colorectal cancer in 2004 and breast cancer in 2008 [135]. This is so far the most clinically successful antiangiogenic inhibitor; however, concerns related to the long-term side effects of its use and development of resistance had already been raised. The safety and efficacy of Avastin in breast cancers was found to be inadequate, which resulted in removal from the market by the FDA in 2011. Several antibodies against VEGFR-2 are still being generated. The idea is that targeting only VEGFR-2 will reduce the side effects associated with anti-VEGF antibodies, since only the angiogenesis-specific pathway will be inhibited, while the VEGF–VEGFR-1 pathway will function normally.
Small-molecule TKIs
TKIs mainly function by binding to the ATP binding site or by binding to the substrate binding site, thereby preventing the enzyme from binding [136]. Currently, there are two FDA-approved TKIs that target VEGFR-2: sorafenib (Nexavar®) and sunitinib (Sutent®), commercialized by Bayer (Leverkusen, Germany) and Pfizer (NY, USA), respectively. Many other kinase inhibitors targeting VEGFR-2 are still in development or in ongoing clinical trials [137]. Sunitinib and sorafenib are both orally administered TKIs, approved by the FDA for advanced renal cancer and gastrointestinal stromal tumors [138,139]. Many drawbacks are associated with these inhibitors; they easily develop resistance by mutation and/or overexpression of the receptor, they are nonspecific because they crossreact with other kinase receptors in the body and they also exhibit greater cytotoxicity effects [136]. Both sunitinib and sorafenib have been associated with serious side effects such as hypertension, stomatitis, neutropenia and thrombocytopenia [140].
Antagonist VEGF polypeptides
Another approach that is used to block VEGF and its receptor interaction is the use of polypeptides. One such example is VEGF Trap (Aflibercept®), produced by Regeneron Pharmaceuticals (NY, USA). VEGF Trap is a recombinant fusion protein that combines VEGF binding domains of VEGFR-1 and VEGFR-2 with an Fc segment of IgG1 [141]. It is currently being evaluated in a Phase II clinical trial as a treatment combination in cancer patients. The combination regimen is 2 mg/kg intravenously of VEGF Trap given every 3 weeks in combination with 7 mg/m2 of 250 cc docetaxel. Although VEGF Trap demonstrated high affinity and specificity for both VEGF receptors, it has been suggested that it has the same problems as VEGF-neutralizing antibodies in long-term treatment [142].
An innovative peptide approach to cancer treatment
The main efforts of our laboratory in the past two decades have been the development of unique peptide vaccines and peptidomimetic therapeutic approaches (Figure 2) for targeting viral, bacterial and cancer antigens. The novelty of our approach resides in a hypothesis-driven basic research in vaccinology with an incremental approach involving the elucidation of several basic immunological and structural concepts that could eventually be translated to the clinic for the benefit of cancer patients. These innovations include:
Figure 2. Combination of HER-2 vaccines and VEGF peptide mimics.

HER-2 vaccines and VEGF peptide inhibitors developed in our laboratories to inhibit signaling pathways. Antibodies elicited by immunization with MVF-HER-2 (266–296) vaccine bind to domain II of HER-2 and, similarly, anti-MVF-HER-2 (597–626) binds domain IV of HER-2, providing dual inhibition of homo/heterodimerization, and consequently downstream signaling, shutting down the PI3K and MAPK pathways, thereby preventing cancer growth and metastasis. On the other hand, VEGF peptide mimics P3 and P4, which are designed to directly block VEGF binding to VEGFR-2, inhibit intracellular phosphorylation of the tyrosine kinase domain, which reduces angiogenesis.
EGFR: EGF receptor; P: Phosphate; VEGFR: VEGF receptor.
The ability to predict biologically active immune epitopes and confirm their properties in vitro and in vivo;
The engineering of unique B-cell epitope peptides that can recapitulate the exquisite native structure of the tumor antigen [143–145];
The design of uniquely inhibitory peptide mimics that can block receptor–ligand interactions [146–148];
The idea of combining the B-cell epitopes into chimeric constructs that incorporate a ‘promiscuous’ T-cell activating species [149]. Vaccination with these novel HER-2 chimeric immunogens results in the production of highly efficacious native-like antipeptide antibodies that can delay, prevent and/or eradicate tumor growth and metastasis with increased efficacy and little or no toxicity [150–153];
In order to complement and enhance our vaccine approach, we have been developing ‘non-immunogenic’ peptide therapeutics that target both HER-2 and VEGF pathways [154,155]. This strategy involves the design and synthesis of conformational VEGF peptide mimics that are aimed at disrupting the receptor–ligand interactions with enhanced stability and efficacy [156];
The concept of ‘tritherapy’, which is the combination of immunotherapy, angiotherapy and metronomic chemotherapeutic drugs.
HER-2 peptide design strategy
There are several strategies to design immunogenic B-cell epitopes of unknown proteins (reviewed by Kaumaya et al. [143]). First, we have pioneered a unique approach of identifying antigenic epitopes on the surface of proteins whose 3D structure and whose antigen–antibody complex is unknown. It relies on predicting epitopes using correlates of antigenicity such as hydrophilicity, flexibility, hydrophobic index, amphiphilicity and secondary structural propensities. Computer algorithms are available to predict potential B-cell epitopes and we have successfully applied this technology to several targets including HTLV-1, HER-2, VEGF and others. For a large protein, several antigenic epitopes can be prioritized and these peptides are then synthesized by solid-phase peptide chemistry as chimeric immunogens with an appropriate T-cell epitope.
Design of conformation-dependent antigenic determinants
Second, if the 3D structure of the antigen is unknown, prediction of the secondary structures by several criteria, such as α-helices, β-turn and sheet, and loop regions, can be made to aid the design. In that respect, knowledge of protein folding, structure and dynamics can be used to design the B-cell epitopes to fold into stable supersecondary structures (αα, αβ, βαβ and β-turns and loops) [143–146]. When the crystallographic structures of the antigen–antibody complex are known, the engineering of a selective B-cell epitope to mimic the structure of the immunogenic binding site on the tumor antigen is greatly facilitated.
Active immunotherapy with chimeric B-cell peptide & promiscuous T-cell epitopes
The third aspect is to construct a chimeric peptide containing a promiscuous T-cell epitope that could be used as an immunogen to elicit high-titered and high-affinity antibodies that are able to recognize the protein. We have pioneered a strategy that is accomplished by selecting an appropriate T-cell epitope. Several of these promiscuous T-cell epitopes are known, and in our previous in-depth studies [149,157] we have found the measles virus fusion (MVF) protein and the TT epitopes to be the most efficacious ones. The most important consideration in the successful design of a peptide vaccine is that the B-cell epitope must be covalently linked to the ‘helper’ T-cell epitope at either the N- or C-terminus (Figure 3). In our vaccine approach, we preferentially used the T-cell epitope at the N-terminus [143].
Figure 3. Chimeric B- and T-cell epitopes.
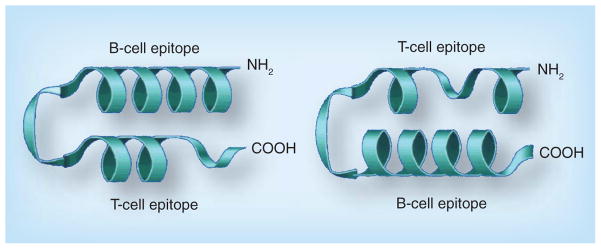
B- and T-cell epitopes are colinearly synthesized with a GPSL flexible linker. The linker is flexible, allowing the two epitopes to fold or adopt different conformations independent of each other.
The size of the B-cell epitope should minimally be approximately 18 amino acid residues (~900 Å2), the size of an antigen–antibody binding region, to ensure the peptide propensity to adopt a defined structure. The helper T-cell should ideally be a promiscuous epitope (~18 residues), the linker should be approximately four residues and the final construct should consist of between 40 and 60 amino acid residues. In some cases, the B-cell epitope can be greater than 18 residues to accommodate a larger binding site, as demonstrated by the HER-2–trastzumab or the HER-2–pertuzumab interface. The chimeric B- and T-cell epitope strategy can fully stimulate the immune system to produce high-affinity antibodies that can specifically target cancer cells and, most importantly, establish an immunological memory in order to prevent cancer relapse.
Advantages of chimeric peptides as vaccines
Traditionally, peptide sequences are coupled to larger carrier proteins, such as bovine serum albumin, TT or keyhole limpet hemocyanin, in order to induce immunogenicity. This approach is fraught with difficulties as the resulting conjugate is subject to processing by the immune cells and the resulting B-cell epitope may not represent the exact epitope identity or configuration, resulting in low-affinity antibodies. Additionally, the coupling procedures are not an exact science in the sense that they cannot be duplicated from batch to batch, resulting in an unpredictable immune response. Other problems are epitope suppression by the carrier protein and poorly characterized constructs due to ambiguity during the chemical coupling reaction. A strategy that involves B- and helper T-cell epitopes is MHC-restricted by the fact that T-helper epitopes are recognized by only a few MHC class II alleles at most. We overcame those problems by proposing the use of ‘universal’ or promiscuous T-helper epitopes [158,159] that bind to several MHC haplotypes [149,158,159].
The chimeric peptide is presented intact to the immune cells without processing, yielding high-titered and high-affinity protective polyclonal antibodies. This strategy avoids the proteolysis by APC, normally associated with peptides coupled to large carrier proteins in which the authenticity of the epitopes is destroyed resulting in a poorly immunogenic vaccine. Unlike antigens presented to MHC class I, which are restricted to peptides 8–10-mers, the ends of the groove of MHC class II are open, allowing larger peptides to bind. This is the major advantage over other strategies that focus on generating antibodies in vaccine development. Other active immunization regimens, such as activation of CTLs for many different types of cancer, have been exclusively and extensively used without much success so far, but there is hope that newer methods of delivery and adjuvants may lead to improved vaccines activating the cellular arm of the immune system.
Enhancing immunogenicity of peptide vaccines
An ideal peptide vaccine should consist of an appropriate tumor antigen B-cell epitope or CTL epitope covalently linked to a universally immunogenic T-helper epitope. In addition, the peptide vaccine must be formulated with an adjuvant or cytokines, which are essential in the development of a good immune response. Adjuvants are usually defined as compounds that increase and/or moderate the intrinsic immunogenicity of an antigen. Generally speaking, adjuvants function in three basic ways: they cause the slow release of an antigen; they modulate the immune response; and they increase the presentation of the antigen to immune cells such as antigen-presenting cells [160]. Poorly soluble aluminum salts (aluminum phosphate, aluminum hydroxide and alum [KAl(SO4)2·12H2O]), as well as calcium phosphate and AS04 (FENDrix®, GlaxoSmithKline, Brentford, UK), are the only vaccine adjuvants currently licensed in the USA. While generally considered quite safe, there are several areas in which these adjuvants are inadequate. Despite the wide use and acceptance of alum, there are a series of problems. Adjuvants that can stimulate Th1 cytokine-dependent IgG2a and IgG2b antibodies have been developed. Oil emulsions, such as MF59, which is composed of squalene, polyoxyethylene and muramyl tripeptide, have been shown to dominantly induce IgG2a production via Th1 immune response [161]. QS-21, which is a purified saponin, also induces IgG2a responses via cell-mediated immunity [162]. Another group of adjuvants that are microbial-derived has also been shown to activate Th1 responses and cause IgG2 production. A TLR-2 ligand known as macrophage-activating protein-2, which is a purified derivative from mycoplasma, causes IgG2a production [163], while virosomes, which are stabilized lipid complexes with viral proteins, can also activate IgG2a and IgA responses [164]. A bacterial DNA CpG, which is also a TLR-9 ligand, can also cause Ig2a production when delivered with TT [165]. Some cytokines that are used as adjuvants include IL-2, IL-6 and IL-12, which are all known to stimulate T-cell production and cause the production of IgG2a antibodies [162,166].
In most of our work, dating back to 2000, we have adopted the use of muramyl dipeptide (N-acetyl-glucosamine-3-yl-acetyl L-alanyl-D-isoglutamine [nor-MDP]) as an adjuvant as it was used in a WHO vaccine. Nor-MDP, a ubiquitous constituent of bacterial cell walls, is recognized by APCs and activates many different cell types, including macrophages, leukocytes, mastocytes, endothelial cells and fibroblasts, inducing the secretion of cytokines such as IL-1, B-cell growth factor and fibroblast-activating factor. The muramyl dipeptide, when emulsified in a squalene-in-water emulsion ISA 720 (SEPPIC, Paris, France), preferentially activates the humoral arm of the immune system.
Advantages of active immunotherapy
The final puzzle is solved by vaccination with the chimeric immunogen that will elicit endogenous production of high-titer anti-HER-2 polyclonal antibodies. Such polyclonal antibodies may effectively inhibit function of the target molecule without the risk of tumor escape, as may occur with a mAb directed at a single epitope. The goal of the strategy is active specific immunotherapy providing prolonged therapeutic benefit by eliciting antibodies that could delay, prevent and/or eradicate tumor growth and metastasis with potentially increased efficacy and the generation of immunologic memory. Additional advantages include the avoidance of the need for multiple and expensive infusions that usually lead to severe toxicities.
The advantage of active immunotherapy over passive immunotherapy with mAbs, such as trastuzumab, pertuzumab and bevacizumab, is a fundamental aspect of our work. The half-life of IgG, administered intravenously, can range from 5 to 21 days. Thus, repeated treatments with trastuzumab are necessary – patients typically receive the mAb every 3 weeks. The repeated treatment with trastuzumab raises the cost of passive immunotherapy with this mAb to approximately US$140,000 per year.
Development of a combination of two HER-2/neu B-cell epitopes as potential vaccine candidates
We identified (reviewed by Kaumaya et al. [167]) several B-cell epitopes of HER-2 ECD using antigenicity algorithms. These peptide B-cell sequences were assembled by solid-phase peptide synthesis together with a MVF protein (amino acids 288–302) promiscuous T-cell epitope. The vaccination protocol consisted of emulsifying the peptide with nor-MDP adjuvant in ISA 720 (oil-in-water mixture). Outbred rabbits immunized individually with the eight different chimeric MVF-HER-2 peptide vaccine constructs, elicited in each case exceptionally high antibody titers that bind the native HER-2 protein, as assessed by immunoprecipitation, flow cytometry and indirect ELISA. We extended our in vivo studies in transgenic mice and demonstrated that the 628–647 epitope had the highest activity by eliciting antibodies were able to cause ADCC, as measured by lyzing overexpressing cell lines BT474 and SKBR3. More importantly, the vaccine was effective in preventing mammary tumors in neu transgenic mice [125,150].
Epitope specificity
For vaccines eliciting a biologically relevant immune response, the specificity of each epitope is crucial. We extended our studies to all seven identified HER-2 epitopes, and characterized their biological activities in vitro and in vivo. In so doing, we were able to demonstrate that antipeptide antibodies against the 316–339 epitope bound the native protein – as measured by indirect ELISA, flow cytometry and immunoprecipitation – but also caused ADCC and inhibition of phosphorylation. We next used a combination of the two best epitopes to determine the best candidate for translating our vaccine to the clinic. We found that the combination vaccine regimen of 316–339 and 628–647 elicited the best titers and also caused the highest receptor downmodulation, similar to that produced by the control antibody HER-2 mAb L26. Combination vaccines (all three possible combinations) were able to induce higher levels of IFN-γ in the presence of effector human peripheral blood mononuclear cells compared with single-epitope vaccines. We determined the efficacy of our vaccine by first immunizing BALB/c mice three times with the combination peptides and challenging with syngeneic tumor cells transfected with human HER-2 (RENCA/lacZ/HER-2), and we enumerated the pulmonary metastases. We observed the greatest reduction (>45%) in the number of pulmonary metastases with the peptide 316–339 alone and the combination of 316–339 and 628–647 in the presence of IL-12.
The most effective combination vaccine was found to be HER-2 sequences 316–339 and 628–647 [150], identified through our peptide strategy. It turns out that these two sequences overlapped with the pertuzumab and trastuzumab binding sites of HER-2. We initiated a Phase I clinical trial with a combination of these two peptides in 2002 at the Ohio State University Comprehensive Cancer Center (OH, USA). Recently, several combination strategies for pertuzumab and trastuzumab with different chemotherapeutic agents have been conducted in clinical trials and a recent randomized Phase III clinical trial with docetaxel (CLEOPATRA, NCT00567190 [302]) showed promise [168].
From bench to clinic
A National Cancer Institute (NCI)-funded (CA84356), OSU Cancer IRB-approved (2001C0108) and FDA-approved (BB-IND-9803) Phase I clinical trial with a first-generation combination of two HER-2 chimeric B-cell MVF 316–339 epitopes and MVF 628–647 [169–171] emulsified with nor-MDP as adjuvant and ISA 720 vehicle was recently successfully completed at the James Cancer Hospital (OH, USA). The goals of the trial were to determine the safety and toxicity of the vaccine, as well as the maximum tolerated dose. Patients with confirmed metastatic and/or recurrent solid tumors were screened and only those meeting all the eligibility criteria were accrued. The trial consisted of four cohorts with increasing doses of peptide vaccines ranging from 0.5 to 3.0 mg. Eligible patients received three inoculations 3 weeks apart. A total of 24 patients (six per cohort) received their vaccinations at the intended dose level. The patients had different types of cancer ranging from breast (five), colon (three), squamous cell carcinoma (one), ovarian (five), endometrial (two), adrenal (one), pancreas (one), rectal (two), leiomyosarcoma (one), gastrointestinal stromal tumor (one), cervical (one) and non-small-cell lung cancer (one). HER-2 expression was not required for enrolment since the primary aim of the Phase I study was to evaluate toxicity and immunogenicity. Nonetheless, all patients were evaluated for HER-2 status and this was performed by FISH or immunohistochemistry.
All patients had an immune response to the vaccine eliciting HER-2-specific antibodies. These antibodies inhibited signal transduction pathways in vitro, as exhibited by inhibition of proliferation of HER-2-expressing cell lines and phosphorylation of the HER-2 protein. The highest dose level of 1.5 mg of each component of the combination vaccine was established as the maximum tolerated dose. This corresponds to 3.0 mg of the combination vaccine. HER-2-specific antibodies increased with larger doses of the vaccine from 0.25 to 1.5 mg. The vaccine was well tolerated and the maximum tolerated dose was identified as the highest dose level, 1.5 mg of each peptide. Additionally, patients produced antibodies of the IgG isotype against the vaccine, and patients receiving the highest dose level had a statistically significant increase in the IgG antibody response compared with patients receiving the lowest dose level. Six patients had clinical benefit: four patients (adrenal, colon, ovarian and squamous cell carcinoma of unknown primary) were deemed to have stable disease; two patients (endometrial and ovarian cancer) had partial responses; and 11 patients had progressive disease. Patients showing stable disease received 6-month booster immunization and one patient received a 20-month booster immunization.
These results demonstrate that tumor antigen-specific immune responses are reproducibly induced in stage IV cancer patients; that is, tolerance can be reversed, without the induction of serious adverse events or autoimmune disorders. Additionally, several patients received booster immunization 6 months after the initial immunization and one patient received a 2-year booster. The combination vaccines elicited HER-2-specific antibody responses in a number of patients (62.5%) and preliminary evidence of clinical activity in several patients was detected. There were no serious adverse events reported for this trial, and the vaccination did not induce an autoimmune response or any cardiotoxic events. In conclusion, we have successfully translated our work into a Phase I clinical trial where we showed that our vaccine was immunogenic in more than 65% of the patients and six out of 24 showed clinical responses to the vaccine [172].
Developing the second-generation HER-2 vaccines
In our continuing efforts to rationally design effective vaccines, we have taken advantage of information garnered from antigen–antibody complexes of HER-2 protein with clinically important antibodies. As such, we can design efficacious functional vaccines that mimic the 3D epitopes.
Structural studies of HER-2 with pertuzumab & trastuzumab
Numerous x-ray crystallographic structural studies revealing how the HER family initiates signal transduction have been published [169,170]. The structure of soluble HER-2–trastuzumab Fab complex shows that the trastuzumab binding region is located on the C-terminus of the HER-2 ECD domain IV and this complex buries 1350 Å2 of the HER-2 surface with three loops residues 579–583, 615–625 and 592–595. On the other hand, the crystal structure of the HER-2 ECD bound to pertuzumab shows a different binding epitope focused on domain II residues 266–333 [171]. This structure provides a model in which pertuzumab sterically interferes with HER-2 dimerizing with other members of the HER family.
Our first-generation HER-2 peptide vaccines (316–339 and 628–647) were identified prior to the publications of the crystal structure of the HER-2 ECD [169–171] using computer-aided analysis. The structures of the HER-2–trastuzumab and HER-2–pertuzumab complexes have led to new understandings of the mechanistic and biological activities of HER-2 antibodies as well as the process of ligand-induced receptor dimerization, which, in turn, has empowered us to rationally design more effective HER-2 conformational epitopes; the trastuzumab-binding epitope (597–626) and the pertuzumab-binding epitope (266–296).
Design & evaluation of novel pertuzumab-binding conformational B-cell epitopes
The crystal structure of pertuzumab bound to the ECD of HER-2/neu revealed the details of interacting region of residues 266–333 (Figure 4) [171]. We designed and studied the important binding sequences spanning residues 266–296, 298–333 and 315–333 in order to define the most biologically relevant conformational epitope to mimic the pertuzumab-binding conformational region for effective vaccination (Table 2).
Figure 4. Binding interface of pertuzumab and HER-2.
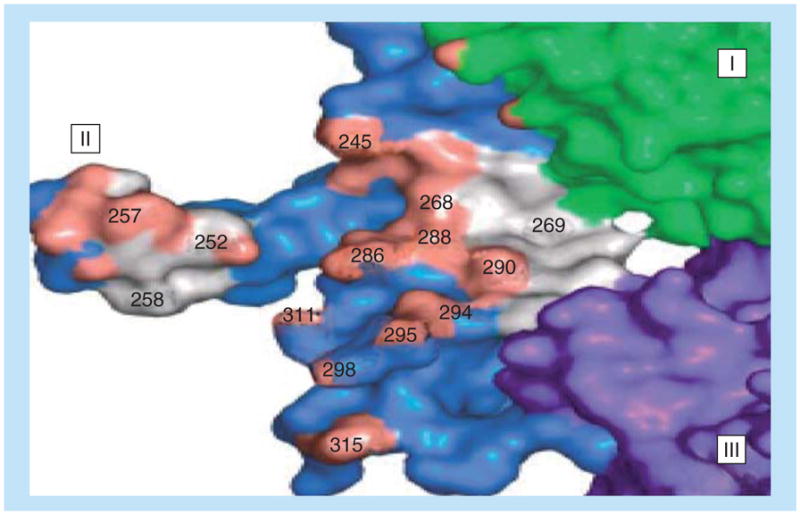
Important binding residues at the interface of pertuzumab and HER-2 show the key amino acid residues that are critical for binding. (I–III) are the HER-2 domains.
Table 2.
HER-2 sequences mimicking the pertuzumab region.
| Designation | Peptide | Sequence | M.Wt. |
|---|---|---|---|
| MVF 266 Cyc. | 266–296, one disulfide bond | H2N-KLLSLIKGVIVHRLEGVE-GPSL-LHCPALVTYNTDTFESMPNPEGRYTFGASCV-COOH | 5423 |
| MVF 298 Cyc. | 298–333, two disulfide bonds | H2N-KLLSLIKGVIVHRLEGVE-GPSL-ACPYNYLSTDVGSCTLVCPLHNQEVTAEDGTQRCEK-COOH | 6297 |
| MVF 315 Cyc. | 315–333, one disulfide bond | H2N-KLLSLIKGVIVHRLEGVE-GPSL-CPLHNQEVTAEDGTQRCEK-COOH | 4493 |
Cyc.: Cyclized; M.Wt.: Molecular weight.
We reported on the extensive in vitro and in vivo results obtained by the various constructs having complex disulfide bonds, as well as the noncyclized (non-Cyc.) linear peptides [152]. We vaccinated both mice and rabbits in order to elucidate the immunogenicity of the cyclized (Cyc.) and non-Cyc. constructs. We found that each of the epitopes was able to elicit high-affinity, high-titer antibody responses that bound the native HER-2/neu receptor. Additionally, only three of the six putative constructs – 266–296 (Cyc.), 266–296 (non-Cyc.) and 315–333 (Cyc.) – were able to reduce the phosphorylation of the HER-2/neu tyrosine kinase domain and to mediate ADCC. Additionally, we were able to show that epitope 266–296 was able to suppress cellular proliferation in heregulin-stimulated MCF-7 cells.
Our experiments in two transplantable tumor mice models (Balb/c and FVB/n) show that only the 266–296 engineered epitopes had statistically reduced tumor onset. We also demonstrated that there was significant reduction in tumor development in Balb-neuT and VEGF+/−Neu2–5+/− transgenic mouse tumor models. In conclusion, we were able to show that the epitope spanning sequences 266–296 was by far the best candidate to elicit an efficacious antitumor immune response. This signifies that the 266–296 peptide vaccine could duplicate the antitumor effects of pertuzumab in vivo without the concomitant harmful side effects associated with mAb therapy.
Design & evaluation of novel trastuzumab-binding conformational B-cell epitopes
The 3D structure of the complex (Figure 5) between human HER-2 and trastuzumab revealed that the region of HER-2 spanning residues 563–626 of the antigen-binding domain harbors a complex disulfide bonding pattern [169,170]. In order to minimally dissect the interacting region of HER-2 binding domain, four synthetic peptides with different levels of structural flexibility were designed and synthesized. The interacting loops in subdomain IV comprise residues in loop 1: 579–583 (two disulfide pairings between C563 and C576, and between C567 and C584); loop 2: 592–595 (cysteine disulfide pairing between C587 and C596); and loop 3: 615–625 (cysteine disulfide between C600 and C623). The chimeric peptides in Table 3 were successfully synthesized, purified and characterized. All conformation-restricted peptides were able to bind to trastuzumab with 563–598 and 597–626 showing higher reactivity.
Figure 5. Binding interface of trastuzumab to HER-2.
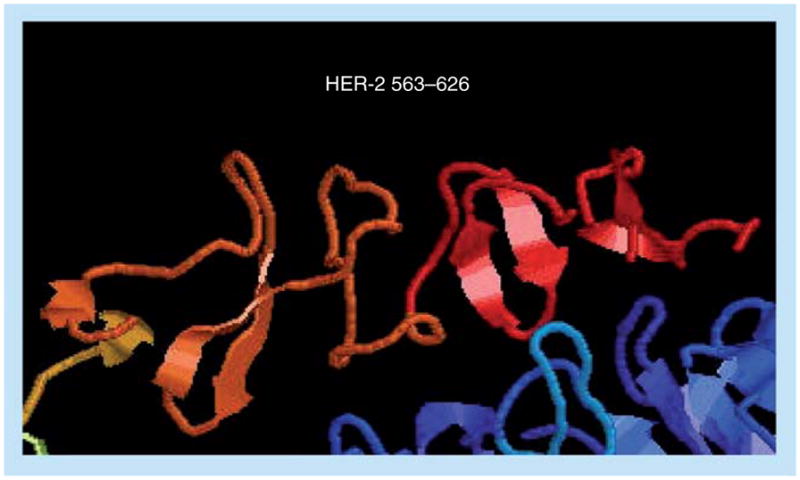
The crystal structure of HER-2 (red) in contact with trastuzumab (blue). The three loops that make direct contact with trastuzumab are clearly shown, depicting important binding residues.
Table 3.
HER-2 sequences mimicking trastuzumab-like region.
| Designation | Peptide | Sequence |
|---|---|---|
| MVF 563 Cyc. | 563–598 peptide, three disulfide bonds | H2N-KLLSLIKGVIVHRLEGVE-GPSL-CHPECQPQNGSVTCFGPEADQCVACAHYKDPPFCVA-COOH |
| MVF 585 Cyc. | 585–598 peptide, one disulfide bond | H2N-KLLSLIKGVIVHRLEGVE-GPSL-VACAHYKDPPFCVA-COOH |
| MVF 597 Cyc. | 597–626 peptide, one disulfide bond | H2N-KLLSLIKGVIVHRLEGVE-GPSL-VARCPSGVKPDLSYMPIWKFPDEEGACQPL |
| MVF 613 | 613–626 peptide | H2N-KLLSLIKGVIVHRLEGVE-GPSL-IWKFPDEEGACQPL |
Cyc.: Cyclized.
All four peptide sequences inhibited tumor cell proliferation. Although all four sequences were immunogenic in FVB/N mice, only anti-597–626 and anti-613–626 were able to bind HER-2. We further examined the immunogenicity of the 597–626 epitope in outbred rabbits and high-titered antibodies were elicited that recognized HER-2 at the HER-2–trastuzumab interface, inhibited proliferation of HER-2-expressing breast cancer cells in vitro and mediated ADCC. Antibodies against the 597–626 construct were able to mediate both direct and indirect mechanisms of antitumor activity against HER-2 in vitro.
We also demonstrated in transgenic BALB-neuT mice that immunization with the 597–626 epitope significantly reduced tumor burden. In conclusion, our results imply that the 597–626 peptide could be used as a vaccine for HER-2-overexpressing cancers since the resulting antibodies show similar biological activities to trastuzumab.
New clinical trial peptides in the clinic
We are currently conducting an FDA-approved (investigational new drug 14633) and NCI-funded trial at the Ohio State University James Cancer Hospital (2010C0075; OSU 09138) entitled ‘Phase I Active Immunotherapy Trial with a Combination of Two Chimeric HER-2 B Cell Peptide Vaccines emuslified in ISA 720 and nor-MDP in Patients with Advanced Solid Tumors’ (NCT01376505) [203]. The combination peptide vaccine from the trastuzumab-like (MVF-HER-2 [597–626]) and pertuzumab-like (MVF-HER-2 [266–296]) binding sites have demonstrated efficacy in preclinical studies [152,153]. The vaccine targets two different epitopes of the HER-2 ECD (II and IV), making it beneficial to both HER-2-positive and HER-2-negative (EGFR overexpressing) cancer patients. The dose-escalation trial was opened for accrual in July 2011; we have completed two cohorts and the trial is still open for accrual (cohorts 3 and 4).
The main objectives of this study will be to perform an early-phase clinical trial assessing safety and clinical toxicity of immunization with two HER-2 multivalent vaccines in patients with advanced solid tumors, as well as to establish a biologically optimum dose of combination vaccines with nor-MDP as adjuvant emulsified in ISA 720. We will also evaluate whether the combination of HER-2 epitopes shows therapeutic benefit, provides synergistic and/or additive effects and enumerate mechanisms of action. Once the biologically optimum dose has been established, the protocol calls for extending the trial in a Phase IIb efficacy trial in two indications. After completion of the Phase I trial (four cohorts, 24 patients), we will decide, based on evaluation of the results of the trial, to select two appropriate indications to extend the trial.
VEGFR & the design of VEGF inhibitors
Many studies have shown that peptides are the preferred candidates for drug and vaccine development owing to an improvement in engineering techniques and an increased knowledge of the crystal structure of proteins that play a role in diseases such as cancer [173]. Protein–protein interactions are very important in cancer cell growth and development. Thus, designing smart peptide mimics aimed at disrupting these interactions is an innovative strategy to target growth factor signaling pathways.
Peptides & peptidomimetics
Peptides are small protein-like chains of amino acids and a peptide mimic is a peptide or a peptide-like molecule that mimics one portion of the entire protein, which is usually a binding or active site of an enzyme [174]. Peptides that can block receptor–ligand interaction can be obtained by screening a combinatorial library of compounds or by the use of structure-based design. The main goal is to maintain the conformational integrity and flexibility of the bioactive surface for cooperative binding to a given receptor [175]. Peptide mimics offer the benefits of being water soluble, nonimmunogenic and able to easily cross tissue barriers [176]. One of the major drawbacks of using peptides as therapy is their high susceptibility to proteosomal degradation [177]. This obstacle can be overcome with the use of pseudo and modified peptides. The retro-inverso (RI) modification is a reversal of the peptide backbone by inverting the amino acid sequence, as well as reversal of the amino acid chirality by utilizing D-amino acids (Figure 6). The resulting product is a topographical equivalent of the parent peptide with the amino side chain in similar orientation. Because RI peptides are synthesized with D-amino acids and proteases usually recognize L-amino acids, they should be resistant to proteosomal degradation, and, therefore, will increase the bioavailability of the peptidomimetic in vivo [148,177,178]. By reversing the direction of the peptide backbone as well as inverting the chirality of the amino acid (from L to D), the resultant peptide mirrors both the structure of the protein as well as its side-chain conformation, adopting a stable ‘mirror image’ of the corresponding 3D structure of its parent all-L protein.
Figure 6. Design of retro-inverso peptide mimics.

The schematics show the strategy used to design RI peptides. The side-chain conformation structure (mirror image) between the parent peptide and the RI peptide is maintained when D-amino acids are used and the synthesis is carried out in the reverse direction.
RI: Retro-inverso.
VEGF peptide mimics
Unlike other growth receptors, which only trigger mitogen pathways, VEGF signaling via VEGFR-2 activates pathways leading to survival, 3D tube organization and vascular permeability [179]. VEGF–VEGFR-2 interaction is therefore very important for angiogenesis and blocking this interaction is the most attractive model in the development of antiangiogenic drugs [180]. Our strategy to develop angiogenic inhibitors involves the synthesis of conformational peptides that can mimic the VEGF binding region to VEGFR-2, which will inhibit angiogenesis by competing with VEGF for binding to its receptor.
Design & synthesis of VEGF peptide mimics
The interface between VEGF and VEGFR-2 has been mapped by alanine scanning, which showed that the binding site is localized at the loop between β5 and β6 [181]. This loop is within the region containing the biologically active site (also called ‘hot spot’) in the VEGF protein, which includes monoclonal epitope binding (Avastin, G-6 and B20-4) and VEGFR binding. Both Muller et al. [182,183], who analyzed the contact residues on both sides of the interface of the complex between VEGF and the Fab fragment of a humanized antibody, and Zilberberg et al. [184], who analyzed the VEGF sequence interacting with VEGFR-2 have concluded that residues spanning sequence 79–93 are important. Thus, we selected the VEGF sequence 102–122 (detailed in Figure 7) to design our VEGF peptide mimic; this region corresponds to sequence 76–96 in the VEGF crystal structure and includes the entire loop region between β5 and β6 [185].
Figure 7. VEGF peptide mimics.
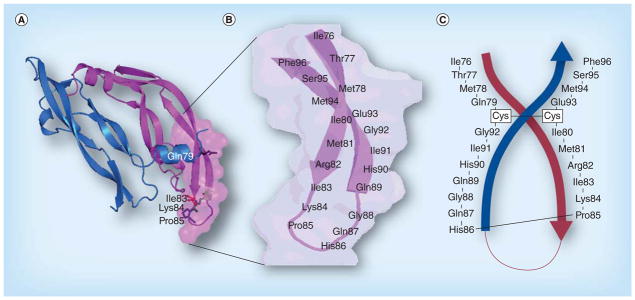
(A) The structure of VEGF shows the critical amino acid residues that are involved in binding of VEGF to its receptor VEGFR-2. (B) The anti-parallel β-sheet structure of VEGF 76–96 connected by loop residues 83–89. (C) Shows the engineered VEGF peptide mimic with two artificial cysteine residues inserted to form a cyclized anti-parallel β-structure. The complex peptide is synthesized from Phe96 to Glu93 followed by Cys and the synthesis continues with Ile80 through Gly92. The other Cys is placed between Gly92 and Gly79 and ends with Ile76.
Our strategy for creating the conformational cyclic peptide VEGF-P3 (Cyc.), which displays an antiparallel β-sheet (shown in Table 4 and Figure 7) was to introduce two artificial cysteine residues between residues Gln79 and Gly92, and also between residues Ile80 and Glu93. The VEGF-P3 (Cyc.) was synthesized with cysteines at position 80 and 92 to allow cyclization to yield a peptide mimicking its native conformation – that of an antiparallel β-sheet contained within a loop structure. The VEGF-P3 non-Cyc. peptide after synthesis, cleavage and high-performance liquid chromatography purification was oxidized using iodine and characterized by mass spectrometry. The VEGF-RI-P4 peptide mimic required the use of D-amino acids to yield the RI peptidomimetic [186–188], resulting in a more stable peptide than its L-amino acid counterpart, exhibiting similar activity with higher bioavailability.
Table 4.
VEGF peptide sequences.
| Designation | Peptide sequence |
|---|---|
| VEGF 102–122 | 76ITMQ79–80IMRIKPHQGQHIG92–EMSF96 |
| VEGF-P3 (non-Cyc.) | 76ITMQ79–C–92GIHQGQH PKIRMI80–C–EMSF96 |
| VEGF-P3 (Cyc.) | 76ITMQ79–C–92GIHQGQH PKIRMI80–C–EMSF96 |
Cyc.: Cyclized; non-Cyc.: Noncyclized.
We initially evaluated the inhibitory effect of the peptide mimic in a number of different in vitro assays. First, we showed, by surface plasmon resonance, that the VEGF peptide mimic demonstrated the highest affinity for VEGFR-2. Second, we demonstrated that the VEGF mimics were able to inhibit VEGFR-2 phosphorylation with VEGF-P3 (Cyc.) showing the greatest inhibition. In order to delineate whether the VEGF mimics were effective angiogenic inhibitors, we conducted a series of angiogenesis assays [126] such as human umbilical vein endothelial cell proliferation, tube formation and migration in Matrigel™ (BD Biosciences, CA, USA), mouse aortic ring assay and Matrigel plug assay [155]. Our results show that all the VEGF mimics inhibited endothelial cell proliferation, migration and network formation with the conformational VEGF-P3 (Cyc.) being the most effective. In addition, in vivo studies in a transgenic model of VEGF+/−Neu2–5+/− demonstrated the efficacy of VEGF-P3 (Cyc.) in delaying tumor formation. We concluded that the peptide mimic had significant antiangiogenic effects in vitro and in vivo [126,189], validating our design principles.
Animal models & human cancer immunotherapy
In order to evaluate the efficacy of our vaccines, and therapeutic peptide mimics of HER-2 and VEGF, several different types of animal models were deemed necessary. Despite the fact that several cancer mouse models can be used to study breast cancer, no individual model available can cover the entire variety of human disease [190,191] and the molecular heterogeneity of specific cancers. In our studies we have utilized many different mouse models to assess the validity of our approaches. Overall, each model has its advantages and disadvantages, and is differentially suited for studies of immunotherapy and/or pre-clinical testing of peptide vaccines and peptide therapy. The strategy can be divided into both transgenic and transplantable mouse models.
Transgenic mouse models
Initially, in our published studies [125], we utilized the mouse mammary tumor virus (MMTV)/neu202 model, in which approximately 50% of the transgenic female mice will express tumors by day 205 [192]. We were able to use this rat c-neu (N202) transgenic mouse model because our human HER-2 epitopes had >90% homology and antibodies to the epitopes were able to immunoprecipitate the rat neu- receptor. However, the use of any of these genetic models of cancer is not free from pitfalls, requiring a compromise between their features, their usefulness and the specific problem to be addressed. These studies are cumbersome because the tumor has a long latency period and arises in a small percentage of mice. We also studied two different but commonly used transgenic models. First, the BALB-neuT mice on the BALB/c background, which is one of the most aggressive models of rat HER-2/neu carcinogenesis, develop invasive carcinomas in all ten mammary glands by the age of 33 weeks [193]. A lobular carcinoma from this transgenic line was cloned and these TUBO cells [90] were kindly provided to us by J Morris (NIH/NCI). The onset of tumor growth (20.5 weeks of age) is due to a mutation in the transmembrane domain of HER-2 causing it to be easily dimerized with itself and other HER receptors [194]. We have used the BALB-neuT transgenic mice to evaluate vaccine efficacy [153].
The second model we have used is the MMTV-neu on the FVB/n background. The Neu2–5+/− transgenic mice develop multifocal mammary tumors at approximately 177 days [195] due to a mutation in the neu gene. The murine VEGF+/− overexpresses VEGF under the promoter (MMTV) and, when these two transgenic models are crossed together, the double transgenic mouse VEGF+/− Neu2–5+/− can be produced, in which tumor development begins at approximately 6 weeks of age because of the increased vascularization due to overexpression of VEGF [129]. We have also utilized this double-transgenic VEGF+/−Neu2–5+/− model in studies to test VEGF peptide mimics [154].
The polyma middle T oncoprotein transgenic mouse model is clearly established as an excellent model for understanding human breast cancer progression. This is owing to its great similarities with human breast cancer based on its morphological characteristics and biomarker expression. Tumor growth and development in this mouse model are mainly due to the over-expression of HER-2/neu, which later results in the turning on of the angiogenic switch and increased metastasis. This transgenic mouse model (Supplementary Figures 1 & 2 [see online www.futuremedicine.com/doi/suppl/10.2217/FON.12.95]) starts to develop mammary tumors at approximately 5 weeks of age and the different stages of development are similar to those of human breast cancer, making it a very useful model for the disease [196] and useful for studying HER family inhibitors. One disadvantage of this model is that tumor onset starts very early (between 4 and 5 weeks) making it difficult to use this model in the study of preventive vaccines.
Transplantable mouse models
Transplantable mouse models have been extensively used to study cancer drugs and, in most cases, this involves the use of immunocompromised mice that are challenged with human cancer cells. Generally speaking, these models do not represent the full spectrum of the disease because the tumors are very sensitive owing to the absence of an intact immune system in the mice. Tumors that develop in wild-type mice are more resistant because they have undergone serious immune editing and are able to overcome the immune system. Also, the study of vaccines cannot be adequately carried out in immunocompromised mice because all arms of the immune system are not functional.
In view of the above-mentioned difficulties, we have also utilized transplantable tumors as a means of evaluating in vivo efficacy with the added advantage of being able to screen multiple epitopes in a relatively short period of time (3–4 months). We initially used the BALB/c model followed by challenge with syngeneic tumor cells (RENCA-lacZ/HER-2) and enumerated pulmonary metastases at 3–4 months, finding significant reduction in metastases in vaccinated animals [150]. Another model is the NT2.5 transplantable mouse model, in which wild-type FVB/n mice are challenged with NT2.5 cells and tumor onset occurs at approximately 1 week of age. This model can also be used to study vaccines and therapeutic agents. We have also utilized the NT2.5 cell line derived from a spontaneous mammary tumor for challenge in FVB/n mice to determine the immunoprotective effect of our vaccines [151]. However, there are also several disadvantages to using these challenge models thus necessitating a combined approach for determining efficacy of treatment.
The transplantable BALB/c TUBO model requires the use of wild-type BALB/c mice that are challenged with TUBO cells (derived from BALB-neuT transgenic mice). The TUBO cells are very aggressive and all the mice develop tumors at less than 2 weeks of age [155]. This is an excellent model to study both vaccines and inhibitors of HER-2-positive cancers. Finally, we have utilized the wild-type FVB/n Met-1 transplantable mouse model, in which female FVB/n mice are challenged with Met-1 cells derived from the FVB/N-Tg polyma middle T model [197]. In this model, mice develop tumors at approximately 10 days after challenge, and the model can be used for evaluating both vaccines and therapeutic agents. Notwithstanding their limited usefulness, some xenograft models have been shown to correlate with clinical activity to some degree.
Overview of vaccine strategies versus immunotherapy with peptide mimics
The idea of using a chimeric B- and T-cell peptide vaccine to engage the immune system to elicit memory-like antibodies represents a therapy with great potential, which has merit over the infusion of humanized mAbs to treat cancer without the significant toxicities associated with the latter. The HER-2 vaccine involves the elicitation of high-titered, high-affinity polyclonal antibodies that interfere with binding to the ECD of HER-2. The antibody-mediated antitumor mechanisms are generally poorly understood but one can assume that a number of different parameters may be effective. We routinely perform in vitro assays that include ADCC, inhibition of downstream signal transduction pathways, induction of apoptosis and cell cycle arrest, downregulation of HER-2, internalization of HER-2, inhibition of receptor dimerization and angiogenesis. On the other hand, peptide mimics represent a safe and viable therapeutic goal for blocking aberrant signaling pathways. The VEGF peptide mimic strategy involves direct binding to the VEGFR-2 receptors with high affinity and strong potency. Similarly, VEGF-targeted therapy is complex and also involves multiple mechanisms. Generally, one would expect that peptide mimic binding to VEGFR-2 would be likely to inhibit new blood vessel growth by preventing nutrients and oxygen from reaching the tumors through a series of actions such as enhanced endothelial cell proliferation and survival, migration and invasion, as well as increased vascular permeability of existing vessels.
Combination therapy targeting both HER-2 & VEGF in cancer
An authoritative perspective on the principles of and challenges to the development of combination targeted therapies as well as their tolerability and the efficacy of combination reagents has been published [198]. There are many potentially beneficial combinations applicable in cancer immunotherapy, ranging from antibodies to growth factors with small TKI molecules to CTL vaccines with inhibition of Tregs with or without radiation and/or chemotherapy. The overexpression of HER-2 is associated with increased expression of VEGF at both the RNA and protein level in human breast cancer cells [199]. In addition, exposure of HER-2 overexpressing cells to trastuzumab significantly decreases VEGF expression [200]. Taken together, these data suggest that VEGF is a downstream target of the HER-2 signaling pathway. A positive association between HER-2 and VEGF expression in breast cancer patients has been identified [201]. A two-pronged approach of targeting cancer cells by co-immunizing with defined tumor-associated antigens and angiogenesis-associated antigens has been shown to have synergistic effects [202–204]. Combination treatments with defined tumor-associated and angiogenesis-associated antigens are well known to produce synergistic effects [205]. Combining antiangiogenic therapies with other direct anticancer agents has been shown to be beneficial in various malignancies [206].
It is still unclear how to combine these targeted agents into routine oncology practice to produce long-term clinical benefits with minimal toxicities. We have developed several HER-2 vaccines designed to elicit functional antibodies that can inhibit tumor progression. Alternatively, we have created several important VEGF peptide mimics that block ligand (VEGF) –receptor (VEGFR-2) interactions, thereby inhibiting angiogenesis. A combination approach targeting these two receptors is therefore considered a better option because two separate compartments of the tumor are targeted; that is, the tumor cells and the tumor microenvironment.
Because of the potential synergy between therapy with HER-2 inhibitors and antiangiogenic therapy with VEGF inhibitors in vitro and in vivo, we evaluated the effects of combination treatment with these two peptides. From our previous studies, the peptide epitope HER-2 266–296 was shown to produce the best antitumor effects both in vitro and in vivo. We therefore decided to combine this novel HER-2 peptide mimic with our antiangiogenic VEGF peptide mimic.
Combination treatment with HER-2 & VEGF peptide mimics induces potent antitumor & antiangiogenic responses in vitro & in vivo
We demonstrated that combination treatment with both peptide mimics using different cancer cells in vitro produced superior antitumor effects, as determined by cell viability, proliferation and HER-2 phosphorylation assays. We also demonstrated in vivo in a transplantable BALB/c mouse tumor model of HER-2-positive breast cancer that treatment with the peptide mimics resulted in a greater delay in tumor growth and development. Similarly, treatment with the peptide mimics inhibited angiogenesis in vivo as assessed by a Matrigel plug assay. To address the problem of degradability of L-amino acid peptides in vivo, we synthesized the RI D-peptide mimics, which resulted in higher efficacy in treatment. In conclusion, our study demonstrated that combination treatment with HER-2 and VEGF peptide mimics provides greater efficacy than individual treatments, validating the peptide approach as a cancer therapeutic [189].
Combined vaccination with HER-2 peptide followed by therapy with VEGF peptide mimics show effective antitumor & angiogenic effects in vitro & in vivo
We have completed new studies [207] evaluating the efficacy of vaccination with HER-2 peptides followed by treatment with VEGF peptide mimics in two animal models: wild-type BALB/c followed by challenge with TUBO cells; and a polyma middle T transgenic mouse. Initially, we conducted a series of in vitro assays – proliferation, HER-2 phosphorylation and ADCC – to validate whether combining HER-2 and VEGF would be effective.
We have previously raised rabbit antibodies to both the MVF-HER-2 (266–296) and MVF-VEGF peptides. HER-2 and VEGF antipeptide antibodies were used to inhibit cancer cell growth in different in vitro assays. Our results pointed to the benefits of using a combination strategy targeting both HER-2 and VEGF in HER-2-dependent cancers. Therefore, we were interested in testing the hypothesis in vivo – active immunotherapy with MVF-HER-2 B-cell epitope vaccines; and antiangiogenic therapy with structured peptide mimics of VEGF. A transplantable Balb/c mouse model challenged with TUBO cells was used to test the in vivo combined effects of both immunization with HER-2 peptide vaccine and treatment with the VEGF peptide mimics. The results obtained showed that immunization with a HER-2 peptide epitope elicited high-affinity HER-2 native antibodies that are effective in inhibiting tumor growth in vivo; treatment with VEGF peptide mimics provided enhanced efficacy in preventing tumor growth by inhibiting angiogenesis. A combination treatment with HER-2 and VEGF showed enhanced efficacy with additive/synergistic effects. We demonstrated that this novel combined approach induces potent antitumor and antiangiogenic responses, pointing to a strategy that is cheap, and that has the potential of being safe and nontoxic.
Immunotherapy with HER-2 & VEGF peptide mimics plus metronomic paclitaxel causes superior inhibition of tumor growth in both transplantable & transgenic mouse models of human breast cancer
One of the greatest challenges of using most chemotherapeutic agents today is to minimize toxicity, which has led to the suggestion that combination treatment with low-dose chemotherapy and antiangiogenic agents will reduce toxicity and augment antitumor activity. Antiangiogenic agents cause normalization of the tumor vasculature, thereby increasing drug accessibility to the tumor and leading to better efficacy [208]. Many studies have shown improved response rates with the use of a combination approach in many preclinical settings and paclitaxel is one of the most widely used chemotherapeutic agents in the treatment of various types of cancers [209]. Based on these data, we hypothesized that combining low doses of paclitaxel with our peptide mimics may increase response rates with minimal toxicity [210].
We investigated the in vivo effects of combining minimal doses of paclitaxel with the HER-2 or VEGF peptide mimics in two different models of human breast cancer. We first evaluated the toxicity, if any, that is associated with our peptides, paclitaxel chemotherapy or trastuzumab. The results obtained showed that both paclitaxel and trastuzumab caused significantly higher levels of serum cardiac troponin I, which is an indicator of cardiac toxicity. The HER-2 peptides showed no significant increase in cardiac troponin. We therefore decided to combine low doses of the paclitaxel with the peptide mimics and we observed better antitumor responses and no increase in cardiac troponin I. Low concentrations of paclitaxel in combination with our peptide mimics caused a greater delay in onset of tumor growth and development in both transplantable and transgenic mouse models of cancer. This shows that combining low doses of chemotherapy with other treatment options may increase patient survival and minimize toxicity.
Immediate parallel & compensatory pathways
A major concern in targeted therapies is the problem of resistance whereby patients become refractory to continued treatment. Recently, it has become clearer that we need to develop strategies to selectively inhibit multiple receptors and their signaling pathways because of the extensive crosstalk that exists in the tumor microenvironment. A case in point is the many patients with HER-2-overexpressing meta-static breast cancer who initially respond to trastuzumab but ultimately develop disease progression. Several molecular mechanisms (reviewed by Nahta [211]) are operative within the HER family of receptors, such as HER-2–HER-3, as well as compensatory signaling from outside the HER family, such as VEGF, IGF-1 receptor (IGF-1R) [211] and other intracellular downstream signaling pathway. Roop and Ma recently reviewed endocrine resistance in breast cancer, the molecular pathways involved and the development of targeted therapies [212]. Having explored the HER-2/VEGF combination therapy as described in this article, we are now focusing on the HER-3 and IGF-1R axis.
IGF-1R crosstalk with HER-2
The IGF receptor has been implicated in the growth and development of several malignancies [213]. The main signaling pathway implicated in cancer is the IGF-1R–IGF-1 pathway, which supports the growth of many different types of cancer [214,215]. This pathway not only contributes to tumorigenesis but is highly implicated in the development of resistance to anticancer drugs that are available in the clinic [214,216]. The IGF-1–IGF-1R pathway has been shown to play critical roles in many cancers including lung [217], breast [218], pancreas [219], colorectal [220], prostate [221] and head and neck [222] cancers. Increased expression of IGF-1R causes induction of EGFR (HER-1) and requires the activity of the receptor for downstream signaling and EGFR has also been shown to form heterodimers with IGF-1R [223,224]. There is also significant evidence that shows that crosstalk occurs between IGF-1R and HER-2 and both receptors form heterodimers in many breast cancers [225–227]. IGF-1 is also known to be able to stimulate angiogenesis by upregulating VEGF expression [228].
Conclusion
HER family receptors remain a crucial target in many epithelial cancers despite efforts that have led to the FDA approval of some drugs to target this group of receptors. Most of the drugs in the clinic have detrimental side effects and unacceptable safety profiles that overshadow the minimal benefits most patients derive from treatment with them. Patients on these drugs are also likely to progress due to development of acquired resistance. We have developed several HER-2 vaccines designed to elicit functional antibodies that can inhibit tumor progression. We have translated our basic studies to two Phase I clinical trials, one of which is ongoing. We have created several important VEGF peptide mimics that block the ligand (VEGF) –receptor (VEGFR-2) interactions, thereby inhibiting angiogenesis. A combination approach targeting these two receptors has been a major effort in our laboratories because of the potential synergy between HER-2 vaccination and antiangiogenic therapy with VEGF inhibitors. In our recently published studies, we have demonstrated that dual inhibition of HER-2 and VEGF offers enhanced antitumor effects; shutting down these pathways could help in preventing resistance to most drugs and overcome mechanisms of resistance. The mechanism by which these pathways operate is quite complex, gradually evolving during treatment owing to signaling through alternative pathways. It is clear that disappointing clinical outcomes may be owing to the development of escape mechanisms. It is still unclear how to combine these targeted agents into routine oncology practice to produce long-term clinical benefits with minimal toxicities. This serves as a focus to developing additional safe inhibitors of several alternative pathways and combining them in innovative ways. Studies designed to develop vaccines and peptide mimics of HER-1, HER-3 and IGF-1R are a major emphasis in our laboratories. The goal has been to develop a paradigm to guide the rational and effective advancement of targeted innovative therapies for the ultimate benefit of patients with cancer.
Future perspective
An enormous amount of data indicate that current cancer treatment is greatly hampered by toxicity and serious adverse effects. Most of this is as a result of the nonspecificity of existing agents and the development of resistance. The cost of cancer treatment is also a great concern for obvious reasons. There is, therefore, an unmet need to develop better, safer and less toxic anticancer agents that will selectively target cancer-specific pathways without interrupting normal signaling processes. Efforts to design a cancer vaccine that is either preventive or therapeutic will go a long way to increasing the quality of life of most cancer patients. The use of peptides as either vaccines or therapeutic agents looks promising and could provide a better alternative to current treatment options. Efforts in our laboratory are focused towards developing smart medicine to be used in combination therapy.
Ongoing studies are focused on:
Targeting both HER-1 and HER-3 and defining important epitopes in order to unravel mechanisms of crosstalk and resistance;
Developing novel HER-3 targeting agents to provide insights into the relevance and function of this receptor;
Developing combination regimens using low doses of chemotherapy with HER-2 and VEGF inhibitors to determine the best synergistic partners;
Developing novel peptide inhibitors of the IGF axis, which play a major role in cancer cell proliferation, survival and resistance to anticancer therapies in human cancers;
Determining and developing the best peptide combinatorial approach for targeted therapies focused on the HER-1, HER-2, HER-3, VEGF and IGF pathways;
Translating new peptide-based cancer vaccines and peptide mimic therapies to the clinic.
Supplementary Material
Executive summary.
Cancer is a multifactorial disease and a complete understanding of what drives metastasis is still elusive.
Many diverse and potentially useful cancer immunotherapy strategies and combinations are being tested in the clinic but none offer a cure.
The HER family receptors form a signaling network that contributes to cancer progression.
Angiogenesis plays a major role in cancer and VEGF-targeted agents have been shown to be useful in advanced-stage malignancies.
Current antitumor cancer drugs targeting the HER and VEGF receptors have some beneficial effects but are accompanied by unacceptable safety profiles and low quality of life experienced by most patients; in addition, they do not cure the cancer.
There is an unmet need to develop more specific drugs that will selectively target the HER family receptors with few or no toxic effects.
Members of the HER family receptors can form homo- and heterodimers and blocking just one receptor may lead to stimulation of alternative partnering among the receptors.
Peptides provide a good platform to develop efficient cancer vaccines or therapeutic agents that can specifically disrupt the network of signaling in this group of receptors.
Peptide epitopes must be engineered to adopt the 3D structure of the corresponding antigen in order to elicit high-affinity antibodies that can kill tumor cells effectively.
‘Promiscuous’ T-cell epitopes offer the best alternative in the design of chimeric vaccines able to function as potential cancer vaccines.
Peptide mimics must also be designed to adopt structures of the corresponding ligand or receptor to function as effective inhibitors of signaling pathways.
Combining minimal doses of chemotherapy with combination peptide vaccines or peptide mimics can produce additive/synergistic effects with minimal toxicity.
Smart and innovative strategies with rationally engineered peptide vaccines and/or peptide mimics could establish a new paradigm in cancer immunotherapy.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
The work described in this article was funded in part by the NIH, OSU Pelotonia funds and Ob/Gyn Peptide Research Funds. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 2.Kim JY, Sun Q, Oglesbee M, Yoon SO. The role of ErbB2 signaling in the onset of terminal differentiation of oligodendrocytes in vivo. J Neurosci. 2003;23:5561–5571. doi: 10.1523/JNEUROSCI.23-13-05561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinter-Jensen L. Pharmacological effects of epidermal growth factor (EGF) with focus on the urinary and gastrointestinal tracts. APMIS Suppl. 1999;93:1–42. [PubMed] [Google Scholar]
- 4.Arteaga CL, Moulder SL, Yakes FM. HER (erbB) tyrosine kinase inhibitors in the treatment of breast cancer. Semin Oncol. 2002;29:4–10. doi: 10.1053/sonc.2002.34047. [DOI] [PubMed] [Google Scholar]
- 5.Barbacci EG, Guarino BC, Stroh JG, et al. The structural basis for the specificity of epidermal growth factor and heregulin binding. J Biol Chem. 1995;270:9585–9589. doi: 10.1074/jbc.270.16.9585. [DOI] [PubMed] [Google Scholar]
- 6.Tzahar E, Waterman H, Chen X, et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996;16:5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beerli RR, Graus-Porta D, Woods-Cook K, Chen X, Yarden Y, Hynes NE. Neu differentiation factor activation of ErbB-3 and ErbB-4 is cell specific and displays a differential requirement for ErbB-2. Mol Cell Biol. 1995;15:6496–6505. doi: 10.1128/mcb.15.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graus-Porta D, Beerli RR, Hynes NE. Single-chain antibody-mediated intracellular retention of ErbB-2 impairs Neu differentiation factor and epidermal growth factor signaling. Mol Cell Biol. 1995;15:1182–1191. doi: 10.1128/mcb.15.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hynes NE, Stern DF. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim Biophys Acta. 1994;1198:165–184. doi: 10.1016/0304-419x(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 12.Scholl S, Beuzeboc P, Pouillart P. Targeting HER2 in other tumor types. Ann Oncol. 2001;12(Suppl 1):S81–S87. doi: 10.1093/annonc/12.suppl_1.s81. [DOI] [PubMed] [Google Scholar]
- 13.Colomer R, Shamon LA, Tsai MS, Lupu R. Herceptin: from the bench to the clinic. Cancer Invest. 2001;19:49–56. doi: 10.1081/cnv-100000074. [DOI] [PubMed] [Google Scholar]
- 14.Press MF, Cordon-Cardo C, Slamon DJ. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. 1990;5:953–962. [PubMed] [Google Scholar]
- 15.Paik S, Hazan R, Fisher ER, et al. Pathologic findings from the National Surgical Adjuvant Breast and Bowel Project: prognostic significance of erbB-2 protein overexpression in primary breast cancer. J Clin Oncol. 1990;8:103–112. doi: 10.1200/JCO.1990.8.1.103. [DOI] [PubMed] [Google Scholar]
- 16.Niehans GA, Singleton TP, Dykoski D, Kiang DT. Stability of HER-2/neu expression over time and at multiple metastatic sites. J Natl Cancer Inst. 1993;85:1230–1235. doi: 10.1093/jnci/85.15.1230. [DOI] [PubMed] [Google Scholar]
- 17.Mimura K, Kono K, Hanawa M, et al. Frequencies of HER-2/neu expression and gene amplification in patients with oesophageal squamous cell carcinoma. Br J Cancer. 2005;92:1253–1260. doi: 10.1038/sj.bjc.6602499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrison C, Zanagnolo V, Ramirez N, et al. HER-2 is an independent prognostic factor in endometrial cancer: association with outcome in a large cohort of surgically staged patients. J Clin Oncol. 2006;24:2376–2385. doi: 10.1200/JCO.2005.03.4827. [DOI] [PubMed] [Google Scholar]
- 19.Yano T, Doi T, Ohtsu A, et al. Comparison of HER2 gene amplification assessed by fluorescence in situ hybridization and HER2 protein expression assessed by immunohistochemistry in gastric cancer. Oncol Rep. 2006;15:65–71. [PubMed] [Google Scholar]
- 20.Cirisano FD, Karlan BY. The role of the HER-2/neu oncogene in gynecologic cancers. J Soc Gynecol Invest. 1996;3:99–105. [PubMed] [Google Scholar]
- 21.Berchuck A, Rodriguez G, Kinney RB, et al. Overexpression of HER-2/neu in endometrial cancer is associated with advanced stage disease. Am J Obstet Gynecol. 1991;164:15–21. doi: 10.1016/0002-9378(91)90615-x. [DOI] [PubMed] [Google Scholar]
- 22.Kern JA, Schwartz DA, Nordberg JE, et al. p185neu expression in human lung adenocarcinomas predicts shortened survival. Cancer Res. 1990;50:5184–5187. [PubMed] [Google Scholar]
- 23.Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. 1992;89:4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24▪▪.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–280. doi: 10.1038/ncponc0509. Outlines the reasons for the development of resistance to HER-2-targeted drugs such as trastuzumab because of activation of multiple receptor pathways that include HER-2-related receptors or non-HER receptors such as the IGF-1 receptor. [DOI] [PubMed] [Google Scholar]
- 25.Agus DB, Akita RW, Fox WD, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2:127–137. doi: 10.1016/s1535-6108(02)00097-1. [DOI] [PubMed] [Google Scholar]
- 26.Agus DB, Gordon MS, Taylor C, et al. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J Clin Oncol. 2005;23(11):2534–2543. doi: 10.1200/JCO.2005.03.184. [DOI] [PubMed] [Google Scholar]
- 27.Nahta R, Hung MC, Esteva FJ. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004;64:2343–2346. doi: 10.1158/0008-5472.can-03-3856. [DOI] [PubMed] [Google Scholar]
- 28.Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009;24:9330–9336. doi: 10.1158/0008-5472.CAN-08-4597. [DOI] [PubMed] [Google Scholar]
- 29.Walshe JM, Denduluri N, Berman AW, Rosing DR, Swain SM. A Phase II trial with trastuzumab and pertuzumab in patients with HER2-overexpressed locally advanced and metastatic breast cancer. Clin Breast Cancer. 2006;6:535–539. doi: 10.3816/CBC.2006.n.009. [DOI] [PubMed] [Google Scholar]
- 30.Heymach JV, Nilsson M, Blumenschein G, Papadimitrakopoulou V, Herbst R. Epidermal growth factor receptor inhibitors in development for the treatment of non-small cell lung cancer. Clin Cancer Res. 2006;12:4441S–4445S. doi: 10.1158/1078-0432.CCR-06-0286. [DOI] [PubMed] [Google Scholar]
- 31.Gordon MS, Matei D, Aghajanian C, et al. Clinical activity of pertuzumab (rhuMAb 2C4), a HER dimerization inhibitor, in advanced ovarian cancer: potential predictive relationship with tumor HER2 activation status. J Clin Oncol. 2006;24:4324–4332. doi: 10.1200/JCO.2005.05.4221. [DOI] [PubMed] [Google Scholar]
- 32.Agus DB, Sweeney CJ, Morris MJ, et al. Efficacy and safety of single-agent pertuzumab (rhuMAb 2C4), a human epidermal growth factor receptor dimerization inhibitor, in castration-resistant prostate cancer after progression from taxane-based therapy. J Clin Oncol. 2007;25:675–681. doi: 10.1200/JCO.2006.07.0649. [DOI] [PubMed] [Google Scholar]
- 33.Chow NH, Liu HS, Lee EI, et al. Significance of urinary epidermal growth factor and its receptor expression in human bladder cancer. Anticancer Res. 1997;17:1293–1296. [PubMed] [Google Scholar]
- 34.Angelucci A. Targeting ERBB receptors to inhibit metastasis: old hopes and new certainties. Curr Cancer Drug Targets. 2009;9:1–18. doi: 10.2174/156800909787314048. [DOI] [PubMed] [Google Scholar]
- 35.Patan S. Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J Neurooncol. 2000;50:1–15. doi: 10.1023/a:1006493130855. [DOI] [PubMed] [Google Scholar]
- 36▪.Mendelsohn J. Targeting the epidermal growth factor receptor for cancer therapy. J Clin Oncol. 2002;20:1S–13S. Describes the role played by EGF receptor (EGFR) in different types of cancer and the efforts that have been made to target EGFR in the clinic. The article illustrates the central role played by EGFR in tumorigenesis and how it can be upregulated when other HER family receptors are targeted. [PubMed] [Google Scholar]
- 37.Johnston JB, Navaratnam S, Pitz MW, et al. Targeting the EGFR pathway for cancer therapy. Curr Med Chem. 2006;13:3483–3492. doi: 10.2174/092986706779026174. [DOI] [PubMed] [Google Scholar]
- 38.Grothey A. Recognizing and managing toxicities of molecular targeted therapies for colorectal cancer. Oncol (Williston Park) 2006;20:21–28. [PubMed] [Google Scholar]
- 39.Schaefer G, Haber L, Crocker LM, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 2011;20:472–486. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 40▪.Sutherland RL. Endocrine resistance in breast cancer: new roles for ErbB3 and ErbB4. Breast Cancer Res. 2011;13(3):106. doi: 10.1186/bcr2878. Discusses the roles played by ErbB-3 and ErbB-4 in the development of resistance, which is a major limitation to the successful treatment of estrogen receptor-positive (ER+) breast cancer, and the EGF receptor and ErbB-2 receptor tyrosine kinases are involved in this process. It summarizes recent studies that now implicate the other two ErbB family members, ErbB-3 and -4, in the resistance and metastasis of cancer cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brennan PJ, Kumagai T, Berezov A, Murali R, Greene MI. HER2/Neu: mechanisms of dimerization/oligomerization. Oncogene. 2002;21:328. doi: 10.1038/sj.onc.1205119. [DOI] [PubMed] [Google Scholar]
- 42.Stern DF. ERBB3/HER3 and ERBB2/HER2 duet in mammary development and breast cancer. J Mammary Gland Biol. 2008;13:215–223. doi: 10.1007/s10911-008-9083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Srinivasan R, Leverton KE, Sheldon H, Hurst HC, Sarraf C, Gullick WJ. Intracellular expression of the truncated extracellular domain of c-erbB-3/HER3. Cell Signal. 2001;13:321–330. doi: 10.1016/s0898-6568(01)00155-3. [DOI] [PubMed] [Google Scholar]
- 44.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 45.Eisenberg D, Singer E, Landgraf R, Horan T, Slamon D. Identification of a heregulin binding site in HER3 extracellular domain. J Biol Chem. 2001;276:44266–44274. doi: 10.1074/jbc.M105428200. [DOI] [PubMed] [Google Scholar]
- 46▪.Sliwkowski MX, Lee-Hoeflich ST, Crocker L, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. Provides significant evidence that HER-3 plays a central role in HER-2-amplified breast cancers and that targeting just HER-2 results in the stimulation of HER-3 signaling mechanisms as an escape pathway to HER-2 targeting agents. The paper concludes that a combined approach targeting both HER-2 and HER-3 is a promising strategy. [DOI] [PubMed] [Google Scholar]
- 47.Pinkas-Kramarski R, Soussan L, Waterman H, et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996;15:2452–2467. [PMC free article] [PubMed] [Google Scholar]
- 48.Alimandi M, Romano A, Curia MC, et al. Cooperative signaling of Erbb3 and Erbb2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10:1813–1821. [PubMed] [Google Scholar]
- 49.Garrett JT, Olivares MG, Rinehart C, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA. 2011;108(12):5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee-Hoeflich ST, Crocker L, Yao E, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 51.Engelman JA, Schoeberl B, Faber AC, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70:2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hynes NE, Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sithanandam G, Anderson LM. The ERBB3 receptor in cancer and cancer gene therapy. Cancer Gene Ther. 2008;15:413–448. doi: 10.1038/cgt.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tzahar E, Waterman H, Chen XM, et al. A hierarchical network of interreceptor interactions determines signal transduction by neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996;16:5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Croce CM, Iorio MV, Casalini P, et al. microRNA-205 regulates HER3 in human breast cancer. Cancer Res. 2009;69:2195–2200. doi: 10.1158/0008-5472.CAN-08-2920. [DOI] [PubMed] [Google Scholar]
- 56.Liu BL, Ordonez-Ercan D, Fan ZY, Edgerton SM, Yang XH, Thor AD. Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer. 2007;120:1874–1882. doi: 10.1002/ijc.22423. [DOI] [PubMed] [Google Scholar]
- 57.Alaoui-Jamali MA, Yen L, Benlimame N, et al. Differential regulation of tumor angiogenesis by distinct ErbB homo- and heterodimers. Mol Biol Cell. 2002;13:4029–4044. doi: 10.1091/mbc.E02-02-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rajkumar T, Stamp GWH, Pandha HS, Waxman J, Gullick WJ. Expression of the type 1 tyrosine kinase growth factor receptors EGF receptor, c-erbB2 and c-erbB3 in bladder cancer. J Pathol. 1996;179:381–385. doi: 10.1002/(SICI)1096-9896(199608)179:4<381::AID-PATH603>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 59.Moasser MM, Sergina NV. The HER family and cancer: emerging molecular mechanisms and therapeutic targets. Trends Mol Med. 2007;13:527–534. doi: 10.1016/j.molmed.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moasser MM, Sergina NV, Rausch M, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 62.Amler L, Makhija S, Januario T, et al. Downregulation of HER3 may predict clinical benefit in ovarian cancer from pertuzumab, a HER2 dimerization-inhibiting antibody. Presented at. ASCO-NCI-EORTC Annual Meeting on Molecular Markers in Cancer. 2008:Abstract 25. [Google Scholar]
- 63.Jordan VC, Osipo C, Meeke K, et al. Role for HER2/neu and HER3 in fulvestrant-resistant breast cancer. Int J Oncol. 2007;30:509–520. [PubMed] [Google Scholar]
- 64.Lykkesfeldt AE, Frogne T, Benjaminsen RV, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 2009;114:263–275. doi: 10.1007/s10549-008-0011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arteaga CL, Miller TW, Perez-Torres M, et al. Loss of phosphatase and tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69:4192–4201. doi: 10.1158/0008-5472.CAN-09-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elenius K, Erjala K, Sundvall M, et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2006;12:4103–4111. doi: 10.1158/1078-0432.CCR-05-2404. [DOI] [PubMed] [Google Scholar]
- 67.Hamburger AW, Zhang YX, Linn D, et al. EBP1, an ErbB3-binding cancer and implicated in protein, is decreased in prostate hormone resistance. Mol Cancer Ther. 2008;7:3176–3186. doi: 10.1158/1535-7163.MCT-08-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber J. Peptide vaccines for cancer. Cancer Invest. 2002;20:208–221. doi: 10.1081/cnv-120001149. [DOI] [PubMed] [Google Scholar]
- 69.Buteau C, Markovic SN, Celis E. Challenges in the development of effective peptide vaccines for cancer. Mayo Clin Proc. 2002;77:339–349. doi: 10.4065/77.4.339. [DOI] [PubMed] [Google Scholar]
- 70.Lazoura E, Apostolopoulos V. Rational peptide-based vaccine design for cancer immunotherapeutic applications. Curr Med Chem. 2005;12:629–639. doi: 10.2174/0929867053202188. [DOI] [PubMed] [Google Scholar]
- 71.Disis ML, Calenoff E, McLaughlin G, et al. Existent T-cell and antibody immunity to HER-2/neu protein in patients with breast cancer. Cancer Res. 1994;54:16–20. [PubMed] [Google Scholar]
- 72.Disis ML, Pupa SM, Gralow JR, Dittadi R, Menard S, Cheever MA. High-titer HER-2/neu protein-specific antibody can be detected in patients with early-stage breast cancer. J Clin Oncol. 1997;15:3363–3367. doi: 10.1200/JCO.1997.15.11.3363. [DOI] [PubMed] [Google Scholar]
- 73.Disis ML, Gralow JR, Bernhard H, Hand SL, Rubin WD, Cheever MA. Peptide-based, but not whole protein, vaccines elicit immunity to HER-2/neu, oncogenic self-protein. J Immunol. 1996;156:3151–3158. [PubMed] [Google Scholar]
- 74.Disis ML, Grabstein KH, Sleath PR, Cheever MA. Generation of immunity to the HER-2/neu oncogenic protein in patients with breast and ovarian cancer using a peptide-based vaccine. Clin Cancer Res. 1999;5:1289–1297. [PubMed] [Google Scholar]
- 75.Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med. 1995;181:2109–2117. doi: 10.1084/jem.181.6.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baxevanis CN, Voutsas IF, Tsitsilonis OE, Gritzapis AD, Sotiriadou R, Papamichail M. Tumor-specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J Immunol. 2000;164:3902–3912. doi: 10.4049/jimmunol.164.7.3902. [DOI] [PubMed] [Google Scholar]
- 77.Lindencrona JA, Preiss S, Kammertoens T, et al. CD4+ T cell-mediated HER-2/neu-specific tumor rejection in the absence of B cells. Int J Cancer. 2004;109:259–264. doi: 10.1002/ijc.11654. [DOI] [PubMed] [Google Scholar]
- 78.Tuttle TM, Anderson BW, Thompson WE, et al. Proliferative and cytokine responses to class II HER-2/neu-associated peptides in breast cancer patients. Clin Cancer Res. 1998;4:2015–2024. [PubMed] [Google Scholar]
- 79.Kobayashi H, Wood M, Song Y, Appella E, Celis E. Defining promiscuous MHC class II helper T-cell epitopes for the HER2/neu tumor antigen. Cancer Res. 2000;60:5228–5236. [PubMed] [Google Scholar]
- 80.Sotiriadou R, Perez SA, Gritzapis AD, et al. Peptide HER2(776-788) represents a naturally processed broad MHC class II-restricted T cell epitope. Br J Cancer. 2001;85:1527–1534. doi: 10.1054/bjoc.2001.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Perez SA, Sotiropoulou PA, Sotiriadou NN, et al. HER-2/neu-derived peptide 884-899 is expressed by human breast, colorectal and pancreatic adenocarcinomas and is recognized by in-vitro-induced specific CD4(+) T cell clones. Cancer Immunol Immunother. 2002;50:615–624. doi: 10.1007/s002620100225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fisk B, Hudson JM, Kavanagh J, et al. Existent proliferative responses of peripheral blood mononuclear cells from healthy donors and ovarian cancer patients to HER-2 peptides. Anticancer Res. 1997;17:45–53. [PubMed] [Google Scholar]
- 83.Mittendorf EA, Clifton GT, Holmes JP, et al. Clinical trial results of the HER-2/neu (E75) vaccine to prevent breast cancer recurrence in high-risk patients: from US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Cancer. 2012;118(10):2594–2602. doi: 10.1002/cncr.26574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sears AK, Perez SA, Clifton GT, et al. AE37: a novel T-cell-eliciting vaccine for breast cancer. Expert Opin Biol Ther. 2011;11:1543–1550. doi: 10.1517/14712598.2011.616889. [DOI] [PubMed] [Google Scholar]
- 85.Mittendorf EA, Storrer CE, Foley RJ, et al. Evaluation of the HER2/neu-derived peptide GP2 for use in a peptide-based breast cancer vaccine trial. Cancer. 2006;106:2309–2317. doi: 10.1002/cncr.21849. [DOI] [PubMed] [Google Scholar]
- 86.Clive KS, Tyler JA, Clifton GT, et al. The GP2 peptide: a HER2/neu-based breast cancer vaccine. J Surg Oncol. 2012;105:452–458. doi: 10.1002/jso.21723. [DOI] [PubMed] [Google Scholar]
- 87.Reilly RT, Gottlieb MB, Ercolini AM, et al. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 2000;60:3569–3576. [PubMed] [Google Scholar]
- 88.Nanni P, Nicoletti G, De Giovanni C, et al. Combined allogeneic tumor cell vaccination and systemic interleukin 12 prevents mammary carcinogenesis in HER-2/neu transgenic mice. J Exp Med. 2001;194:1195–1205. doi: 10.1084/jem.194.9.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cefai D, Morrison BW, Sckell A, et al. Targeting HER-2/neu for active-specific immunotherapy in a mouse model of spontaneous breast cancer. Int J Cancer. 1999;83:393–400. doi: 10.1002/(sici)1097-0215(19991029)83:3<393::aid-ijc16>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 90.Dela Cruz JS, Lau SY, Ramirez EM, et al. Protein vaccination with the HER2/neu extracellular domain plus anti-HER2/neu antibody-cytokine fusion proteins induces a protective anti-HER2/neu immune response in mice. Vaccine. 2003;21:1317–1326. doi: 10.1016/s0264-410x(02)00741-7. [DOI] [PubMed] [Google Scholar]
- 91.Amici A, Venanzi FM, Concetti A. Genetic immunization against neu/erbB2 transgenic breast cancer. Cancer Immunol Immunother. 1998;47:183–190. doi: 10.1007/s002620050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piechocki MP, Pilon SA, Wei WZ. Complementary antitumor immunity induced by plasmid DNA encoding secreted and cytoplasmic human ErbB-2. J Immunol. 2001;167:3367–3374. doi: 10.4049/jimmunol.167.6.3367. [DOI] [PubMed] [Google Scholar]
- 93.Pupa SM, Invernizzi AM, Forti S, et al. Prevention of spontaneous neu-expressing mammary tumor development in mice transgenic for rat proto-neu by DNA vaccination. Gene Ther. 2001;8:75–79. doi: 10.1038/sj.gt.3301360. [DOI] [PubMed] [Google Scholar]
- 94.Yip YL, Smith G, Koch J, Dubel S, Ward RL. Identification of epitope regions recognized by tumor inhibitory and stimulatory anti-ErbB-2 monoclonal antibodies: implications for vaccine design. J Immunol. 2001;166:5271–5278. doi: 10.4049/jimmunol.166.8.5271. [DOI] [PubMed] [Google Scholar]
- 95.Riemer AB, Klinger M, Wagner S, et al. Generation of peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu. J Immunol. 2004;173:394–401. doi: 10.4049/jimmunol.173.1.394. [DOI] [PubMed] [Google Scholar]
- 96.Jasinska J, Wagner S, Radauer C, et al. Inhibition of tumor cell growth by antibodies induced after vaccination with peptides derived from the extracellular domain of Her-2/neu. Int J Cancer. 2003;107:976–983. doi: 10.1002/ijc.11485. [DOI] [PubMed] [Google Scholar]
- 97.Riemer AB, Klinger M, Wagner S, et al. Generation of peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu. J Immunol. 2004;173:394–401. doi: 10.4049/jimmunol.173.1.394. [DOI] [PubMed] [Google Scholar]
- 98.Riemer AB, Kraml G, Scheiner O, Zielinski CC, Jensen-Jarolim E. Matching of trastuzumab (Herceptin) epitope mimics onto the surface of Her-2/neu – a new method of epitope definition. Mol Immunol. 2005;42:1121–1124. doi: 10.1016/j.molimm.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 99.Atzori F, Tabernero J, Cervantes A, et al. A Phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17:6304–6312. doi: 10.1158/1078-0432.CCR-10-3336. [DOI] [PubMed] [Google Scholar]
- 100.Wiedermann U, Wiltschke C, Jasinska J, et al. A virosomal formulated Her-2/neu multi-peptide vaccine induces Her-2/neu-specific immune responses in patients with metastatic breast cancer: a Phase I study. Breast Cancer Res Treat. 2010;119:673–683. doi: 10.1007/s10549-009-0666-9. [DOI] [PubMed] [Google Scholar]
- 101.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251–275. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- 102.Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175:409–416. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 104.Carpenito C, Davis PD, Dougherty ST, Dougherty GJ. Exploiting the differential production of angiogenic factors within the tumor microenvironment in the design of a novel vascular-targeted gene therapy-based approach to the treatment of cancer. Int J Radiat Oncol Biol Phys. 2002;54:1473–1478. doi: 10.1016/s0360-3016(02)03921-4. [DOI] [PubMed] [Google Scholar]
- 105.Casella I, Feccia T, Chelucci C, et al. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood. 2003;101:1316–1323. doi: 10.1182/blood-2002-07-2184. [DOI] [PubMed] [Google Scholar]
- 106.Monsky WL, Mouta Carreira C, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin Cancer Res. 2002;8:1008–1013. [PubMed] [Google Scholar]
- 107.Kim DW, Huamani J, Fu A, Hallahan DE. Molecular strategies targeting the host component of cancer to enhance tumor response to radiation therapy. Int J Radiat Oncol Biol Phys. 2006;64:38–46. doi: 10.1016/j.ijrobp.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 108.Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clin Transl Oncol. 2006;8:77–82. doi: 10.1007/s12094-006-0162-1. [DOI] [PubMed] [Google Scholar]
- 109.Folkman J. Tumor suppression by p53 is mediated in part by the antiangiogenic activity of endostatin and tumstatin. Sci STKE. 2006;2006:PE35. doi: 10.1126/stke.3542006pe35. [DOI] [PubMed] [Google Scholar]
- 110.Naumov GN, Bender E, Zurakowski D, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst. 2006;98:316–325. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- 111.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 112.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 113.Ferrara N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist. 2004;9(Suppl 1):S2–S10. doi: 10.1634/theoncologist.9-suppl_1-2. [DOI] [PubMed] [Google Scholar]
- 114.Saito H, Tsujitani S, Ikeguchi M, Maeta M, Kaibara N. Relationship between the expression of vascular endothelial growth factor and the density of dendritic cells in gastric adenocarcinoma tissue. Br J Cancer. 1998;78:1573–1577. doi: 10.1038/bjc.1998.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McMahon G. VEGF receptor signaling in tumor angiogenesis. Oncologist. 2000;5(Suppl 1):S3–S10. doi: 10.1634/theoncologist.5-suppl_1-3. [DOI] [PubMed] [Google Scholar]
- 116.Ferrara N. The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS. 2005;(94):209–231. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- 117.Moldovan L, Moldovan NI. Role of monocytes and macrophages in angiogenesis. EXS. 2005;(94):127–146. doi: 10.1007/3-7643-7311-3_9. [DOI] [PubMed] [Google Scholar]
- 118.Moldovan NI. Functional adaptation: the key to plasticity of cardiovascular “stem” cells? Stem Cells Dev. 2005;14:111–121. doi: 10.1089/scd.2005.14.111. [DOI] [PubMed] [Google Scholar]
- 119.Anghelina M, Krishnan P, Moldovan L, Moldovan NI. Monocytes/macrophages cooperate with progenitor cells during neovascularization and tissue repair: conversion of cell columns into fibrovascular bundles. Am J Pathol. 2006;168:529–541. doi: 10.2353/ajpath.2006.050255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Anghelina M, Krishnan P, Moldovan L, Moldovan NI. Monocytes and macrophages form branched cell columns in matrigel: implications for a role in neovascularization. Stem Cells Dev. 2004;13:665–676. doi: 10.1089/scd.2004.13.665. [DOI] [PubMed] [Google Scholar]
- 121.Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol. 1991;5:1806–1814. doi: 10.1210/mend-5-12-1806. [DOI] [PubMed] [Google Scholar]
- 122.Tischer E, Mitchell R, Hartman T, et al. The human gene for vascular endothelial growth factor Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- 123.Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161:851–858. doi: 10.1016/0006-291x(89)92678-8. [DOI] [PubMed] [Google Scholar]
- 124.Fuh G, Wu P, Liang WC, et al. Structure–function studies of two synthetic anti-vascular endothelial growth factor Fabs and comparison with the Avastin Fab. J Biol Chem. 2006;281:6625–6631. doi: 10.1074/jbc.M507783200. [DOI] [PubMed] [Google Scholar]
- 125.Dakappagari NK, Douglas DB, Triozzi PL, Stevens VC, Kaumaya PT. Prevention of mammary tumors with a chimeric HER-2 B-cell epitope peptide vaccine. Cancer Res. 2000;60:3782–3789. [PubMed] [Google Scholar]
- 126.Vicari D, Foy KC, Liotta EM, Kaumaya PT. Engineered conformation-dependent VEGF peptide mimics are effective in inhibiting VEGF signaling pathways. J Biol Chem. 2011;286(15):13612–13625. doi: 10.1074/jbc.M110.216812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 128.Zhu Z, Witte L. Inhibition of tumor growth and metastasis by targeting tumor-associated angiogenesis with antagonists to the receptors of vascular endothelial growth factor. Invest N Drugs. 1999;17:195–212. doi: 10.1023/a:1006314501634. [DOI] [PubMed] [Google Scholar]
- 129.Oshima RG, Lesperance J, Munoz V, et al. Angiogenic acceleration of Neu induced mammary tumor progression and metastasis. Cancer Res. 2004;64:169–179. doi: 10.1158/0008-5472.can-03-1944. [DOI] [PubMed] [Google Scholar]
- 130.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 131.Ferrara N. VEGF as a therapeutic target in cancer. Oncology. 2005;69(Suppl 3):S11–S16. doi: 10.1159/000088479. [DOI] [PubMed] [Google Scholar]
- 132.Sereno M, Brunello A, Chiappori A, et al. Cardiac toxicity: old and new issues in anti-cancer drugs. Clin Transl Oncol. 2008;10:35–46. doi: 10.1007/s12094-008-0150-8. [DOI] [PubMed] [Google Scholar]
- 133.Nelson NJ. Angiogenesis research is on fast forward. J Natl Cancer Inst. 1999;91:820–822. doi: 10.1093/jnci/91.10.820. [DOI] [PubMed] [Google Scholar]
- 134.Smith WD, Wells PW, Burrells C, Dawson AM. Immunoglobulins, antibodies and inhibitors of parainfluenza 3 virus in respiratory secretions of sheep. Arch Virol. 1975;49:329–337. doi: 10.1007/BF01318242. [DOI] [PubMed] [Google Scholar]
- 135.Reinacher-Schick A, Pohl M, Schmiegel W. Drug insight: antiangiogenic therapies for gastrointestinal cancers – focus on monoclonal antibodies. Nat Clin Pract Gastroenterol Hepatol. 2008;5:250–267. doi: 10.1038/ncpgasthep1097. [DOI] [PubMed] [Google Scholar]
- 136.Levitzki A. Tyrphostins – potential antiproliferative agents and novel molecular tools. Biochemical Pharmacol. 1990;40:913–918. doi: 10.1016/0006-2952(90)90474-y. [DOI] [PubMed] [Google Scholar]
- 137.Khosravi Shahi P, Fernandez Pineda I. Tumoral angiogenesis: review of the literature. Cancer Invest. 2008;26:104–108. doi: 10.1080/07357900701662509. [DOI] [PubMed] [Google Scholar]
- 138.Le Tourneau C, Raymond E, Faivre S. Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST) Ther Clin Risk Manag. 2007;3:341–348. doi: 10.2147/tcrm.2007.3.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ryan AJ, Wedge SR. ZD6474 – a novel inhibitor of VEGFR and EGFR tyrosine kinase activity. Br J Cancer. 2005;92(Suppl 1):S6–S13. doi: 10.1038/sj.bjc.6602603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bhojani N, Jeldres C, Patard JJ, et al. Toxicities associated with the administration of sorafenib, sunitinib, and temsirolimus and their management in patients with metastatic renal cell carcinoma. Eur Urol. 2008;53:917–930. doi: 10.1016/j.eururo.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 141.Holash J, Davis S, Papadopoulos N, et al. VEGF-trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bianco F, Basini G, Grasselli F. Angiogenic activity of swine granulosa cells: effects of hypoxia and vascular endothelial growth factor Trap R1R2, a VEGF blocker. Domest Anim Endocrinol. 2005;28:308–319. doi: 10.1016/j.domaniend.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 143.Kaumaya PT, VanBuskirk AM, Goldberg E, Pierce SK. Design and immunological properties of topographic immunogenic determinants of a protein antigen (LDH-C4) as vaccines. J Biol Chem. 1992;267:6338–6346. [PubMed] [Google Scholar]
- 144.Kaumaya PT, Berndt KD, Heidorn DB, Trewhella J, Kezdy FJ, Goldberg E. Synthesis and biophysical characterization of engineered topographic immunogenic determinants with alpha alpha topology. Biochemistry. 1990;29:13–23. doi: 10.1021/bi00453a002. [DOI] [PubMed] [Google Scholar]
- 145.Kobs-Conrad S, Lee H, DiGeorge AM, Kaumaya PT. Engineered topographic determinants with alpha beta, beta alpha beta, and beta alpha beta alpha topologies show high affinity binding to native protein antigen (lactate dehydrogenase-C4) J Biol Chem. 1993;268:25285–25295. [PubMed] [Google Scholar]
- 146.Srinivasan M, Wardrop RM, Gienapp IE, Stuckman SS, Whitacre CC, Kaumaya PT. A retro-inverso peptide mimic of CD28 encompassing the MYPPPY motif adopts a polyproline type II helix and inhibits encephalitogenic T cells in vitro. J Immunol. 2001;167:578–585. doi: 10.4049/jimmunol.167.1.578. [DOI] [PubMed] [Google Scholar]
- 147.Srinivasan M, Gienapp IE, Stuckman SS, et al. Suppression of experimental autoimmune encephalomyelitis using peptide mimics of CD28. J Immunol. 2002;169:2180–2188. doi: 10.4049/jimmunol.169.4.2180. [DOI] [PubMed] [Google Scholar]
- 148.Allen SD, Rawale SV, Whitacre CC, Kaumaya PT. Therapeutic peptidomimetic strategies for autoimmune diseases: costimulation blockade. J Pept Res. 2005;65:591–604. doi: 10.1111/j.1399-3011.2005.00256.x. [DOI] [PubMed] [Google Scholar]
- 149.Kaumaya PT, Kobs-Conrad S, Seo YH, et al. Peptide vaccines incorporating a ‘promiscuous’ T-cell epitope bypass certain haplotype restricted immune responses and provide broad spectrum immunogenicity. J Mol Recogn. 1993;6:81–94. doi: 10.1002/jmr.300060206. [DOI] [PubMed] [Google Scholar]
- 150.Dakappagari NK, Pyles J, Parihar R, Carson WE, Young DC, Kaumaya PT. A chimeric multi-human epidermal growth factor receptor-2 B cell epitope peptide vaccine mediates superior antitumor responses. J Immunol. 2003;170:4242–4253. doi: 10.4049/jimmunol.170.8.4242. [DOI] [PubMed] [Google Scholar]
- 151.Dakappagari NK, Lute KD, Rawale S, et al. Conformational HER-2/neu B-cell epitope peptide vaccine designed to incorporate two native disulfide bonds enhances tumor cell binding and antitumor activities. J Biol Chem. 2005;280:54–63. doi: 10.1074/jbc.M411020200. [DOI] [PubMed] [Google Scholar]
- 152.Allen SD, Garrett JT, Rawale SV, et al. Peptide vaccines of the HER-2/neu dimerization loop are effective in inhibiting mammary tumor growth in vivo. J Immunol. 2007;179:472–482. doi: 10.4049/jimmunol.179.1.472. [DOI] [PubMed] [Google Scholar]
- 153.Garrett JT, Rawale S, Allen SD, et al. Novel engineered trastuzumab conformational epitopes demonstrate in vitro and in vivo antitumor properties against HER-2/neu. J Immunol. 2007;178:7120–7131. doi: 10.4049/jimmunol.178.11.7120. [DOI] [PubMed] [Google Scholar]
- 154.Vicari D, Foy KC, Liotta EM, Kaumaya PT. Engineered conformation-dependent VEGF peptide mimics are effective in inhibiting VEGF signaling pathways. J Biol Chem. 2011;286:13612–13625. doi: 10.1074/jbc.M110.216812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Foy KC, Liu Z, Phillips G, Miller M, Kaumaya PT. Combination treatment with HER-2 and VEGF peptide mimics induces potent anti-tumor and anti-angiogenic responses in vitro and in vivo. J Biol Chem. 2011;286:13626–13637. doi: 10.1074/jbc.M110.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kaumaya PT. Could precision-engineered peptide epitopes/vaccines be the key to a cancer cure? Future Oncol. 2011;7:807–810. doi: 10.2217/fon.11.60. [DOI] [PubMed] [Google Scholar]
- 157.Lairmore MD, DiGeorge AM, Conrad SF, Trevino AV, Lal RB, Kaumaya PT. Human T-lymphotropic virus type 1 peptides in chimeric and multivalent constructs with promiscuous T-cell epitopes enhance immunogenicity and overcome genetic restriction. J Virol. 1995;69:6077–6089. doi: 10.1128/jvi.69.10.6077-6089.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Partidos CD, Steward MW. Prediction and identification of a T cell epitope in the fusion protein of measles virus immunodominant in mice and humans. J Gen Virol. 1990;71(Pt 9):2099–2105. doi: 10.1099/0022-1317-71-9-2099. [DOI] [PubMed] [Google Scholar]
- 159.Paniana-Bordignon P, Tan A, Termijtelen A, Demotz S, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes: promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur J Immunol. 1989;19:2237. doi: 10.1002/eji.1830191209. [DOI] [PubMed] [Google Scholar]
- 160.Cox E, Verdonck F, Vanrompay D, Goddeeris B. Adjuvants modulating mucosal immune responses or directing systemic responses towards the mucosa. Vet Res. 2006;37:511–539. doi: 10.1051/vetres:2006014. [DOI] [PubMed] [Google Scholar]
- 161.O’Hagan DT, Valiante NM. Recent advances in the discovery and delivery of vaccine adjuvants. Nat Rev Drug Discov. 2003;2:727–735. doi: 10.1038/nrd1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McNeela EA, Mills KH. Manipulating the immune system: humoral versus cell-mediated immunity. Adv Drug Deliv Rev. 2001;51:43–54. doi: 10.1016/s0169-409x(01)00169-7. [DOI] [PubMed] [Google Scholar]
- 163.Jackson DC, Lau YF, Le T, et al. A totally synthetic vaccine of generic structure that targets Toll-like receptor 2 on dendritic cells and promotes antibody or cytotoxic T cell responses. Proc Natl Acad Sci USA. 2004;101:15440–15445. doi: 10.1073/pnas.0406740101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Daemen T, de Mare A, Bungener L, de Jonge J, Huckriede A, Wilschut J. Virosomes for antigen and DNA delivery. Adv Drug Deliv Rev. 2005;57:451–463. doi: 10.1016/j.addr.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 165.Diwan M, Tafaghodi M, Samuel J. Enhancement of immune responses by co-delivery of a CpG oligodeoxynucleotide and tetanus toxoid in biodegradable nanospheres. J Control Release. 2002;85:247–262. doi: 10.1016/s0168-3659(02)00275-4. [DOI] [PubMed] [Google Scholar]
- 166.Lynch JM, Briles DE, Metzger DW. Increased protection against pneumococcal disease by mucosal administration of conjugate vaccine plus interleukin-12. Infect Immun. 2003;71:4780–4788. doi: 10.1128/IAI.71.8.4780-4788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kaumaya PTP, Kobs-Conrad S, DiGeorge AM, Stevens V. Denovo engineering of protein immunogenic and antigenic determinants. In: Anantharamaiah GM, editor. Peptides. Springer-Verlag; Berlin, Germany: 1994. pp. 133–164. [Google Scholar]
- 168.Baselga J, Cortés J, Kim SB, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169▪▪.Cho HS, Mason K, Ramyar KX, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. Elucidation of the structure of the HER-2 extracellular domain alone and in complex with the humanized monoclonal antibody trastuzumab. [DOI] [PubMed] [Google Scholar]
- 170.Garrett TP, McKern NM, Lou M, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 171▪▪.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–328. doi: 10.1016/s1535-6108(04)00083-2. Provides significant insights into the structural interactions between the pertuzumab antibody and domain II of the HER-2 extracellular domain. Based on these interactions, it is possible to design specific B-cell epitopes that include the key amino acids that are important in binding. [DOI] [PubMed] [Google Scholar]
- 172.Kaumaya PT, Foy KC, Garrett J, et al. Phase I active immunotherapy with combination of two chimeric, human epidermal growth factor receptor 2, B-cell epitopes fused to a promiscuous T-cell epitope in patients with metastatic and/or recurrent solid tumors. J Clin Oncol. 2009;27:5270–5277. doi: 10.1200/JCO.2009.22.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sundaram R, Dakappagari NK, Kaumaya PT. Synthetic peptides as cancer vaccines. Biopolymers. 2002;66:200–216. doi: 10.1002/bip.10258. [DOI] [PubMed] [Google Scholar]
- 174.Mizejewski GJ. Peptides as receptor ligand drugs and their relationship to G-coupled signal transduction. Expert Opin Invest Drugs. 2001;10:1063–1073. doi: 10.1517/13543784.10.6.1063. [DOI] [PubMed] [Google Scholar]
- 175.Hruby VJ. Conformational and topographical considerations in the design of biologically active peptides. Biopolymers. 1993;33:1073–1082. doi: 10.1002/bip.360330709. [DOI] [PubMed] [Google Scholar]
- 176.Taylor EM, Otero DA, Banks WA, O’Brien JS. Retro-inverso prosaptide peptides retain bioactivity, are stable in vivo, and are blood-brain barrier permeable. J Pharmacol Exp Ther. 2000;295:190–194. [PubMed] [Google Scholar]
- 177.Fischer PM. The design, synthesis and application of stereochemical and directional peptide isomers: a critical review. Curr Protein Peptide Sci. 2003;4:339–356. doi: 10.2174/1389203033487054. [DOI] [PubMed] [Google Scholar]
- 178.Fletcher MD, Campbell MM. Partially modified retro-inverso peptides: development, synthesis, and conformational behavior. Chem Rev. 1998;98:763–796. doi: 10.1021/cr970468t. [DOI] [PubMed] [Google Scholar]
- 179.Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gasparini G, Longo R, Toi M, Ferrara N. Angiogenic inhibitors: a new therapeutic strategy in oncology. Nat Clin Pract Oncol. 2005;2:562–577. doi: 10.1038/ncponc0342. [DOI] [PubMed] [Google Scholar]
- 181.Keyt BA, Nguyen HV, Berleau LT, et al. Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors Generation of receptor-selective VEGF variants by site-directed mutagenesis. J Biol Chem. 1996;271:5638–5646. doi: 10.1074/jbc.271.10.5638. [DOI] [PubMed] [Google Scholar]
- 182.Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, de Vos AM. Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc Natl Acad Sci USA. 1997;94:7192–7197. doi: 10.1073/pnas.94.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183▪▪.Muller YA, Chen Y, Christinger HW, et al. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure. 1998;6:1153–1167. doi: 10.1016/s0969-2126(98)00116-6. Maps the VEGF receptor binding site and also VEGF in complex with the Fab-binding antibody bevacizumab. The crystal structure of the complex was solved in this paper and also mutagenesis studies were performed to delineate which amino acids are important in binding. [DOI] [PubMed] [Google Scholar]
- 184.Zilberberg L, Shinkaruk S, Lequin O, et al. Structure and inhibitory effects on angiogenesis and tumor development of a new vascular endothelial growth inhibitor. J Biol Chem. 2003;278:35564–35573. doi: 10.1074/jbc.M304435200. [DOI] [PubMed] [Google Scholar]
- 185.Muller YA, Christinger HW, Keyt BA, de Vos AM. The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: multiple copy flexibility and receptor binding. Structure. 1997;5:1325–1338. doi: 10.1016/s0969-2126(97)00284-0. [DOI] [PubMed] [Google Scholar]
- 186.Goodman M, Ro S, Yamazaki T, et al. Topochemical design of bioactive peptides and peptidomimetics. Bioorg Khim. 1992;18:1375–1393. [PubMed] [Google Scholar]
- 187.Chorev M, Goodman M. Recent developments in retro peptides and proteins--an ongoing topochemical exploration. Trends Biotechnol. 1995;13:438–445. doi: 10.1016/S0167-7799(00)88999-4. [DOI] [PubMed] [Google Scholar]
- 188.Chorev M. The partial retro-inverso modification: a road traveled together. Biopolymers. 2005;80:67–84. doi: 10.1002/bip.20219. [DOI] [PubMed] [Google Scholar]
- 189.Foy KC, Liu Z, Phillips G, Miller M, Kaumaya PT. Combination treatment with HER-2 and VEGF peptide mimics induces potent anti-tumor and anti-angiogenic responses in vitro and in vivo. J Biol Chem. 2011;286:13626–13637. doi: 10.1074/jbc.M110.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cardiff RD, Anver MR, Gusterson BA, et al. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000;19:968–988. doi: 10.1038/sj.onc.1203277. [DOI] [PubMed] [Google Scholar]
- 191.Hennighausen L. Mouse models for breast cancer. Oncogene. 2000;19:966–967. doi: 10.1038/sj.onc.1203346. [DOI] [PubMed] [Google Scholar]
- 192.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Boggio K, Nicoletti G, Di Carlo E, et al. Interleukin 12-mediated prevention of spontaneous mammary adenocarcinomas in two lines of Her-2/neu transgenic mice. J Exp Med. 1998;188:589–596. doi: 10.1084/jem.188.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gullick WJ, Bottomley AC, Lofts FJ, et al. Three dimensional structure of the transmembrane region of the proto-oncogenic and oncogenic forms of the neu protein. EMBO J. 1992;11:43–48. doi: 10.1002/j.1460-2075.1992.tb05025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dankort D, Maslikowski B, Warner N, et al. Grb2 and Shc adapter proteins play distinct roles in Neu (ErbB-2)-induced mammary tumorigenesis: implications for human breast cancer. Mol Cell Biol. 2001;21:1540–1551. doi: 10.1128/MCB.21.5.1540-1551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zabuawala T, Taffany DA, Sharma SM, et al. An ets2-driven transcriptional program in tumor-associated macrophages promotes tumor metastasis. Cancer Res. 2010;70:1323–1333. doi: 10.1158/0008-5472.CAN-09-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pomerantz MM, Shrestha Y, Flavin RJ, et al. Analysis of the 10q11 cancer risk locus implicates MSMB and NCOA4 in human prostate tumorigenesis. PLoS Genet. 2010;6:e1001204. doi: 10.1371/journal.pgen.1001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yen L, You XL, Al Moustafa AE, et al. Heregulin selectively upregulates vascular endothelial growth factor secretion in cancer cells and stimulates angiogenesis. Oncogene. 2000;19:3460–3469. doi: 10.1038/sj.onc.1203685. [DOI] [PubMed] [Google Scholar]
- 200.Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 201.Konecny GE, Meng YG, Untch M, et al. Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin Cancer Res. 2004;10:1706–1716. doi: 10.1158/1078-0432.ccr-0951-3. [DOI] [PubMed] [Google Scholar]
- 202.Sun X, Kanwar JR, Leung E, Lehnert K, Wang D, Krissansen GW. Angiostatin enhances B7.1-mediated cancer immunotherapy independently of effects on vascular endothelial growth factor expression. Cancer Gene Ther. 2001;8:719–727. doi: 10.1038/sj.cgt.7700370. [DOI] [PubMed] [Google Scholar]
- 203.Kuo CJ, Farnebo F, Yu EY, et al. Comparative evaluation of the antitumor activity of antiangiogenic proteins delivered by gene transfer. Proc Natl Acad Sci USA. 2001;98:4605–4610. doi: 10.1073/pnas.081615298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Nair S, Boczkowski D, Moeller B, Dewhirst M, Vieweg J, Gilboa E. Synergy between tumor immunotherapy and antiangiogenic therapy. Blood. 2003;102:964–971. doi: 10.1182/blood-2002-12-3738. [DOI] [PubMed] [Google Scholar]
- 205.Grunstein J, Roberts WG, Mathieu-Costello O, Hanahan D, Johnson RS. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res. 1999;59:1592–1598. [PubMed] [Google Scholar]
- 206.Schneider BP, Wang M, Radovich M, et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol. 2008;26:4672–4678. doi: 10.1200/JCO.2008.16.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Foy KC, Miller MJ, Moldovan N, Carson WE, Kaumaya PTP. Combined vaccination with HER-2 peptide followed by therapy with VEGF peptide mimics exerts effective anti-tumor and anti-angiogenic effects in vitro and in vivo. OncoImmunology. 2012 doi: 10.4161/onci.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208▪▪.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. Discusses the benefits antiangiogenic agents have in the treatment of cancer. The article proposes the concept of tumor vasculature normalization, which results in better delivery of cancer therapeutics, thereby avoiding resistance and preventing tumor growth. [DOI] [PubMed] [Google Scholar]
- 209.Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays. 1991;13:31–36. doi: 10.1002/bies.950130106. [DOI] [PubMed] [Google Scholar]
- 210.Foy KC, Miller MJ, Moldovan N, Bozanovic T, Carson WE, Kaumaya PTP. Immunotherapy with HER2 and VEGF peptide mimics plus metronomic paclitaxel causes superior antineoplastic effects in transplantable and transgenic mouse models of human breast cancer. OncoImmunology. 2012;1:7. doi: 10.4161/onci.21057. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Nahta R. Pharmacological strategies to overcome HER2 cross-talk and trastuzumab resistance. Curr Med Chem. 2012;19:1065–1075. doi: 10.2174/092986712799320691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212▪.Roop RP, Ma CX. Endocrine resistance in breast cancer: molecular pathways and rational development of targeted therapies. Future Oncol. 2012;8:273–292. doi: 10.2217/fon.12.8. Summarizes the development of resistance mechanisms in breast cancer with emphasis on the different molecular pathways and ideas on how future therapeutics should be developed. [DOI] [PubMed] [Google Scholar]
- 213.Yakar S, Leroith D, Brodt P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: lessons from animal models. Cytokine Growth Factor Rev. 2005;16:407–420. doi: 10.1016/j.cytogfr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 214.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 215.Pollak MN. Insulin-like growth factors and neoplasia. Novartis Found Symp. 2004;262:84–98. Discussion 98–107, 265–268. [PubMed] [Google Scholar]
- 216.Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–1093. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case–control analysis. J Natl Cancer Inst. 1999;91:151–156. doi: 10.1093/jnci/91.2.151. [DOI] [PubMed] [Google Scholar]
- 218.Hankinson SE, Willett WC, Colditz GA, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 219.Bergmann U, Funatomi H, Yokoyama M, Beger HG, Korc M. Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 1995;55:2007–2011. [PubMed] [Google Scholar]
- 220.Ma J, Pollak MN, Giovannucci E, et al. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst. 1999;91:620–625. doi: 10.1093/jnci/91.7.620. [DOI] [PubMed] [Google Scholar]
- 221.Chan JM, Stampfer MJ, Giovannucci E, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279:563–566. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- 222.Wu X, Zhao H, Do KA, et al. Serum levels of insulin growth factor (IGF-I) and IGF-binding protein predict risk of second primary tumors in patients with head and neck cancer. Clin Cancer Res. 2004;10:3988–3995. doi: 10.1158/1078-0432.CCR-03-0762. [DOI] [PubMed] [Google Scholar]
- 223.Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006;66:10100–10111. doi: 10.1158/0008-5472.CAN-06-1684. [DOI] [PubMed] [Google Scholar]
- 224.Kuribayashi A, Kataoka K, Kurabayashi T, Miura M. Evidence that basal activity, but not transactivation, of the epidermal growth factor receptor tyrosine kinase is required for insulin-like growth factor I-induced activation of extracellular signal-regulated kinase in oral carcinoma cells. Endocrinology. 2004;145:4976–4984. doi: 10.1210/en.2004-0713. [DOI] [PubMed] [Google Scholar]
- 225.Balana ME, Labriola L, Salatino M, et al. Activation of ErbB-2 via a hierarchical interaction between ErbB-2 and type I insulin-like growth factor receptor in mammary tumor cells. Oncogene. 2001;20:34–47. doi: 10.1038/sj.onc.1204050. [DOI] [PubMed] [Google Scholar]
- 226.Camirand A, Lu Y, Pollak M. Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors causes synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit. 2002;8:BR521–BR526. [PubMed] [Google Scholar]
- 227.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65:11118–11128. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 228.Slomiany MG, Black LA, Kibbey MM, Day TA, Rosenzweig SA. IGF-1 induced vascular endothelial growth factor secretion in head and neck squamous cell carcinoma. Biochem Biophys Res Commun. 2006;342:851–858. doi: 10.1016/j.bbrc.2006.02.043. [DOI] [PubMed] [Google Scholar]
Websites
- 301.American Cancer Society. www.cancer.org.
- 302.A Study to Evaluate Pertuzumab + Trastuzumab + Docetaxel vs. Placebo + Trastuzumab + Docetaxel in Previously Untreated Her2-Positive Metastatic Breast Cancer (CLEOPATRA) http://clinicaltrials.gov/ct2/show/NCT00567190?term=NCT00567190&rank=1.
- 303.Vaccine Therapy in Treating Patients With Metastatic Solid Tumors. http://clinicaltrials.gov/ct2/show/NCT01376505?term=NCT+01376505&rank=1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.