

Esophageal squamous cell carcinoma and esophageal adenocarcinoma are aggressive cancers with poor patient response to conventional chemotherapy and radiation treatment. In this study, comprehensive genomic profiling showed that the frequently altered genes and biological pathways differed between the two subtypes, and a high frequency of clinically relevant genomic alterations was noted as a means of finding a potential targeted therapy to be used in addition or as an alternative to conventional treatment.
Keywords: Comprehensive genomic profiling, Next-generation sequencing, Esophageal cancer, Targeted therapy, Adenocarcinoma, Squamous cell
Abstract
Background.
Esophageal squamous cell carcinomas (ESCCs) and esophageal adenocarcinomas (EACs) account for >95% of esophageal malignancies and represent a major global health burden. ESCC is the dominant histology globally but represents a minority of U.S. cases, with EAC accounting for the majority of U.S. cases. The patient outcomes for advanced ESCC and EAC are poor, and new therapeutic options are needed. Using a sensitive sequencing assay, we compared the genomic profiles of ESCC and EAC with attention to identification of therapeutically relevant genomic alterations.
Methods.
Next-generation sequencing-based comprehensive genomic profiling was performed on hybridization-captured, adaptor ligation-based libraries to a median coverage depth of >650× for all coding exons of 315 cancer-related genes plus selected introns from 28 genes frequently rearranged in cancer. Results from a single sample were evaluated for all classes of genomic alterations (GAs) including point mutations, short insertions and deletions, gene amplifications, homozygous deletions, and fusions/rearrangements. Clinically relevant genomic alterations (CRGAs) were defined as alterations linked to approved drugs and those under evaluation in mechanism-driven clinical trials.
Results.
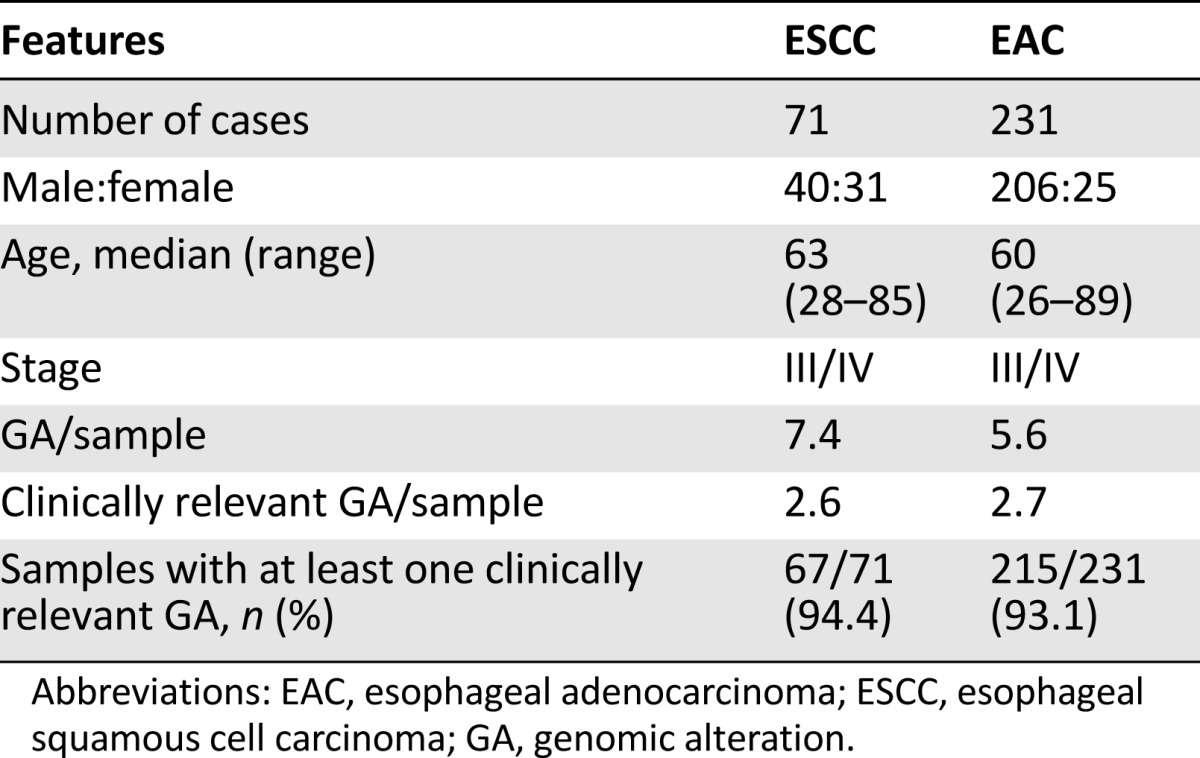
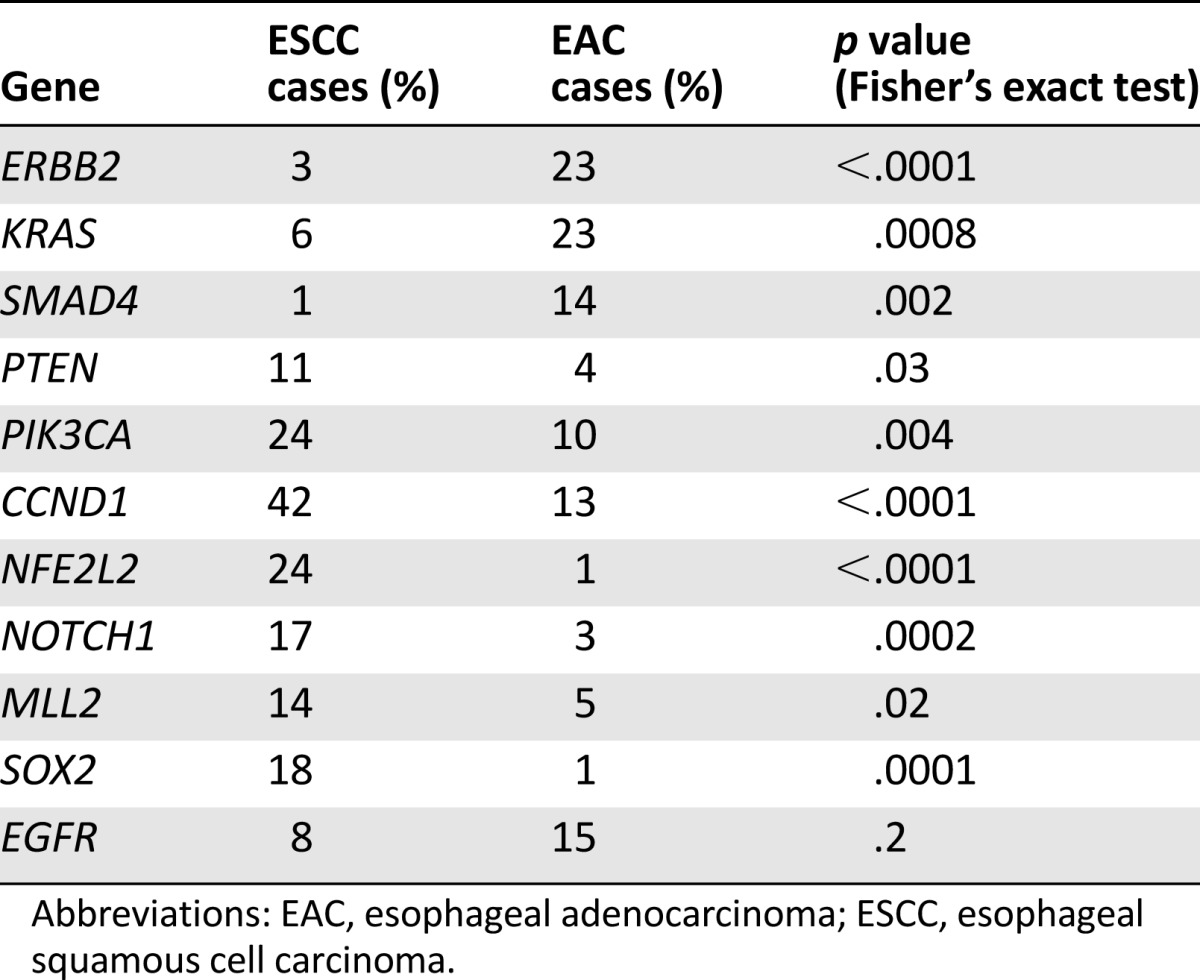
There were no significant differences by sex for either tumor type, and the median age for all patients was 63 years. All ESCCs and EACs were at an advanced stage at the time of sequencing. All 71 ESCCs and 231 EACs featured GAs on profiling, with 522 GAs in ESCC (7.4 per sample) and 1,303 GAs in EAC (5.6 per sample). The frequency of clinically relevant GAs in ESCC was 94% (2.6 per sample) and 93% in EAC (2.7 per sample). CRGAs occurring more frequently in EAC included KRAS (23% EAC vs. 6% ESCC) and ERBB2 (23% EAC vs. 3% ESCC). ESCC samples were enriched for CRGA in PIK3CA (24% ESCC vs. 10% EAC), PTEN (11% ESCC vs. 4% EAC), and NOTCH1 (17% ESCC vs. 3% EAC). Other GAs that differed significantly between histologic tumor types included SMAD4 (14% EAC vs. 1% ESCC), RB1 (14% ESCC vs. 2% EAC), SOX2 (18% ESCC vs. 1% EAC), and NFE2L2 (24% ESCC vs. 1% EAC).
Conclusion.
ESCC and EAC share similarly high frequencies of overall and clinically relevant genomic alterations; however, the profiles of genomic alterations in the two diseases differ widely, with KRAS and ERBB2 far more frequently altered in EAC compared with ESCC and with mammalian target of rapamycin (MTOR) pathway genes (PIK3CA and PTEN) and NOTCH1 more frequently altered in ESCC compared with EAC. Comprehensive genomic profiling highlights the promise of identifying clinically relevant genomic alterations in both ESCC and EAC and suggests new avenues for molecularly directed therapies in esophageal cancer.
Implications for Practice:
Both esophageal squamous cell carcinoma and esophageal adenocarcinoma are aggressive cancers with poor patient response to conventional chemotherapy and radiation treatment. In this study, comprehensive genomic profiling was performed for 302 advanced esophageal cancers, and it was found that the frequently altered genes and biological pathways differed between the two subtypes. Also, a high frequency of clinically relevant genomic alterations was noted for both types of esophageal cancer as a means of finding a potential targeted therapy to be used in addition to or as an alternative to conventional treatment.
Introduction
Esophageal cancers account for more than 400,000 cancer-related deaths worldwide and represent a major global cancer burden. The vast majority of esophageal neoplasms are adenocarcinomas of the distal esophagus/gastroesophageal junction (esophageal adenocarcinoma [EAC]) and squamous cell carcinomas of the proximal and midesophagus (esophageal squamous cell carcinoma [ESCC]) [1–4]. Both histologic subtypes share a common origin, developing from the epithelial lining of the esophagus, and typically present at an advanced clinical stage at the time of diagnosis [1–4]. Determining the exact site of origin for EAC is challenging because visual and pathologic analysis may not confirm origination from metaplastic glandular epithelium of the distal esophagus or directly from gastric cardia epithelium with proximal growth into the distal esophagus [1–4]. For this reason, the term “gastroesophageal” adenocarcinoma has been used to encompass tumors of uncertain site of origin [1–4]. The development of EAC is generally considered to progress through a metaplasia-dysplasia-adenocarcinoma (M-D-A) sequence associated with chronic inflammatory insult linked to gastroesophageal reflux disease and the premalignant condition known as Barrett’s esophagus [1–4]. ESCC has been linked to both smoking and alcohol consumption, with a less substantiated pathophysiologic link to human papillomavirus (HPV) infection [5–7].
The therapeutic approach to ESCC and EAC is similar in both the locoregional and advanced settings, and the histologic subtypes are frequently lumped together in large phase III clinical trials. Generally, the standard of care for patients with locoregional ESCC involves neoadjuvant chemoradiation followed by response assessment and observation or surgical resection [1–4, 8–11]. To date, there are no approved targeted therapies for ESCC, and recurrence rates remain high for locoregional disease [8–11]. Locoregional EAC is approached similarly, although the data supporting surgical resection after neoadjuvant therapy is stronger in EAC.
Although surgical resection is the primary treatment of choice for locoregional esophageal tumors, a large proportion are inoperable or metastatic at diagnosis. Similarly, the recurrence rates and 3- to 5-year survival for locoregional disease remains poor with current therapies [12–14]. Systemic therapy for inoperable and recurrent or metastatic ESCC and EAC using conventional chemotherapy and radiation has achieved only modest success, and the vast majority of patients with advanced disease die of their illness within the first 2 years after treatment has been started [15–17]. There are currently no molecularly directed agents approved for ESCC. In contrast to ESCC, anti-ERBB2 targeted therapy (trastuzumab) for ERBB2 amplified (fluorescence in situ hybridization or immunohistochemistry) is approved worldwide for advanced EAC harboring this molecular target [18, 19]. The trastuzumab-platinum regimen used on label for EAC has significantly improved patient survival for the 15% of EAC that tests positive for HER2 (ERBB2) amplification or overexpression [18, 19]. Currently, ESCCs are not routinely tested for HER2 status, and anti-HER2 therapy has not been widely used for pure squamous esophageal cancers. Although a number of EGFR, HER2, and VEGF therapies are in clinical trial phase II/III for esophageal cancer, to date, practice-changing results have remained elusive [20].
In recent years, promising information has emerged from comprehensive genomic profiling of relapsed and refractory cancers including separate evaluation of major esophageal cancers such as EAC and ESCC [21–24]. These research studies have identified potential genomic targets for patients with esophageal cancers, but comprehensive molecular testing has not been widely adopted in routine clinical practice for esophageal cancer. In this paper, we present a series of EACs and ESCCs subjected to comprehensive genomic profiling and present the respective genomic signatures analyzed for their biological significance and impact for potential targeted therapies. Our series reflects real-world esophageal cancer cases and demonstrates a high frequency of clinically relevant genomic alterations (CRGAs) in EAC and ESCC. The therapeutic impact of differences between profiling results for ESCC and EAC suggests targeted therapies for future consideration in a disease with historically poor outcomes and limited options.
Methods
Next-generation sequencing-based comprehensive genomic profiling was performed on all formalin-fixed paraffin-embedded tissues using a hybrid capture-based next-generation sequencing platform (FoundationOne; Foundation Medicine, Cambridge, MA, http://www.foundationone.com) at a Clinical Laboratory Improvement Amendments-certified, New York State and College of American Pathologists-accredited laboratory (Foundation Medicine) on the Illumina HiSeq2500 instruments (Illumina Inc., San Diego, CA, http://www.illumina.com) [25]. The 50 ng of DNA extracted from 71 clinically advanced ESCCs and 231 EACs was adaptor ligated, and capture was performed for all coding exons of 315 cancer-related genes and selected introns of 28 genes frequently rearranged in cancer. Captured libraries were sequenced to a median exon coverage depth of >650×, and resultant sequences were analyzed for base substitutions, short insertions, deletions, gene copy number alterations (focal amplifications and homozygous deletions), and gene fusions, as previously described [25]. The sequence analysis methods and validation of the comprehensive genomic profiling platform used in this study included extensive comparisons to orthogonal methodologies [25]. Base substitution detection was performed using a Bayesian methodology, which allows detection of novel somatic mutations at low mutant allele frequency (MAF) and increased sensitivity for mutations at hotspot sites through the incorporation of tissue-specific prior expectations [25]. Reads with mapping quality <25 were discarded, as were base calls with quality ≤2. Final calls are made at MAF ≥5% (MAF ≥1% at hotspots) after filtering for strand bias (Fisher’s exact test, p < 16), read location bias (Kolmogorov-Smirnov test, p < 16), and presence in 2 or more normal controls. To detect indels, de novo local assembly in each targeted exon was performed using the de Bruijn approach [26, 27]. After read pairs were collected and decomposed, the statistical support for competing haplotypes was evaluated, and candidate indels were aligned against the reference genome. Filtering of insertion and deletion (indel) candidates was carried out as described for base substitutions. Gene amplifications and homozygous deletions were detected by comparing complete chromosomal copy number maps to reference process-matched normal control samples. Gene fusions and rearrangements were detected by analysis of chimeric read pairs [25]. Finally, unique sequences of nine viruses (hepatitis B virus, hepatitis C virus, human herpesvirus 4 [HHV-4; Epstein-Barr virus], HHV-8, HPV-11, HPV-16, HPV-18, HPV-6, and human T-lymphotropic virus type 1) and Helicobacter pylori were baited, and the sequenced DNA data were mapped to the virus genomes to detect virus DNA.
CRGAs were defined as those genomic alterations that could be targeted using anticancer drugs currently on the market for any tumor type with known primary site or GA required for entry in mechanism-driven registered clinical trials. Local site permissions to use clinical samples and approval by the Albany Medical College institutional review board to analyze and report patient data were obtained for this study.
Results
Patient and sample characteristics are presented in Table 1. A total of 302 samples representing 71 ESCC and 231 EAC cases from stage III and IV esophageal cancers were analyzed. The ESCC cohort was composed of 40 (56%) male and 31 (44%) female patients with a median age of 63 years (range: 28–85 years). For EAC, there were 206 (89%) male and 25 (11%) female patients with a median age of 60 years (range: 26–89 years) (Table 1).
Table 1.
Clinical features and genomic alteration frequencies in 302 cases of esophageal carcinoma
All 71 (100%) ESCCs and 231 (100%) EACs featured at least 1 GA on profiling. A total of 522 GAs were identified in the ESCC group (7.4 per sample), and 1,303 GAs (5.6 per sample) were noted in the EAC group (Table 1). The frequency of CRGAs in ESCC was 94% (67 of 71 cases, mean 2.6 CRGAs per sample), nearly identical to that found in the EAC cohort (93%; 215 of 231 cases, mean 2.7 CRGAs per sample) (Table 1). Distribution of the types of genomic alterations (base substitutions and short indels, truncation mutations, copy number changes, and rearrangements/fusions) was similar for the two tumor types (Figs. 1, 2). Individual CRGAs that were more frequently altered in EAC than ESCC included KRAS, which was altered in 23% of EAC versus 6% of ESCC cases (p = .0008), and ERBB2, which was altered in 23% of EAC versus 3% in ESCC cases (p < .0001). Clinically relevant genomic alterations enriched in the ESCC cohort included PIK3CA, altered in 24% of ESCCs versus 10% of EACs (p = .004); PTEN, altered in 11% of ESCCs versus 4% of EACs (p = .03); and NOTCH1, altered in 17% of ESCCs versus 3% of EACs (p = .0002) (Table 2). Other GAs that differed significantly between the tumor types included SMAD4 (14% EAC vs. 1% ESCC; p = .0018), RB1 (14% ESCC vs. 2% EAC;p = .0003), SOX2 (18% ESCC vs. 1% EAC; p = .0001), MLL2 (14% ESCC vs. 5% EAC; p = .0179), CCND1 (42% ESCC vs. 13% EAC; p < .0001), and NFE2L2 (24% ESCC vs. 1% EAC; p < .0001). HPV-16 was detected in 2 (3%) and HPV-18 in 2 (3%) ESCCs. Neither HPV nor H. pylori were detected in any cases of EAC. In summary, the genes that were altered at a high frequency in ESCC were TP53, CCND1, FGF3/4/19, CDKN2A/B, NFE2L2, PIK3CA, SOX2, and NOTCH1 (Fig. 1, supplemental online Table 1). The genes that were altered at a high frequency in EAC were TP53, CDKN2A, ERBB2, KRAS, MYC, EGFR, SMAD4, FGF3/4/19, and CCND1 (Fig. 2, supplemental online Table 2).
Figure 1.

Long-tail distribution of genomic alterations in 71 cases of esophageal squamous cell carcinoma.
Figure 2.

Long-tail distribution of genomic alterations in 231 cases of esophageal adenocarcinoma.
Table 2.
Contrasting genomic alterations in 71 cases of ESCC and 231 cases of EAC
Genomic alterations were grouped by biological pathway in which the altered gene existed (Fig. 3). Differential GA frequency reflected activation of distinct biological pathways between ESCC and EAC. The ESCC cohort demonstrated enrichment in alterations involving PI3K/AKT/MTOR signaling (45% ESCC vs. 23% EAC), epigenetic regulation pathway (51% ESCC vs. 34% EAC), fibroblast growth factor (FGF) signaling (56% ESCC vs. 20% EAC), the Keap/NRF2 pathway (27% ESCC vs. 0% EAC), and the Notch signaling pathway (23% ESCC vs. 6% EAC) (Fig. 3). The pathways altered at a higher frequency in EAC included ERBB (37% EAC vs. 13% ESCC), transforming growth factor (TGF) β signaling (34% EAC vs. 8% ESCC), and RAS/MEK/MAPK (34% EAC vs. 8% ESCC). Cell cycle pathway genes were found to be equally altered in both subtypes of esophageal cancers, with ESCC cases at 94% and EAC cases at 93% (Fig. 3).
Figure 3.

Frequency of alterations in biologic pathways in esophageal squamous cell carcinoma and esophageal adenocarcinoma.
In the 71 ESCC cases, 2 (3%) tumors featured ERBB2 amplification. In the 231 EAC cases, 54 (23%) featured ERBB2 amplification (Table 2). In two cases of EAC harboring ERBB2 gene amplification, point mutations in ERBB2 were also found involving one kinase domain mutation (V777L) and one extracellular domain mutation (S310F). The 231 EACs also featured 31 (13%) tumors with EGFR amplification (Fig. 2). Two EAC cases with EGFR amplification also featured EGFR point mutations (P596L and K714N). In addition, two EGFR rearrangements were found in the EAC group, which may predict alternative activation of EGFR signaling. In the 71 ESCC cases, 6 (8%) featured EGFR amplification (Table 2).
High-level ERBB2 amplification (>30 copies) was identified in a male patient aged 70 years with de novo metastatic EAC. This patient with multiple hepatic metastases demonstrated a rapid response to 3 months of single-agent trastuzumab (Fig. 4). Concurrent GA in this case included GNAS R165C, SMARCA4 R1135W, TP53 R273H, DNMT3A C586fs*26, and CDK12 gene rearrangement and amplification. Similarly, another EAC patient with metastatic recurrence 2 years after neoadjuvant chemoradiation and surgery was found to harbor high-level ERBB2 amplification with more than 70 copies. This patient demonstrated a prolonged radiographic remission after trastuzumab-containing systemic therapy, followed by 2 years of trastuzumab maintenance monotherapy with continuous remission since 2013 (Fig. 5). The other alterations identified in this patient included CDKN2A V51F, TP53 R213*, CREBBP M1193fs*49, and MCL1 gene amplification with copy number at seven copies.
Figure 4.

Response after 3 months of trastuzumab in a 70-year-old patient with ERBB2 amplified esophageal adenocarcinoma identified by comprehensive genomic profiling. From left to right, extensive hepatic metastases (A) resolved after 3 months of genotype-directed therapy (B).
Figure 5.

Response to FOLFOX + herceptin in a patient with ERBB2-amplified esophageal adenocarcinoma. A male patient aged 66 years showed ERBB2-amplified advanced esophageal adenocarcinoma (A). High-level ERBB2 amplification with >70 copy numbers informed treatment with the FOLFOX regimen plus trastuzumab for 7 months with complete response (B) followed by continuous remission for more than 2 years on single-agent trastuzumab. (A): Right adrenal mass 2.1 × 2.9 cm. Date of computed tomography (CT) scan, June 25, 2012. (B): Right adrenal mass nonmeasurable. Date of CT scan, August 24, 2014.
Discussion
Despite the differing risk factors and pathobiology of ESCC and EAC, both diseases are aggressive malignancies with a propensity to present as advanced disease with substantial patient morbidity [1–4]. Systemic options for metastatic disease are limited and composed primarily of multiagent cytotoxic chemotherapy, which achieves a median overall survival of 12 months. The Anti-ERBB2 monoclonal antibody trastuzumab is the only approved targeted therapy, and use is generally restricted to EAC. Despite the introduction of targeted anti-ERBB2 therapy for EAC [18, 19], advances in patient overall and disease-free survival in the neoadjuvant chemoradiation era have been limited [15–17]. The increasing importance of molecularly directed therapies for advanced cancers has been highlighted by successes in non-small cell lung cancer harboring oncogenic drivers (ALK, EGFR, ROS, cMET, RET) and other malignancies. Extension of this genomically driven therapeutic paradigm to other disease may be possible and has led investigators to seek such options for patients with advanced esophageal carcinoma. Comprehensive genomic profiling technologies have enabled clinical appreciation of molecular targets in advanced esophageal cancer such as the series presented in this paper.
In the current study, the traditional clinicopathologic differences between ESCC and EAC have been expanded by addition of genomic profiling. ESCC and EAC both have high frequencies of overall and clinically relevant GA; however, the specific distribution of GA in the two diseases differs widely with regard to altered genes and pathways, with KRAS and ERBB2 far more frequently altered in EAC compared with ESCC and with mammalian target of rapamycin pathway genes (PIK3CA and PTEN) and NOTCH1 more frequently altered in ESCC compared with EAC (Table 2, Fig. 3). In addition, the more frequent NOTCH1 mutations seen in ESCC versus EAC identified in the current study have also been reported previously in a retrospective analysis of primary tumors, validating the approach used in our study [22]. For EAC, we compared our frequency of substitutions and indels with one previous study [28]. In the current study, a higher frequency of variants in TP53 (83% vs. 71%) and in CDKN2A (24% vs. 13%) were noted. In addition, there was a significantly lower frequency of variants in ERBB4 in the current study at 6% compared with the previously published frequency of 12% [28]. For all other genes that were evaluated in both EAC series with a frequency of greater than 5%, the difference in the frequency of genomic alterations was not statistically significant (supplemental online Tables 1, 2). Finally, we compared our findings in base substitutions and indels for ESCC with three previous studies of genomic alterations in this disease (supplemental online Figs. 1, 2) [29–32]. The current study is essentially in agreement with previous studies on the frequency of genomic alterations in ESCC but also showed differences possibly of note. TP53 mutations were found in 92% of our cases, but only 58% were noted in the NG3076 data set. NFE2L2 was at 26% but at only 6% and 9% in the 2 published data sets (supplemental online Fig. 1). We identified SOX2 gene amplifications at 16%; in contrast; it was 2% and 10% in the 2 published data sets (supplemental online Fig. 2). Our series is representative of U.S. clinical practice, and the prior esophageal series have generally been based on Asian ESCC patients and involved treatment-naïve samples. The altered frequency difference may indicate distinct genetic mechanisms due to stages of disease at the time of sequencing, environment, ethnicity, and pre- or posttreatment factors. Although our series contains some heterogeneity in terms of specimens used for genomic profiling (pre- or posttreatment, primary vs. metastatic), the cases all had advanced disease, and the overall similarity with previously published research series suggests that the methodology used in our series can achieve robust results capable of influencing patient care. The patient examples presented support two interesting observations from our case series. First, genomic profiling produces quantitative estimates of ERBB2 amplifications, and second, higher level ERBB2 copy number may be associated with greater patient benefit from anti-ERBB2 directed therapies. Larger series with clinical outcomes will be needed to determine the influence on treatment response of the co-occurring GA in these patients, and the concept of passenger GA modifying response is an area of active investigation.
In addition to contrasting the genomic alterations in ESCC versus EAC, we sought to identify potential therapeutic targets not traditionally assessed in cases of advanced esophageal cancer. The frequency of clinically relevant alterations in both tumor types was striking at 2.7 clinically relevant genomic alterations per sample in both ESCC and EAC. For ESCC, notable potential therapy-related targets included alterations in PTEN (11%), EGFR (8%), and FGFR1 (7%). Driver genomic alteration prevalence in ESCC shows significant difference from several published results concerning Asian patients [29–31] and may indicate distinct genetic mechanisms due to stage of disease at the time of sequencing, environmental factors such as infection related tumors, ethnicity, and treatment history. Activation of the PI3K/AKT/MTOR pathway associated with loss of PTEN is a common contributor to ESCC development and progression [33]. PTEN loss or mutation may predict sensitivity to inhibitors of the PI3K/AKT/MTOR pathway [34]. EGFR amplification and/or overexpression has been detected in a wide variety of ESCC samples, ranging from 7% to greater than 60%, and has been associated with adverse prognosis [35, 36]. EGFR activating mutations or amplification may predict sensitivity to EGFR inhibitors including kinase inhibitors and anti-EGFR antibody therapeutics [37]. Nonetheless, the prior suggestion has not held true in an unstratified population of EAC cases, as shown in the phase III clinical trials EXPAND and REAL3 [38, 39]. Such therapy could yield clinical benefit if given to a population known to harbor relevant GA in EGFR. FGFR1 amplification has been reported to occur twice as frequently in ESCC than in EAC and has been associated with an unfavorable prognosis [40]. Tumors with FGFR1 amplification or activating mutations may be sensitive to FGFR inhibitors such as pazopanib and ponatinib and to other kinase inhibitors currently in clinical trials [41]. For EAC, notable potential therapy targets included the well-known ERBB2 target in 23% of cases and the EGFR (14%) and FGFR2 (5%) targets described above in the ESCC patient cohort. Targeting ERBB2 is now well established as a major approach in the treatment of EAC [18, 19]. The EGFR and FGFR2 alterations in this study were more frequently identified in EAC than in ESCC. Notable additional targets included PIK3CA (10% EAC) and MET (4% EAC). One EAC patient with MET amplification responded to crizotinib and had stable disease for more than 2 months and then progressed. The MET gene had a focal change of >50 copies, and this patient also harbored CDKN2A/B gene homozygous deletions, TP53 E286K, SMAD4 R361C, and APC M314fs*23 truncation, so perhaps MET amplification was not the only dominant driving gene for this tumor. In a recent study of MET inhibitor AMG 337, 62% of 13 patients with MET amplification showed responses to AMG 337 [42]. A MET amplified gastroesophageal junction patient, for example, responded significantly to MET inhibitor, and the tumor size decreased 40% in 5 weeks, reached complete response in 40 weeks, and continued response for more than 38 months [42]. More investigation is needed on targeted therapy for patients with MET amplification.
Conclusion
Comprehensive genomic profiling shows significant promise in identifying clinically relevant genomic alterations in both ESCC and EAC and informs the potential use of targeted therapies in both major types of esophageal cancer. The frequent identification of clinically relevant genomic alterations in our series underscores the need for increased molecularly directed clinical trials to confirm the improved patient outcomes observed in our cases.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgment
The contents of our manuscript have not been previously submitted for publication, but the data were presented as an oral presentation at the 2015 American Society of Clinical Oncology Gastrointestinal Cancers Symposium, January 15–17, 2015, in San Francisco, California.
Footnotes
For Further Reading: Patrick M. Forde, Ronan J. Kelly. Genomic Alterations in Advanced Esophageal Cancer May Lead to Subtype-Specific Therapies. The Oncologist 2013;18:823–832.
Implications for Practice: The disease burden of esophageal cancer is increasing in the United States and worldwide, primarily driven by higher rates of adenocarcinoma risk factors, including obesity and Barrett's esophagus. Chemotherapy has moderate efficacy for locally advanced and metastatic esophageal cancer, but new approaches to treatment are urgently needed. This article focuses on potential oncogenic targets in esophageal cancer and comprehensively reviews the current state of the art in targeted therapy for esophageal and gastroesophageal junction tumors. Anti-human epidermal growth factor receptor-2 therapy has provided benefit for a small proportion of patients; however, despite signs of efficacy in early phase clinical trials, results with anti-epidermal growth factor receptor and anti-vascular endothelial growth factor therapy have been generally disappointing. Experience to date with targeted agents suggests that collaborative trials of target-specific agents in those subgroups of patients who have potential oncogenic drivers represent the best opportunity for bringing novel agents to the clinic.
Author Contributions
Conception/Design: Kai Wang, Adrienne Johnson, Siraj M. Ali, Vincent A. Miller, Philip J. Stephens, Jeffrey S. Ross
Provision of study material or patients: Siraj M. Ali, Samuel J. Klempner, Tanios Bekaii-Saab, Jeffrey L. Vacirca, Jeffrey S. Ross
Collection and/or assembly of data: Kai Wang, Adrienne Johnson, Samuel J. Klempner, Tanios Bekaii-Saab, Jeffrey L. Vacirca, Depinder Khaira, Roman Yelensky, Juliann Chmielecki, Julia A. Elvin, Doron Lipson, Jeffrey S. Ross
Data analysis and interpretation: Kai Wang, Adrienne Johnson
Manuscript writing: Kai Wang, Siraj M. Ali, Samuel J. Klempner, Jeffrey S. Ross
Final approval of manuscript: Kai Wang, Adrienne Johnson, Siraj M. Ali, Samuel J. Klempner, Tanios Bekaii-Saab, Jeffrey L. Vacirca, Depinder Khaira, Roman Yelensky, Juliann Chmielecki, Julia A. Elvin, Doron Lipson, Vincent A. Miller, Philip J. Stephens, Jeffrey S. Ross
Disclosures
Kai Wang: Foundation Medicine (E, OI); Adrienne Johnson: Foundation Medicine (E, OI); Siraj M. Ali: Foundation Medicine (E, OI, IP); Samuel J. Klempner: Foundation Medicine (H); Tanios Bekaii-Saab: Genentech (C/A); Jeffrey L. Vacirca: NSHOA Cancer Center (E), Spectrum Pharmaceuticals (C/A, H), Heron Therapeutics (OI); Depinder Khaira: Foundation Medicine (E, OI); Roman Yelensky: Foundation Medicine (E, OI); Juliann Chmielecki: Foundation Medicine (E, OI); Julia A. Elvin: Foundation Medicine (E, OI); Doron Lipson: Foundation Medicine (E, OI); Vincent A. Miller: Foundation Medicine (E, OI, IP); Philip J. Stephens: Foundation Medicine (E, OI); Jeffrey S. Ross: Foundation Medicine (RF, E, OI).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 2.Engel LS, Chow WH, Vaughan TL, et al. Population attributable risks of esophageal and gastric cancers. J Natl Cancer Inst. 2003;95:1404–1413. doi: 10.1093/jnci/djg047. [DOI] [PubMed] [Google Scholar]
- 3.Lagergren J, Lagergren P. Recent developments in esophageal adenocarcinoma. CA Cancer J Clin. 2013;63:232–248. doi: 10.3322/caac.21185. [DOI] [PubMed] [Google Scholar]
- 4.Pennathur A, Gibson MK, Jobe BA, et al. Oesophageal carcinoma. Lancet. 2013;381:400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 5.Al-Haddad S, El-Zimaity H, Hafezi-Bakhtiari S, et al. Infection and esophageal cancer. Ann N Y Acad Sci. 2014;1325:187–196. doi: 10.1111/nyas.12530. [DOI] [PubMed] [Google Scholar]
- 6.Michaelsen SH, Larsen CG, von Buchwald C. Human papillomavirus shows highly variable prevalence in esophageal squamous cell carcinoma and no significant correlation to p16INK4a overexpression: A systematic review. J Thorac Oncol. 2014;9:865–871. doi: 10.1097/JTO.0000000000000166. [DOI] [PubMed] [Google Scholar]
- 7.Hardefeldt HA, Cox MR, Eslick GD. Association between human papillomavirus (HPV) and oesophageal squamous cell carcinoma: A meta-analysis. Epidemiol Infect. 2014;142:1119–1137. doi: 10.1017/S0950268814000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tepper J, Krasna MJ, Niedzwiecki D, et al. Phase III trial of trimodality therapy with cisplatin, fluorouracil, radiotherapy, and surgery compared with surgery alone for esophageal cancer: CALGB 9781. J Clin Oncol. 2008;26:1086–1092. doi: 10.1200/JCO.2007.12.9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh TN, Noonan N, Hollywood D, et al. A comparison of multimodal therapy and surgery for esophageal adenocarcinoma. N Engl J Med. 1996;335:462–467. doi: 10.1056/NEJM199608153350702. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11–20. doi: 10.1056/NEJMoa055531. [DOI] [PubMed] [Google Scholar]
- 11.Sjoquist KM, Burmeister BH, Smithers BM, et al. Survival after neoadjuvant chemotherapy or chemoradiotherapy for resectable oesophageal carcinoma: An updated meta-analysis. Lancet Oncol. 2011;12:681–692. doi: 10.1016/S1470-2045(11)70142-5. [DOI] [PubMed] [Google Scholar]
- 12.Jang R, Darling G, Wong RK. Multimodality approaches for the curative treatment of esophageal cancer. J Natl Compr Canc Netw. 2015;13:229–238. doi: 10.6004/jnccn.2015.0029. [DOI] [PubMed] [Google Scholar]
- 13.Little AG, Lerut AE, Harpole DH, et al. The Society of Thoracic Surgeons practice guidelines on the role of multimodality treatment for cancer of the esophagus and gastroesophageal junction. Ann Thorac Surg. 2014;98:1880–1885. doi: 10.1016/j.athoracsur.2014.07.069. [DOI] [PubMed] [Google Scholar]
- 14.Paul S, Altorki N. Outcomes in the management of esophageal cancer. J Surg Oncol. 2014;110:599–610. doi: 10.1002/jso.23759. [DOI] [PubMed] [Google Scholar]
- 15.Kleinberg L, Forastiere AA. Chemoradiation in the management of esophageal cancer. J Clin Oncol. 2007;25:4110–4117. doi: 10.1200/JCO.2007.12.0881. [DOI] [PubMed] [Google Scholar]
- 16.Duan XF, Tang P, Yu ZT. Neoadjuvant chemoradiotherapy for resectable esophageal cancer: An in-depth study of randomized controlled trials and literature review. Cancer Biol Med. 2014;11:191–201. doi: 10.7497/j.issn.2095-3941.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lloyd S, Chang BW. Current strategies in chemoradiation for esophageal cancer. J Gastrointest Oncol. 2014;5:156–165. doi: 10.3978/j.issn.2078-6891.2014.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 19.Almhanna K, Meredith KL, Hoffe SE, et al. Targeting the human epidermal growth factor receptor 2 in esophageal cancer. Cancer Contr. 2013;20:111–116. doi: 10.1177/107327481302000204. [DOI] [PubMed] [Google Scholar]
- 20.Forde PM, Kelly RJ. Genomic alterations in advanced esophageal cancer may lead to subtype-specific therapies. The Oncologist. 2013;18:823–832. doi: 10.1634/theoncologist.2013-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belkhiri A, El-Rifai W. Advances in targeted therapies and new promising targets in esophageal cancer. Oncotarget. 2015;6:1348–1358. doi: 10.18632/oncotarget.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agrawal N, Jiao Y, Bettegowda C, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2:899–905. doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Streppel MM, Lata S, DelaBastide M, et al. Next-generation sequencing of endoscopic biopsies identifies ARID1A as a tumor-suppressor gene in Barrett’s esophagus. Oncogene. 2014;33:347–357. doi: 10.1038/onc.2012.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dulak AM, Schumacher SE, van Lieshout J, et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012;72:4383–4393. doi: 10.1158/0008-5472.CAN-11-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forbes SA, Bindal N, Bamford S, et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Compeau PE, Pevzner PA, Tesler G. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol. 2011;29:987–991. doi: 10.1038/nbt.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–486. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song Y, Li L, Ou Y, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 30.Lin DC, Hao JJ, Nagata Y, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–473. doi: 10.1038/ng.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao YB, Chen ZL, Li JG, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 32.Bandla S, Pennathur A, Luketich JD, et al. Comparative genomics of esophageal adenocarcinoma and squamous cell carcinoma. Ann Thorac Surg. 2012;93:1101–1106. doi: 10.1016/j.athoracsur.2012.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagatys E, Garrett CR, Boulware D, et al. Activation of the serine/threonine protein kinase Akt during the progression of Barrett neoplasia. Hum Pathol. 2007;38:1526–1531. doi: 10.1016/j.humpath.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanawa M, Suzuki S, Dobashi Y, et al. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int J Cancer. 2006;118:1173–1180. doi: 10.1002/ijc.21454. [DOI] [PubMed] [Google Scholar]
- 36.Kato H, Arao T, Matsumoto K, et al. Gene amplification of EGFR, HER2, FGFR2 and MET in esophageal squamous cell carcinoma. Int J Oncol. 2013;42:1151–1158. doi: 10.3892/ijo.2013.1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reardon DA, Wen PY, Mellinghoff IK. Targeted molecular therapies against epidermal growth factor receptor: Past experiences and challenges. Neuro-oncol. 2014;16(suppl 8):viii7–viii13. doi: 10.1093/neuonc/nou232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lordick F, Kang YK, Chung HC, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomised, open-label phase 3 trial. Lancet Oncol. 2013;14:490–499. doi: 10.1016/S1470-2045(13)70102-5. [DOI] [PubMed] [Google Scholar]
- 39.Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial [published correction appears in Lancet Oncol 2013;14:e254] Lancet Oncol. 2013;14:481–489. doi: 10.1016/S1470-2045(13)70096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugiura K, Ozawa S, Kitagawa Y, et al. Co-expression of aFGF and FGFR-1 is predictive of a poor prognosis in patients with esophageal squamous cell carcinoma. Oncol Rep. 2007;17:557–564. [PubMed] [Google Scholar]
- 41.Dienstmann R, Rodon J, Prat A, et al. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25:552–563. doi: 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwak EL, LoRusso P, Hamid O, et al. Clinical activity of AMG 337, an oral MET kinase inhibitor, in adult patients with MET-amplified gastroesophageal junction, gastric, or esophageal cancer. J Clin Oncol. 2015;33(suppl 3):1a. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.