

Bone marrow fibrosis (BMF) plays a central role in the pathophysiology of primary myelofibrosis (PMF). This article reviews therapeutic strategies for BMF, and issues associated with BMF in PMF, including the pathophysiology and impact of abnormal deposition of excess collagen and reticulin fibers in bone marrow spaces, grading scales, and the prognostic impact of BMF on the overall outcome of patients with PMF.
Keywords: Bone marrow fibrosis, Primary myelofibrosis, Reticulin fibrosis, Collagen fibrosis
Abstract
Primary myelofibrosis is a stem cell-derived clonal malignancy characterized by unchecked proliferation of myeloid cells, resulting in bone marrow fibrosis, osteosclerosis, and pathologic angiogenesis. Bone marrow fibrosis (BMF) plays a central role in the pathophysiology of the disease. This review describes current issues regarding BMF in primary myelofibrosis, including the pathophysiology and impact of abnormal deposition of excess collagen and reticulin fibers in bone marrow spaces, the modified Bauermeister and the European Consensus grading systems of BMF, and the prognostic impact of BMF on the overall outcome of patients with primary myelofibrosis. The impact of novel therapeutic strategies, including JAK-STAT inhibitors and allogeneic stem cell transplant, on BMF is discussed.
Implications for Practice:
Bone marrow fibrosis (BMF) plays an important role in the pathophysiology and the clinical outcomes of patients with primary myelofibrosis. The severity of BMF correlates with the clinical manifestations of the disease and impacts the survival in patients with myelofibrosis. Treatment with ruxolitinib has been shown to reverse BMF and to continue that trend with ongoing treatment. Further studies to fully understand the mechanisms of fibrosis, to further explore the ability of currently available agents (e.g., JAK-STAT inhibitors) to stabilize and/or reverse fibrosis, and to develop additional fibrosis-targeted therapies are warranted.
Introduction
The myeloproliferative neoplasm (MPN) primary myelofibrosis (PMF), previously known as chronic idiopathic myelofibrosis or agnogenic myeloid metaplasia, is a heterogeneous, clonal, hematopoietic stem cell malignancy characterized by progressive stromal fibrosis with an inversely proportional reduction in myeloproliferative capacity involving predominantly the megakaryocytic and granulocytic lineages [1, 2]. Inflammation, disease-associated changes in the bone marrow stroma, altered angiogenesis, and an abnormal cytokine profile are characteristic pathophysiological features of PMF. Bone marrow fibrosis (BMF) in PMF results from abnormal reactive deposition of stromal reticulin and collagen fibers [2]. Several studies have shown that the degree of collagen fibrosis is strongly associated with the severity of the PMF and the degree of myelosuppression [3]. This correlation has not been shown with reticulin fibrosis [4–6]. Moreover, reticulin fibrosis is often reversible with treatment, whereas collagen fibrosis is less likely to respond to treatment [7].
The World Health Organization revised the diagnostic criteria for PMF in 2008 to include new features such as megakaryocyte atypia and the degree of fibrosis [8], thereby placing a renewed emphasis on bone marrow trephine biopsy sampling as an essential tool to establish the diagnosis of PMF and assess the severity of BMF (Fig. 1). Several grading systems have been developed over the years to evaluate the severity of BMF. The two most widely used systems include the Bauermeister system [9], subsequently modified by Manoharan et al. [10], which assesses fibrosis on a scale of 0–4, and the revised European Consensus system, which uses a 0–3 scale to provide a semiquantitative assessment of BMF [3].
Figure 1.
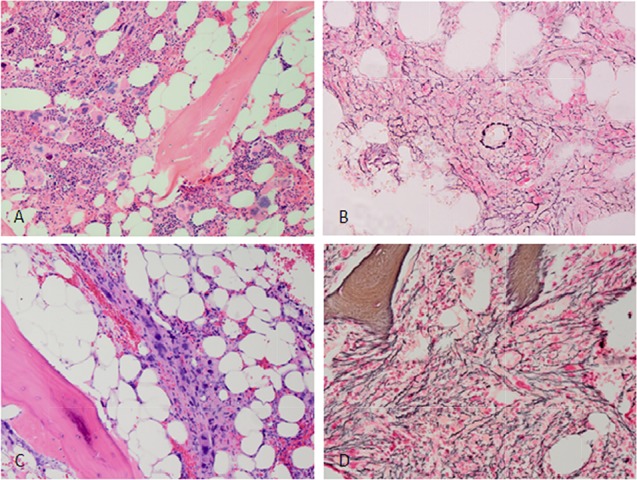
Micrographs of bone marrow biopsy specimens from a patient with primary myelofibrosis harboring the JAK2 V617F mutation. (A): Micrograph of a diagnostic bone marrow core biopsy specimen demonstrating more megakaryocytes with nuclear atypia. Note the presence of background hematopoiesis. (B): Reticulin stain demonstrating moderate reticulin fibrosis at presentation. (C): Micrograph of a bone marrow biopsy specimen from the same patient 5 years after diagnosis. Note confluent aggregates of atypical megakaryocytes and reduction in background hematopoiesis. (D): Reticulin stain shows severe reticulin fibrosis at 5 years after diagnosis.
Two distinct pathogenic processes have been implicated in the initiation and progression of PMF: stem cell-derived clonal myeloproliferation and a reactive cytokine-driven inflammatory fibrosis. BMF also plays a central role in the clinical manifestations of PMF, including extramedullary hematopoiesis, which may result in hepatosplenomegaly that causes abdominal pain, weight loss, and bone marrow failure with subsequent anemia and thrombocytopenia. Furthermore, it has been suggested that the severity of myelofibrosis may also impact the overall survival of PMF patients. Traditionally, allogeneic stem cell transplant (ASCT) has been the only therapeutic modality known to reverse fibrosis in patients with PMF [11]. Although it is well known that ruxolitinib reduces the clinical stigmata associated with PMF, including improvements in spleen size, weight, performance status, and symptom control to prolonged survival, the impacts of ruxolitinib on BMF were only recently defined [12–16]. An exploratory analysis of BMF data from an ongoing, phase I/II, single-arm study of ruxolitinib provided the first insight that JAK-inhibitor therapy meaningfully retards the advancement of BMF [17]. In this study, BMF was shown to stabilize or reverse, after 24 and 48 months of ruxolitinib treatment in the majority of patients, a magnitude of effect not seen with long-term hydroxyurea treatment [17].
In this review, we discuss BMF with an emphasis on the pathophysiology and clinical implications of marrow fibrosis in PMF, therapies that stabilize and reverse fibrosis in patients with PMF (with a focus on JAK-inhibitors and antifibrotic proteins), and the impact of fibrosis reversal in patients with PMF.
Pathophysiology of Fibrosis in PMF
BMF results from the abnormal and excessive deposition of collagen and reticulin fibers derived from marrow fibroblasts [18–20]. Elevation of cytokines such as interleukin (IL)-6, IL-2, IL-8, tumor necrosis factor-α, γ-interferon, and profibrogenic growth factors such as transforming growth factor β (TGF-β), basic fibroblast growth factor (bFGF), and vascular endothelial growth factor (VEGF), are thought to mediate BMF in patients with PMF [21–24] (Fig. 2). Platelet-derived growth factor (PDGF) was one of the first cytokines to be identified as a potential cause of BMF in patients with PMF [18, 25]. PDGF is the primary mediator of the growth and proliferation of marrow fibroblasts [19]; however, it has been demonstrated to have a limited role in the production and deposition of collagen fibers and fibronectin in primary myelofibrosis [19, 20]. Further, the megakaryocyte growth and development factor (MGDF) has also been shown to play a role in megakaryocyte production and the development of fibrosis. MGDF overexpression in mice results in more rapid platelet recovery than seen in control mice after transplantation [26]. Prolonged overexpression of MGDF in mice can lead to decreased marrow hematopoiesis, especially erythropoiesis with a shift to extramedullary hematopoiesis in the spleen and liver [26]. More importantly, all the MGDF-overexpressing mice developed myelofibrosis and osteosclerosis, possibly induced by megakaryocyte- and platelet-produced cytokines. This stimulatory effect of MGDF in vivo was restricted to the megakaryocyte lineage, with no effect on the other hematopoietic lineages.
Figure 2.

A working model summarizing the pathophysiology of bone marrow fibrosis in primary myelofibrosis.
Abbreviations: bFGF, basic fibroblast growth factor; PDGF, platelet-derived growth factor; TGF-B, transforming growth factor β.
Elevated levels of another cytokine, TGF-β, found in megakaryocytes, platelets, and monocytes [27–29], may also play a central role in inciting and propagating BMF in MPNs [30]. Studies have shown a significant correlation between TGF-β and the severity of BMF in PMF and hairy cell leukemia [28, 31]. The interaction between TGF-β and thrombopoietin (TPO) precipitates BMF in animal models [32, 33]. In rats, the injection of a suprapharmacologic dose (100 μg/kg) of pegylated recombinant human megakaryocyte growth and development factor (daily for 5 days was associated with an increase in marrow megakaryocytes and platelet counts at 6–8 days. Myelofibrosis with a predominance of reticulin fibers was noticed on day 10. The level of TGF-β in the extracellular fluid of the marrow and platelets was also increased, suggesting that TGF-β may be required for TPO-induced BMF [33]. Other growth factors that may also contribute to the fibrotic reaction but play a less central role include epidermal growth factor [19], bFGF-2, VEGF, calmodulin, and matrix metalloproteinase-9 [34–37].
Another potential mechanism of fibroblast activation and fibrosis deposition in PMF is via emperipolesis (Fig. 2). Emperipolesis is a physiologic process by which a cell, typically a neutrophil, penetrates into and through the cytoplasm of a megakaryocyte. Emperipolesis has been described with an increased frequency in patients with MPN [38, 39]. Schmitt et al. suggested that the abnormal P-selectin distribution in megakaryocytes induces selective sequestration of eosinophils, resulting in the release of α-granular proteins and assorted growth factors in the megakaryocyte cytoplasm, with subsequent fibroblast activation and fibrous tissue deposition in PMF [22].
It is now generally accepted that the BMF observed in patients with PMF is a “reactive” inflammatory phenomenon affected by non-neoplastic cells in the bone marrow microenvironment. Indeed, facets of the pathophysiology of BMF bear resemblance to reactive bone marrow changes seen with infections [40] or after exposure to chemicals or toxins (the latter frequently leading to agranulocytosis and neutropenia) [41]. Moreover, therapies that eradicate the PMF clone, such as ASCT, may reverse the fibrosis [42]. In addition, several observations have suggested that myelofibrosis may be the result of an imbalance between the microenvironmental niches that orchestrate hematopoietic homeostasis. Alterations of this balance may lead to uncontrolled cellular proliferation and, ultimately, to the promotion of leukemias and MPNs [43].
Several observations have suggested that myelofibrosis may be the result of an imbalance between the microenvironmental niches that orchestrate hematopoietic homeostasis. Alterations of this balance may lead to uncontrolled cellular proliferation and, ultimately, to the promotion of leukemias and MPNs.
Differential Diagnosis of PMF
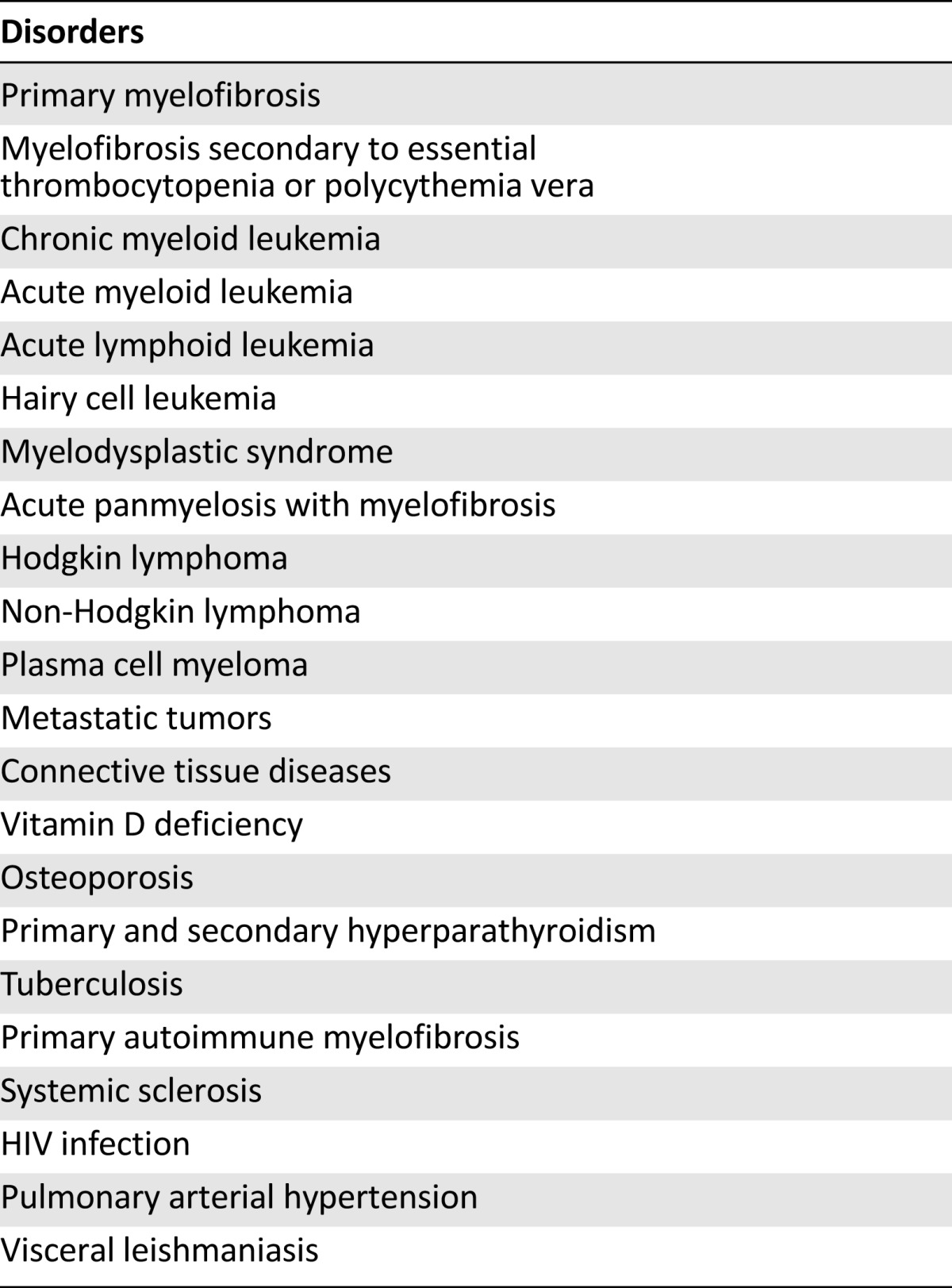
Several benign and malignant diseases are associated with BMF. Some are only associated with reticulin fibrosis without concurrent collagen fibrosis, whereas others are associated with both (Table 1). Furthermore, BMF is seen in other hematologic malignancies, including acute myeloid leukemia (AML) [10], acute lymphocytic leukemia (ALL) [4], myelodysplastic syndrome (MDS) [45, 46], and chronic myeloid leukemia (CML) [47, 48]. In adults with AML and ALL, high-grade fibrosis was found in 9% and 33%, respectively. Although fibrosis in these conditions usually resolves with therapy of the underlying disease and re-emerges at relapse, the presence or degree of fibrosis was not associated with survival or remission rates [10]. In addition, BMF associated with CML is reversible with imatinib mesylate therapy but is usually not affected by interferon [47, 48].
Table 1.
Disorders associated with bone marrow fibrosis [44]
An autoimmune syndrome, primary autoimmune myelofibrosis [AIMF], distinct from primary myelofibrosis has been described [49, 50]. AIMF is characterized by cytopenias, absent or mild splenomegaly, and high grades of BMF with concurrent lymphocytic infiltration of the bone marrow without significant associated osteosclerosis. In a retrospective analysis of seven patients who met the diagnostic criteria for this disease, six patients responded to corticosteroid therapy with normalization of their counts and partial resolution of BMF [50]. In a larger study of 29 patients with primary and secondary AIMF (secondary was defined as marrow reticulin fibrosis and lymphocytic infiltration in the context of an established autoimmune disorder), 20 patients (69%) were considered to have secondary AIMF [51]. Primary and secondary AIMF were pathologically indistinguishable except for an increased incidence of granulocytic hyperplasia in primary AIMF. Peripheral smears showed rare to absent teardrop cells, nucleated red blood cells, and blasts in 96% of the patients [51]. Overall, 72% progressed to a hypercellular marrow, including 87% with MF-1, 10% with MF-2, and 4% with MF-3. Among the 12 patients with follow-up, 4 achieved complete response with resolution of cytopenias and 3 achieved partial response with immunosuppression treatment [51].
Surprisingly, BMF has been noted in diseases that traditionally are not thought to be associated with bone marrow changes. These include HIV infection [5], pulmonary hypertension [52], and visceral leishmaniasis [52]. In a retrospective analysis of 35 patients with HIV, BMF was found in the majority of the patients and was not associated with the CD4 cell count, suggesting no correlation between BMF and the severity of HIV infection [5]. Similarly, the BMF associated with visceral leishmaniasis was not associated with either response to therapy or overall outcome [52]. The exact mechanism of BMF in these diseases remains unknown.
In addition, BMF has been observed in patients receiving treatment with hematopoietic growth factors. In a retrospective analysis of 12 females patients with breast cancer without bone marrow involvement, treatment with interleukin 11 and recombinant human granulocyte-macrophage colony-stimulating factors (GM-CSF) was associated with increase in BMF in 7 (58%) patients [53]. The administration of GM-CSF to patients with MDS with BMF was associated with increased fibrosis in half of the patients and decreased fibrosis in the other half [54]. In a study of nine patients with AML treated with TPO and GM-CSF, eight had a transient increase in the degree of BMF resembling MPN [55].
BMF Grading Systems in PMF
The distinction between increased bone marrow reticulin and collagen fibrosis is essential for accurately evaluating the bone marrow of patients with PMF. A number of scoring systems have been developed to evaluate the normal values of bone marrow cellularity and to grade the amount of reticulin and collagen fibers in patients with myelofibrosis. These scoring systems are subjective and heterogeneous, as they are based on a pathologist’s histomorphometric evaluation, which may require implementation of a variety of bone marrow processing and staining techniques (Table 2). Furthermore, quantitative changes in bone marrow cellularity and fibrosis related to age or concomitant therapy must be taken into consideration at the time of evaluation.
Table 2.
Comparison between the modified Bauermeister and the European Consensus grading systems of bone marrow fibrosis
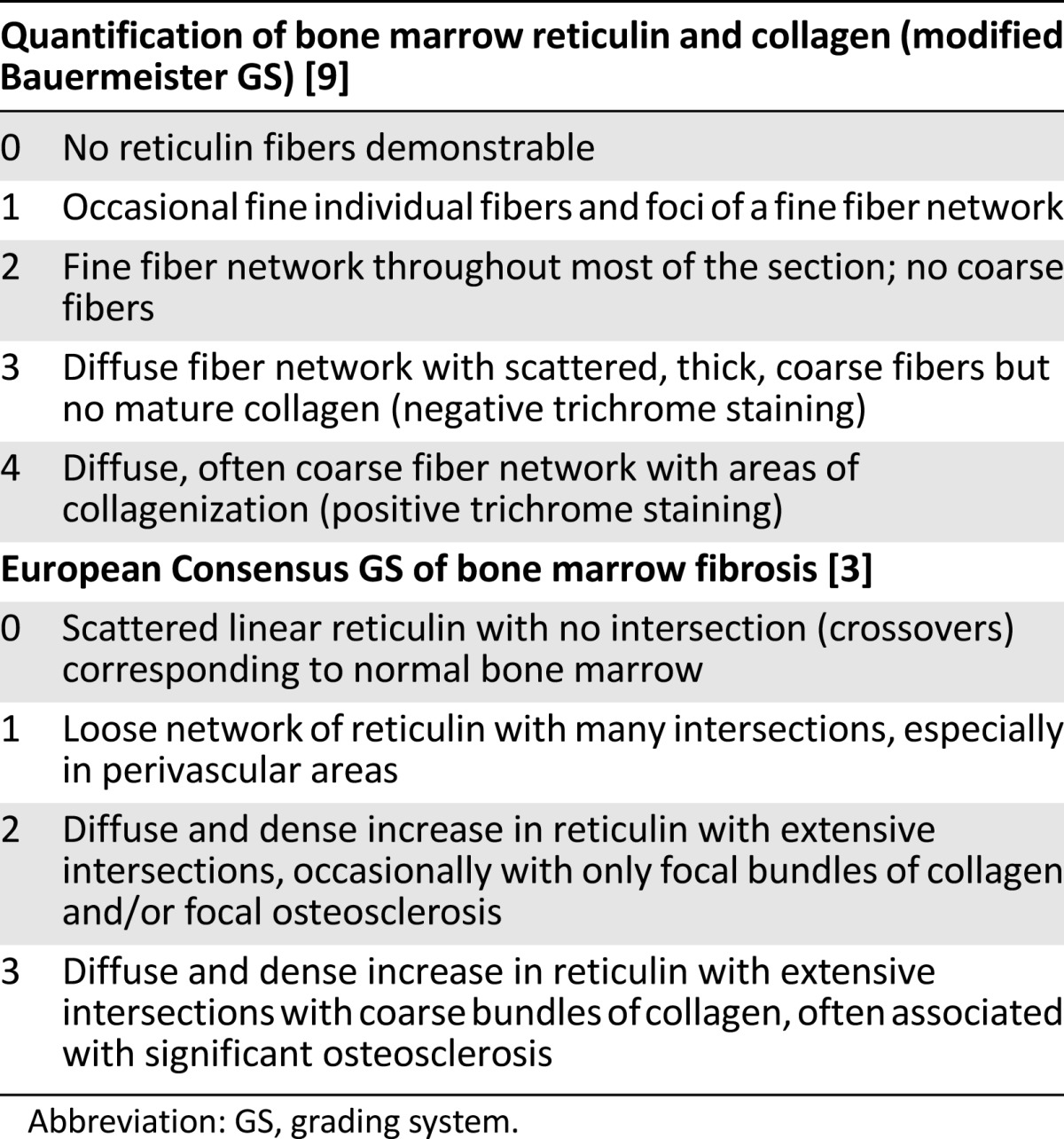
In 2005, a panel of experienced hematopathologists retrospectively reviewed more than 150 myelofibrosis trephine biopsy specimens from various medical institutions in an attempt to reach a consensus on grading cellularity and fibrosis in patients with MPN [3]. The trephine biopsy specimens predominantly included PMF and essential thrombocythemia samples from adult patients before and after therapy [3]. The grading of myelofibrosis was simplified into four categories including, for the first time, a formal differentiation between reticulin and collagen fibers [3]. The panel also pointed out the importance of evaluating the density of fibers in relation to the hematopoietic tissue to avoid a false (spurious) impression of reduced fiber content in fatty and/or edematous bone marrow samples that may be encountered after treatment. Their scoring system has become the most widely used grading system for bone marrow fibrosis [3]. Table 2 demonstrates the comparison between the modified Bauermeister grading system and the European Consensus grading system of bone marrow fibrosis.
In an attempt to minimize the subjectivity of evaluating BMF under the microscope, a computer-assisted digital image analysis study was conducted in 101 patients with newly diagnosed MPNs [56]. Thirty-four of these patients had primary myelofibrosis. Adequate reticulin- and hematoxylin-and-eosin-stained bone marrow core biopsies were digitally scanned with the ScanScope XT digital system (Leica Biosystems Nussloch GmbH, Buffalo Grove, IL, http://www.leicabiosystems.com) to evaluate the amount of osteosclerosis [56]. Subjective scores of BMF by three independent pathologists were obtained by using the Bauermeister system and the revised European grading system and were subsequently correlated with the objective numerical score obtained by the digital imaging. Correlation of the objective score was similar for both grading systems [56]. While the use of computer-assisted digital image analysis might improve the reproducibility of BMF assessment, it remains to be determined whether its use is associated with improved risk stratification of PMF patients.
Surprisingly, BMF has been noted in diseases that traditionally are not thought to be associated with bone marrow changes. These include HIV infection, pulmonary hypertension, and visceral leishmaniasis. In a retrospective analysis of 35 patients with HIV, BMF was found in the majority of the patients and was not associated with the CD4 cell count, suggesting no correlation between BMF and the severity of HIV infection.
BMF and Clinical Manifestations in PMF
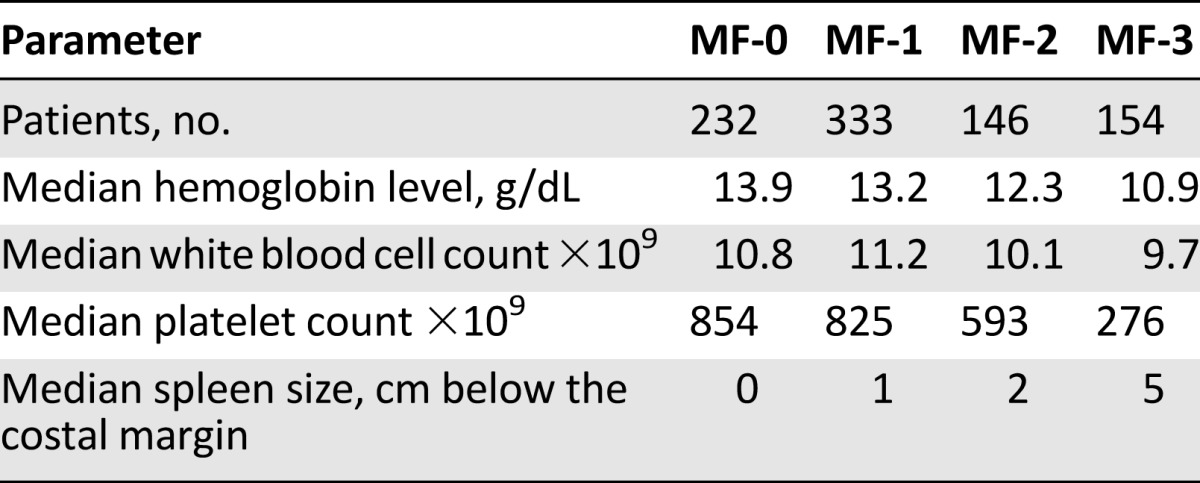
The most common clinical manifestations of the PMF are anemia, leukocytosis, leukopenia, thrombocytosis, constitutional symptoms, and marked splenomegaly with extramedullary hematopoiesis [57, 58]. Several studies have evaluated the relationship between the degree of BMF and the clinical symptoms in patients with PMF. In a large retrospective analysis of 865 bone marrow biopsy specimens from patients with newly diagnosed PMF, low-grade BMF (MF-0/1) was present in 565 patients and was associated with mild anemia and no or mild splenomegaly [59]. Higher grades of BMF (MF-2/3) were associated with marked anemia, large splenomegaly, increased number of peripheral blasts, and low platelets and leukocyte counts (Table 3) [59]. Another multicenter observational study of 309 patients with PMF, with long follow-up, included 822 bone marrow biopsy specimens obtained at different stages of the disease. A quarter of the patients had grade MF-0 disease at diagnosis, 32% had grade MF-1, 17% had grade MF-2, and 26% had grade MF-3 disease [60]. With a median interval of 32 months between biopsy procedures, 67% of the patients with low-grade fibrosis had progressed to higher grades, 42% were stable, and 6% had a regression of myelofibrosis independent of the therapy they received, suggesting that the degree of BMF changes over the course of the disease [60]. This may also explain the varied clinical manifestations seen in later stages of PMF as compared with diagnosis. More importantly, with a median interval of 20 months, 15% of the patients treated with busulfan had a regression in their BMF, 49% were stable, and 36% had progressive BMF. Comparing patients treated with hydroxyurea with those treated with interferon a-2b, 5% versus 17% had a regression in their BMF, 55% versus 44% had stable BMF, and 40% versus 39% had progressive BMF, respectively [60]. These observations confirm the dynamic nature of BMF during the course of the disease, which may be related to either disease progression or treatment.
Table 3.
Correlation between clinical data and bone marrow fibrosis grading in 865 patients with primary myelofibrosis
Prognostic Impact of Fibrosis in PMF
The relationship between the degree of BMF and the outcome of patients with PMF has been reviewed in retrospective studies [59, 61, 62]. Thiele et al. evaluated 865 bone marrow biopsies from patients with primary myelofibrosis over a 25-year period [59]. Although, the p value was not reported, the overall survival (OS) for patients with MF-0/1 (according to the European Consensus grading system) was superior to that of patients with MF-2 and MF-3, respectively (10-year OS was 78/71 months in MF-0/1, 67 months in MF-2, and 35 months in MF-3). Vener et al. evaluated the impact of the degree of BMF in 113 newly diagnosed patients with primary and secondary myelofibrosis [61] including MF-0 (n = 33), MF-1 (n = 27), MF-2 (n = 26), and MF-3 (n = 12). With a median follow-up of 19 months, the OS of patients with MF-0 was longer than that of those with MF-3 (p = .0011). Similarly, the OS of patients with MF-1/2 was longer than that of those with MF-3 (p = .003). Barosi et al. conducted a retrospective analysis of 683 patients with PMF [62]. Bone marrow biopsy specimens at diagnosis were reviewed. Patients with grade MF-0 disease were categorized as having prefibrotic myelofibrosis (pre-MF) and patients with MF-1/2/3 were categorized as having myelofibrosis fibrotic (PMF-fibrotic). The authors defined pre-MF based on the presence of three bone marrow characteristics: dual myeloid megakaryocyte dominance, megakaryocyte morphology (anisocytosis, dense nuclei with plump lobation) with clustering, and a BMF grade of 0 or less than 1. Patients with bone marrow morphological features of pre-MF but having at least grade 1 fibrosis were placed in the PMF-fibrotic category [62]. Patients with pre-MF were more likely to be female, under the age of 50 years at diagnosis, and to have a milder clinical phenotype. Neither the JAK2V617F allele burden nor the percentage of patients who carried the mutation was different in patients with pre-MF compared with PMF-fibrotic type [62]. Pre-MF patients displayed distinct hematological presentations and disease-related complications as compared with the patients with PMF-fibrosis. Seventy percent of the pre-MF patients manifested an isolated thrombocytosis simulating ET, and 30% were diagnosed with splanchnic vein thrombosis without other features of myelofibrosis. The median OS was not reached in pre-MF patients and was 16.6 years in patients with PMF-fibrotic type (p < .001) [62]. Ninety-eight percent, 81%, and 56% of patients with pre-MF, PMF fibrotic type with early BMF, and PMF-fibrotic-type with advanced BMF, respectively, were alive at 10 years from diagnosis (p < .001) [62].
Therapeutic Strategies that Target BMF in PMF
ASCT remains the only potential curative option for patients with PMF. It may also reverse BMF. To evaluate the impact of ASCT on BMF, Thiele et al. performed an immunohistochemical and morphometric evaluation of BM biopsy specimens from 20 patients before and after their transplant [60]. Regression of BMF was observed up to 6 months after transplant in responding patients, but osteosclerosis did not change significantly during the time of observation [60]. The impact of other treatment modalities on BMF in PMF was evaluated in a multicenter observational study of 309 patients with PMF with long follow-up. The study included 822 bone marrow biopsy specimens obtained from patients at different stages of their disease [63]. Among patients who received busulfan, 15% had a regression in BMF, 49% were stable, and 36% had progressive BMF. Five percent of patients treated with hydroxyurea had regression of their BMF, 55% remained stable, and 40 progressed as compared with 17%, 55%, and 39% of patients treated with interferon a-2b, respectively [63].
Several JAK-STAT inhibitors have been evaluated in PMF, polycythemia vera, and ET [64]. Ruxolitinib is an oral JAK-1/2 inhibitor and is the only drug approved for the therapy of myelofibrosis in the U.S. and Europe [12, 14, 16, 65, 66]. In two large randomized trials of ruxolitinib versus best supportive care in patients with PMF (Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment [COMFORT]-1, and COMFORT-2), treatment with ruxolitinib was associated with significant reduction in symptomatic splenomegaly, and improved constitutional symptoms and quality of life [12, 66]. More importantly, patients who received ruxolitinib had a longer OS compared with patients who received best supportive care [12, 14]. Interestingly, JAK2V617F allele burdens have not been shown to be affected by ruxolitinib treatment; however, clinical benefit was achieved irrespective of JAK2V617F mutational status [12, 66]. In a subsequent analysis of the COMFORT-1 trial with median follow-up of 2 years, 100 of 155 patients randomized to ruxolitinib were still receiving treatment [67]. All patients randomized to placebo crossed over to ruxolitinib or discontinued within 3 months of the primary analysis [67]. More importantly, improved survival was continued to be observed in patients who received ruxolitinib compared with placebo (hazard ratio: 0.58; 95% confidence interval: 0.36–0.95; p = .03 [67]. In a longer follow-up of the same trial, OS continued to favor ruxolitinib even though the majority of patients receiving placebo crossed over to ruxolitinib (hazard ratio: 0.69; 95% confidence interval: 0.46–1.03; p = .067) [16]. Although the initial analysis of the COMFORT-2 study did not show any survival advantage in patients who received ruxolitinib compared with best available therapy (BAT), a 3-year follow-up showed that patients randomized to ruxolitinib had a longer OS than those randomized to BAT (hazard ratio: 0.48; 95% confidence interval: 0.28–0.85; log-rank test, p = .009) [68]. In an exploratory analysis of the BM biopsy specimens of patients enrolled in a phase I/II trial of ruxolitinib, 68 patients were evaluated at 24 and 48 months [17]. At 24 months after ruxolitinib treatment, 15% of patients had an improvement in their degree of BMF, 57% had stabilization, and 37% had progression. At 48 months, 22% of patients who continued ruxolitinib had improvement in their degree of BMF, 56% remained stable, and 25% progressed [17]. This study demonstrated the ability of ruxolitinib to reverse BMF in a small subset of patients with PMF. A longer follow-up is still needed to evaluate whether more patients will have improvement in their BMF while continuing treatment with ruxolitinib.
Acknowledgment
This study was supported in part by MD Anderson Cancer Center Leukemia Support Grant CA016672.
Author Contributions
Manuscript Writing: Aziz Nazha, Joseph D. Khoury, Raajit K. Rampal, Naval Daver
Final Approval of Manuscript: Aziz Nazha, Joseph D. Khoury, Raajit K. Rampal, Naval Daver
Disclosures
Raajit K. Rampal: Incyte Corp. (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT) Leuk Res. 2007;31:737–740. doi: 10.1016/j.leukres.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342:1255–1265. doi: 10.1056/NEJM200004273421706. [DOI] [PubMed] [Google Scholar]
- 3.Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128–1132. [PubMed] [Google Scholar]
- 4.Hann IM, Evans DI, Marsden HB, et al. Bone marrow fibrosis in acute lymphoblastic leukaemia of childhood. J Clin Pathol. 1978;31:313–315. doi: 10.1136/jcp.31.4.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Malley DP, Sen J, Juliar BE, et al. Evaluation of stroma in human immunodeficiency virus/acquired immunodeficiency syndrome-affected bone marrows and correlation with CD4 counts. Arch Pathol Lab Med. 2005;129:1137–1140. doi: 10.5858/2005-129-1137-EOSIHI. [DOI] [PubMed] [Google Scholar]
- 6.Thiele J, Grashof K, Fisher R. Follow-up study on bone marrow reticulin fibrosis in AML. Anal Cell Pathol. 1991;3:225–231. [PubMed] [Google Scholar]
- 7.Kuter DJ, Bain B, Mufti G, et al. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol. 2007;139:351–362. doi: 10.1111/j.1365-2141.2007.06807.x. [DOI] [PubMed] [Google Scholar]
- 8.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 9.Bauermeister DE. Quantitation of bone marrow reticulin—a normal range. Am J Clin Pathol. 1971;56:24–31. doi: 10.1093/ajcp/56.1.24. [DOI] [PubMed] [Google Scholar]
- 10.Manoharan A, Horsley R, Pitney WR. The reticulin content of bone marrow in acute leukaemia in adults. Br J Haematol. 1979;43:185–190. doi: 10.1111/j.1365-2141.1979.tb03740.x. [DOI] [PubMed] [Google Scholar]
- 11.Deeg HJ, Gooley TA, Flowers ME, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood. 2003;102:3912–3918. doi: 10.1182/blood-2003-06-1856. [DOI] [PubMed] [Google Scholar]
- 12.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 13.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verstovsek S, Kantarjian HM, Estrov Z, et al. Long-term outcomes of 107 patients with myelofibrosis receiving JAK1/JAK2 inhibitor ruxolitinib: Survival advantage in comparison to matched historical controls. Blood. 2012;120:1202–1209. doi: 10.1182/blood-2012-02-414631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: Results of a median 3-year follow-up of COMFORT-I. Haematologica. 2015;100:479–488. doi: 10.3324/haematol.2014.115840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kvasnicka MH TJ, Bueso-Ramos CE, et al. Exploratory analysis of the effect of ruxolitinib on bone marrow morphology in patients with myelofibrosis. J Clin Oncol. 2013;31 (suppl);abstr 7030. [Google Scholar]
- 18.McCarthy DM. Annotation. Fibrosis of the bone marrow: Content and causes. Br J Haematol. 1985;59:1–7. doi: 10.1111/j.1365-2141.1985.tb02956.x. [DOI] [PubMed] [Google Scholar]
- 19.Kimura A, Katoh O, Hyodo H, et al. Transforming growth factor-beta regulates growth as well as collagen and fibronectin synthesis of human marrow fibroblasts. Br J Haematol. 1989;72:486–491. doi: 10.1111/j.1365-2141.1989.tb04310.x. [DOI] [PubMed] [Google Scholar]
- 20.Terui T, Niitsu Y, Mahara K, et al. The production of transforming growth factor-beta in acute megakaryoblastic leukemia and its possible implications in myelofibrosis. Blood. 1990;75:1540–1548. [PubMed] [Google Scholar]
- 21.Spivak JL. The chronic myeloproliferative disorders: Clonality and clinical heterogeneity. Semin Hematol. 2004;41(Suppl 3):1–5. doi: 10.1053/j.seminhematol.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 22.Schmitt A, Jouault H, Guichard J, et al. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood. 2000;96:1342–1347. [PubMed] [Google Scholar]
- 23.Xu M, Bruno E, Chao J, et al. Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105:4508–4515. doi: 10.1182/blood-2004-08-3238. [DOI] [PubMed] [Google Scholar]
- 24.Tefferi A. Pathogenesis of myelofibrosis with myeloid metaplasia. J Clin Oncol. 2005;23:8520–8530. doi: 10.1200/JCO.2004.00.9316. [DOI] [PubMed] [Google Scholar]
- 25.Groopman JE. The pathogenesis of myelofibrosis in myeloproliferative disorders. Ann Intern Med. 1980;92:857–858. doi: 10.7326/0003-4819-92-6-857. [DOI] [PubMed] [Google Scholar]
- 26.Yan XQ, Lacey D, Fletcher F, et al. Chronic exposure to retroviral vector encoded MGDF (mpl-ligand) induces lineage-specific growth and differentiation of megakaryocytes in mice. Blood. 1995;86:4025–4033. [PubMed] [Google Scholar]
- 27.Martyré MC, Magdelenat H, Bryckaert MC, et al. Increased intraplatelet levels of platelet-derived growth factor and transforming growth factor-beta in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. 1991;77:80–86. doi: 10.1111/j.1365-2141.1991.tb07952.x. [DOI] [PubMed] [Google Scholar]
- 28.Martyré MC, Romquin N, Le Bousse-Kerdiles MC, et al. Transforming growth factor-beta and megakaryocytes in the pathogenesis of idiopathic myelofibrosis. Br J Haematol. 1994;88:9–16. doi: 10.1111/j.1365-2141.1994.tb04970.x. [DOI] [PubMed] [Google Scholar]
- 29.Rameshwar P, Denny TN, Stein D, et al. Monocyte adhesion in patients with bone marrow fibrosis is required for the production of fibrogenic cytokines. Potential role for interleukin-1 and TGF-beta. J Immunol. 1994;153:2819–2830. [PubMed] [Google Scholar]
- 30.Dong M, Blobe GC. Role of transforming growth factor-beta in hematologic malignancies. Blood. 2006;107:4589–4596. doi: 10.1182/blood-2005-10-4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shehata M, Schwarzmeier JD, Hilgarth M, et al. TGF-beta1 induces bone marrow reticulin fibrosis in hairy cell leukemia. J Clin Invest. 2004;113:676–685. doi: 10.1172/JCI19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chagraoui H, Tulliez M, Smayra T, et al. Stimulation of osteoprotegerin production is responsible for osteosclerosis in mice overexpressing TPO. Blood. 2003;101:2983–2989. doi: 10.1182/blood-2002-09-2839. [DOI] [PubMed] [Google Scholar]
- 33.Yanagida M, Ide Y, Imai A, et al. The role of transforming growth factor-beta in PEG-rHuMGDF-induced reversible myelofibrosis in rats. Br J Haematol. 1997;99:739–745. doi: 10.1046/j.1365-2141.1997.4843288.x. [DOI] [PubMed] [Google Scholar]
- 34.Ciurea SO, Merchant D, Mahmud N, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007;110:986–993. doi: 10.1182/blood-2006-12-064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalley A, Smith JM, Reilly JT, et al. Investigation of calmodulin and basic fibroblast growth factor (bFGF) in idiopathic myelofibrosis: Evidence for a role of extracellular calmodulin in fibroblast proliferation. Br J Haematol. 1996;93:856–862. doi: 10.1046/j.1365-2141.1996.d01-1739.x. [DOI] [PubMed] [Google Scholar]
- 36.Di Raimondo F, Azzaro MP, Palumbo GA, et al. Elevated vascular endothelial growth factor (VEGF) serum levels in idiopathic myelofibrosis. Leukemia. 2001;15:976–980. doi: 10.1038/sj.leu.2402124. [DOI] [PubMed] [Google Scholar]
- 37.Martyré MC, Le Bousse-Kerdiles MC, Romquin N, et al. Elevated levels of basic fibroblast growth factor in megakaryocytes and platelets from patients with idiopathic myelofibrosis. Br J Haematol. 1997;97:441–448. doi: 10.1046/j.1365-2141.1997.292671.x. [DOI] [PubMed] [Google Scholar]
- 38.Cashell AW, Buss DH. The frequency and significance of megakaryocytic emperipolesis in myeloproliferative and reactive states. Ann Hematol. 1992;64:273–276. doi: 10.1007/BF01695470. [DOI] [PubMed] [Google Scholar]
- 39.Thiele J, Kuemmel T, Sander C, et al. Ultrastructure of bone marrow tissue in so-called primary (idiopathic) myelofibrosis-osteomyelosclerosis (agnogenic myeloid metaplasia). I. Abnormalities of megakaryopoiesis and thrombocytes. J Submicrosc Cytol Pathol. 1991;23:93–107. [PubMed] [Google Scholar]
- 40.Resende LS, Mendes RP, Bacchi MM, et al. Infiltrative myelopathy by paracoccidioidomycosis. A review and report of nine cases with emphasis on bone marrow morphology. Histopathology. 2006;48:377–386. doi: 10.1111/j.1365-2559.2006.02354.x. [DOI] [PubMed] [Google Scholar]
- 41.van Staa TP, Boulton F, Cooper C, et al. Neutropenia and agranulocytosis in England and Wales: Incidence and risk factors. Am J Hematol. 2003;72:248–254. doi: 10.1002/ajh.10295. [DOI] [PubMed] [Google Scholar]
- 42.Kröger N, Thiele J, Zander A, et al. Rapid regression of bone marrow fibrosis after dose-reduced allogeneic stem cell transplantation in patients with primary myelofibrosis. Exp Hematol. 2007;35:1719–1722. doi: 10.1016/j.exphem.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 43.Lataillade JJ, Pierre-Louis O, Hasselbalch HC, et al. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood. 2008;112:3026–3035. doi: 10.1182/blood-2008-06-158386. [DOI] [PubMed] [Google Scholar]
- 44.Rocha Filho FD, Ferreira FV, Mendes FO, et al. Bone marrow fibrosis (pseudo-myelofibrosis) in human kala-azar. Rev Soc Bras Med Trop. 2000;33:363–366. doi: 10.1590/s0037-86822000000400005. [DOI] [PubMed] [Google Scholar]
- 45.Ohyashiki K, Sasao I, Ohyashiki JH, et al. Clinical and cytogenetic characteristics of myelodysplastic syndromes developing myelofibrosis. Cancer. 1991;68:178–183. doi: 10.1002/1097-0142(19910701)68:1<178::aid-cncr2820680131>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 46.Maschek H, Georgii A, Kaloutsi V, et al. Myelofibrosis in primary myelodysplastic syndromes: A retrospective study of 352 patients. Eur J Haematol. 1992;48:208–214. [PubMed] [Google Scholar]
- 47.Beham-Schmid C, Apfelbeck U, Sill H, et al. Treatment of chronic myelogenous leukemia with the tyrosine kinase inhibitor STI571 results in marked regression of bone marrow fibrosis. Blood. 2002;99:381–383. doi: 10.1182/blood.v99.1.381. [DOI] [PubMed] [Google Scholar]
- 48.Hasserjian RP, Boecklin F, Parker S, et al. ST1571 (imatinib mesylate) reduces bone marrow cellularity and normalizes morphologic features irrespective of cytogenetic response. Am J Clin Pathol. 2002;117:360–367. doi: 10.1309/NR81-VCU0-CKW1-4HT9. [DOI] [PubMed] [Google Scholar]
- 49.Bass RD, Pullarkat V, Feinstein DI, et al. Pathology of autoimmune myelofibrosis. A report of three cases and a review of the literature. Am J Clin Pathol. 2001;116:211–216. doi: 10.1309/6Q99-VRNL-7BTP-W1G8. [DOI] [PubMed] [Google Scholar]
- 50.Pullarkat V, Bass RD, Gong JZ, et al. Primary autoimmune myelofibrosis: definition of a distinct clinicopathologic syndrome. Am J Hematol. 2003;72:8–12. doi: 10.1002/ajh.10258. [DOI] [PubMed] [Google Scholar]
- 51.Vergara-Lluri ME, Piatek CI, Pullarkat V, et al. Autoimmune myelofibrosis: An update on morphologic features in 29 cases and review of the literature. Hum Pathol. 2014;45:2183–2191. doi: 10.1016/j.humpath.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 52.Popat U, Frost A, Liu E, et al. New onset of myelofibrosis in association with pulmonary arterial hypertension. Ann Intern Med. 2005;143:466–467. doi: 10.7326/0003-4819-143-6-200509200-00017. [DOI] [PubMed] [Google Scholar]
- 53.Orazi A, Cooper RJ, Tong J, et al. Effects of recombinant human interleukin-11 (Neumega rhIL-11 growth factor) on megakaryocytopoiesis in human bone marrow. Exp Hematol. 1996;24:1289–1297. [PubMed] [Google Scholar]
- 54.Antin JH, Weinberg DS, Rosenthal DS. Variable effect of recombinant human granulocyte-macrophage colony-stimulating factor on bone marrow fibrosis in patients with myelodysplasia. Exp Hematol. 1990;18:266–270. [PubMed] [Google Scholar]
- 55.Douglas VK, Tallman MS, Cripe LD, et al. Thrombopoietin administered during induction chemotherapy to patients with acute myeloid leukemia induces transient morphologic changes that may resemble chronic myeloproliferative disorders. Am J Clin Pathol. 2002;117:844–850. doi: 10.1309/09NP-3DFG-BLM9-E5LE. [DOI] [PubMed] [Google Scholar]
- 56.Teman CJ, Wilson AR, Perkins SL, et al. Quantification of fibrosis and osteosclerosis in myeloproliferative neoplasms: A computer-assisted image study. Leuk Res. 2010;34:871–876. doi: 10.1016/j.leukres.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tefferi A. Primary myelofibrosis. Cancer Treat Res. 2008;142:29–49. [PubMed] [Google Scholar]
- 58.Tefferi A. New insights into the pathogenesis and drug treatment of myelofibrosis. Curr Opin Hematol. 2006;13:87–92. doi: 10.1097/01.moh.0000208469.48614.2e. [DOI] [PubMed] [Google Scholar]
- 59.Thiele J, Kvasnicka HM. Grade of bone marrow fibrosis is associated with relevant hematological findings—a clinicopathological study on 865 patients with chronic idiopathic myelofibrosis. Ann Hematol. 2006;85:226–232. doi: 10.1007/s00277-005-0042-8. [DOI] [PubMed] [Google Scholar]
- 60.Thiele J, Kvasnicka HM, Dietrich H, et al. Dynamics of bone marrow changes in patients with chronic idiopathic myelofibrosis following allogeneic stem cell transplantation. Histol Histopathol. 2005;20:879–889. doi: 10.14670/HH-20.879. [DOI] [PubMed] [Google Scholar]
- 61.Vener C, Fracchiolla NS, Gianelli U, et al. Prognostic implications of the European consensus for grading of bone marrow fibrosis in chronic idiopathic myelofibrosis. Blood. 2008;111:1862–1865. doi: 10.1182/blood-2007-09-112953. [DOI] [PubMed] [Google Scholar]
- 62.Barosi G, Rosti V, Bonetti E, et al. Evidence that prefibrotic myelofibrosis is aligned along a clinical and biological continuum featuring primary myelofibrosis. PLoS One. 2012;7:e35631. doi: 10.1371/journal.pone.0035631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thiele J, Kvasnicka HM, Schmitt-Graeff A, et al. Bone marrow histopathology following cytoreductive therapy in chronic idiopathic myelofibrosis. Histopathology. 2003;43:470–479. doi: 10.1046/j.1365-2559.2003.01732.x. [DOI] [PubMed] [Google Scholar]
- 64.Santos FP, Verstovsek S. Therapy with JAK2 inhibitors for myeloproliferative neoplasms. Hematol Oncol Clin North Am. 2012;26:1083–1099. doi: 10.1016/j.hoc.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verstovsek S, Kantarjian H, Pardanani A, et al. INCB018424, an orals Selective JAK2 inhibitor, shows significant clinical activity in a phase I/II study in patients with primary myelofibrosis (PMF) and post polycythemia vera/essential thrombocythemia myelofibrosis (Post-PV/ET MF) Blood. 2007;110 abstract 558. [Google Scholar]
- 66.Verstovsek S, Mesa R, Gotlib J, et al. Results of COMFORT-I, a randomized double-blind phase III trial of JAK1/2 inhibitor INCB18424 (424) vs placebo (PB) for patients with myelofibrosis (MF) J Clin Oncol. 2011;29 (suppl):abstract 6500. [Google Scholar]
- 67.Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: Results of a median 2-year follow-up of COMFORT-I. Haematologica. 2013;98:1865–1871. doi: 10.3324/haematol.2013.092155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122:4047–4053. doi: 10.1182/blood-2013-02-485888. [DOI] [PubMed] [Google Scholar]