

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Fibrodysplasia ossificans progressiva (FOP), Myositis ossificans progressiva.
1.2 OMIM# of the disease
135100.
1.3 Name of the analysed genes or DNA/chromosome segments
Activin A type I receptor/activin-like kinase 2 (ACVR1/ALK2) a bone morphogenetic protein (BMP) type I receptor, chromosome 2q23-24.1, 2, 3
1.4 OMIM# of the gene(s)
102576.
1.5 Mutational spectrum
The spectrum described in this paragraph is based on RefSeq NM_001105.4.
All patients have heterozygous ACVR1 missense mutations in conserved amino acids. This disease-causing variant is a de novo mutation and therefore referred to as a mutation.
Patients with classic clinical features of FOP (great toe malformations and progressive heterotopic ossification) have previously been found to carry the same heterozygous mutation (c.617G>A; p.(Arg206His)) in the ACVR1 gene leading to an over-activation of the BMP signalling pathway. Only recently a new heterozygous ACVR1 mutation at codon 207 (c.619C>G, p.(Gln207Glu)) located in a codon adjacent to the c.617G>A, p.(Arg206His) of the ACVR1 was reported in two FOP patients with the classical phenotype.4 Among patients with FOP-like heterotopic ossification and/or toe malformation, there are patients with clinical features unusual for FOP. These atypical FOP patients form two classes: FOP-plus (classic defining features of FOP plus one or more atypical features, predominantly associated with the classical p.(Arg206His) mutation) and FOP variants (major variations in one or both of the two classic defining features of FOP, associated with non-Arg206His mutations within the ACVR1 receptor). Novel ACVR1 mutations occur mainly in FOP variants and some cases of FOP plus.4, 5, 6 A public list of disease causing variants is not available yet.
1.6 Analytical methods
DNA sequence analysis of protein-coding exons and splice junctions.2
1.7 Analytical validation
When a new mutation is found, functional testing will be necessary, like a BMP reporter assay.
1.8 Estimated frequency of the disease (incidence disease at birth (‘birth prevalence') or population prevalence)
1:2 000 000.5
1.9 If applicable, prevalence in the ethnic group of investigated person:
No ethical, racial, gender or geographic prediliction.5
1.10 Diagnostic setting:

Comment: ad A: To differentiate from other forms of heterotopic ossification (different forms of myositis ossificans (MO), progressive osseous heteroplasia (POH) or other forms that might be confused with atypical FOP).6, 7, 8 There are at least three other forms of MO of which the pathology is largely unknown, including MO Circumscripta, characterized by dystrophic calcification generally following severe trauma leading to heterotopic ossifications of a single intramuscular connective tissue, MO pseudo-malignant, which is limited to soft tissue and is not associated to any trauma, and a MO associated with paraplegia, closed head injury or severe trauma (non-hereditary heterotopic ossification).7, 9 POH is characterized by progressive ossification of cutaneous, subcutaneous, and deep connective tissues and caused by an inactivation of GNAS in most cases.10 In early stages misdiagnosis, aggressive fibromatosis or sarcoma may be suspected.
Comment: ad C: Risk assessment in first generation relatives, including brothers and sisters, could be considered due to a so-called 'variant FOP' presenting with normal great toes and late-onset heterotopic ossification11 or when one of the parents has a germ line mosaicism.12
2. TEST CHARACTERISTICS
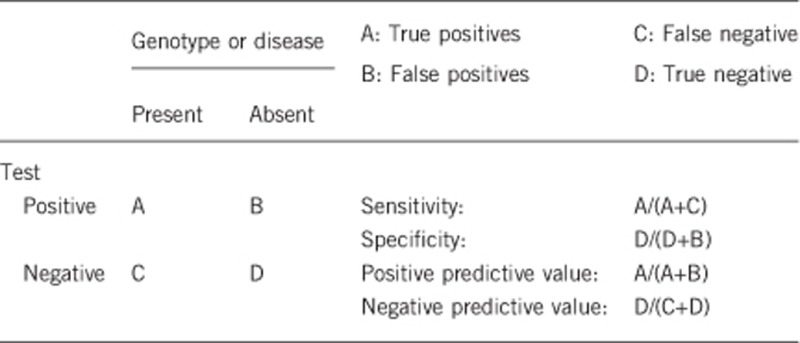
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
100%.2
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
100%.2
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
100%.2
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
100%, although we are aware of few rare cases of FOP with negligible progression.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative).
If the index case in the family has been tested positive for a causative mutation:
100%.
If the index case in the family has not been tested:
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
3. CLINICAL UTILITY
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
100% in the classical mutation, although there is a clinical variability/expressivity.1
3.1.1 Can a diagnosis be made other than through a genetic test?

A diagnosis based on clinical findings (malformed great toes in association with either soft tissue swelling or heterotopic ossification in characteristic anatomic patterns could be made by very experienced doctors,14 but in approximately 87% there is a long delay before awareness or before the appropriate diagnosis has been established.11, 15
3.1.2 Describe the burden of alternative diagnostic methods to the patient
No alternative affirmative methods are available.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
No alternative affirmative methods are available.
On the basis of the clinical and radiologic findings the diagnosis of FOP can be highly suspected, even prior to heterotopic ossifications. Characteristic toe malformations and cervical spine fusions may be diagnosed by X-ray. However, because FOP is infrequently seen by most clinicians and onset of progressive heterotopic ossification may be variable in the first decade of life, clinical misdiagnosis is common.14, 15
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe): see 3.1.4 prognosis.
If the test result is negative (please describe): based on current knowledge no risk.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
The lifestyle and prevention will be the same in patients with a clinical diagnosis, but with or without a genetic diagnosis.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes (if negative).
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
Prenatal diagnosis should only be done for FOP patients (they have 50% risk to transmit the disease) or for parents of FOP patients, if they expect new children (risk of mosaicism in an unaffected parent).12
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, although rare, up to three successive generations of transmissions of FOP have been described.20
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
NA.
Acknowledgments
This work was supported by ZonMw, EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (grant 261469) and the European Society of Human Genetics. GSD is supported by the AO Foundation start-up-grant (S-12-27S) and The Leducq Foundation, The International FOP Association (IFOPA), the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine, the Cali–Weldon Professorship of FOP Research and the National Institute's of Health (R01-AR41916).
The authors declare no conflict of interest.
References
- 1Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L et al: Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat 2009; 30: 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH et al: A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006; 38: 525–527. [DOI] [PubMed] [Google Scholar]
- 3Kaplan FS, Pignolo RJ, Shore EM: The FOP metamorphogene encodes a novel type I receptor that dysregulates BMP signaling. Cytokine Growth Factor Rev 2009; 20: 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4Haupt J, Deichsel A, Stange K, Ast C, Bocciardi R, Ravazzolo R et al: ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum Mol Genet 2014; 23:5364–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5Pignolo RJ, Shore EM, Kaplan FS: Fibrodysplasia ossificans progressiva: clinical and genetic aspects. Orphanet J Rare Dis 2011; 6: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6Shore EM, Kaplan FS: Inherited human diseases of heterotopic bone formation 72. Nat Rev Rheumatol 2010; 6: 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Piombino P, Dell'Aversana OG, Abbate V, Fini G, Liberatore GM, Mici E et al: Circumscribed myositis ossificans of the masseter muscle: report of a case 1. G Chir 2013; 34: 271–274. [PMC free article] [PubMed] [Google Scholar]
- 8Kartal-Kaess M, Shore EM, Xu M, Schwering L, Uhl M, Korinthenberg R et al: Fibrodysplasia ossificans progressiva (FOP): watch the great toes!. Eur J Pediatr 2010; 169: 1417–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9Pignolo RJ, Foley KL: Non-hereditary heterotopic ossification. Implications for injury, arthropathy, and aging. Clin Rev Bone Miner Metabol 2005; 3: 261–266. [Google Scholar]
- 10Adegbite NS, Xu M, Kaplan FS, Shore EM, Pignolo RJ: Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification. Am J Med Genet A 2008; 146A: 1788–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11Barnett CP, Dugar M, Haan EA: Late-onset variant fibrodysplasia ossificans progressiva leading to misdiagnosis of ankylosing spondylitis. Am J Med Genet A 2011; 155A: 1492–1495. [DOI] [PubMed] [Google Scholar]
- 12Janoff HB, Muenke M, Johnson LO, Rosenberg A, Shore EM, Okereke E et al: Fibrodysplasia ossificans progressiva in two half-sisters: evidence for maternal mosaicism. Am J Med Genet 1996; 61: 320–324. [DOI] [PubMed] [Google Scholar]
- 13Kaplan FS, Seemann P, Haupt J, Xu M, Lounev VY, Mullins M et al: Investigations of activated ACVR1/ALK2, a bone morphogenetic protein type I receptor, that causes fibrodysplasia ossificans progressiva. Methods Enzymol 2010; 484: 357–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14Kaplan FS, Xu M, Glaser DL, Collins F, Connor M, Kitterman J et al: Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics 2008; 121: e1295–e1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15Kitterman JA, Kantanie S, Rocke DM, Kaplan FS: Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005; 116: e654–e661. [DOI] [PubMed] [Google Scholar]
- 16Pignolo RJ, Shore EM, Kaplan FS: Fibrodysplasia ossificans progressiva: diagnosis, management, and therapeutic horizons. Pediatr Endocrinol Rev 2013; 10 (Suppl 2): 437–448. [PMC free article] [PubMed] [Google Scholar]
- 17Kaplan FS, Zasloff MA, Kitterman JA, Shore EM, Hong CC, Rocke DM: Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am 2010; 92: 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18Connor JM, Evans DA: Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg Br 1982; 64: 76–83. [DOI] [PubMed] [Google Scholar]
- 19Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A et al: The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am 1993; 75: 215–219. [DOI] [PubMed] [Google Scholar]
- 20Kaplan FS, McCluskey W, Hahn G, Tabas JA, Muenke M, Zasloff MA: Genetic transmission of fibrodysplasia ossificans progressiva. Report of a family. J Bone Joint Surg Am 1993; 75: 1214–1220. [DOI] [PubMed] [Google Scholar]