

Abstract
RMND1 is an integral inner membrane mitochondrial protein that assembles into a large 240 kDa complex to support translation of the 13 polypeptides encoded on mtDNA, all of which are essential subunits of the oxidative phosphorylation (OXPHOS) complexes. Variants in RMND1 produce global defects in mitochondrial translation and were first reported in patients with severe neurological phenotypes leading to mortality in the first months of life. Using whole-exome sequencing, we identified compound heterozygous RMND1 variants in a 4-year-old patient with congenital lactic acidosis, severe myopathy, hearing loss, renal failure, and dysautonomia. The levels of mitochondrial ribosome proteins were reduced in patient fibroblasts, causing a translation defect, which was rescued by expression of the wild-type cDNA. RMND1 was almost undetectable by immunoblot analysis in patient muscle and fibroblasts. BN-PAGE analysis showed a severe combined OXPHOS assembly defect that was more prominent in patient muscle than in fibroblasts. Immunofluorescence experiments showed that RMND1 localizes to discrete foci in the mitochondrial network, juxtaposed to RNA granules where the primary mitochondrial transcripts are processed. RMND1 foci were not detected in patient fibroblasts. We hypothesize that RMND1 acts to anchor or stabilize the mitochondrial ribosome near the sites where the mRNAs are matured, spatially coupling post-transcriptional handling mRNAs with their translation, and that loss of function variants in RMND1 are associated with a unique constellation of clinical phenotypes that vary with the severity of the mitochondrial translation defect.
Introduction
Mitochondria have a dedicated translation apparatus, resembling that of prokaryotes, for the synthesis of 13 polypeptides that are essential structural subunits of the oxidative phosphorylation (OXPHOS) complexes. As the first report of mutations in a mitochondrial translation elongation factor,1 investigation of patients with defects in mitochondrial protein synthesis have identified mutations in many known translation factors,2 and some accessory proteins that were not known to be a part of the core translation machinery.3, 4, 5 In some instances, mutations in specific genes have been linked to particular clinical phenotypes,6, 7, 8 but the molecular basis for these patterns remains an enduring mystery, and there is generally a great deal of unexplained clinical heterogeneity in patients with defects in mitochondrial protein synthesis.
Most patients with mutational defects in the mitochondrial translation machinery have deficiencies in the assembly of more than one OXPHOS complex. In this study, we investigated the molecular basis for a combined OXPHOS deficiency in a patient with congenital lactic acidosis, severe myopathy, hearing loss, renal failure, and dysautonomia.
Materials and methods
Mitochondrial enzyme measurements
The respiratory chain enzyme measurements on patient muscle were performed at the Biochemical Genetics lab (Vancouver, BC, Canada) as previously described.9
Whole-exome sequencing
As part of the TIDEX gene discovery project (UBC IRB approval H12-00067) whole-exome sequencing was performed for the patient and his unaffected parents using the Agilent SureSelect kit and Illumina HiSeq 2000 (PerkinElmer, Waltham, MA, USA). The sequencing reads (30X coverage) were aligned to the human reference genome version hg19 and rare variants were identified and assessed for their potential to disrupt protein function. Identified variants in the RMND1 gene and transcript (NM_017909.3) were submitted to ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/).
Measurement of mitochondrial transcripts levels
RNA was isolated using RNeasy mini kit (Qiagen, Mississauga, ON, Canada). Mitochondrial and RMND1 transcipts were quantified by quantitative RT-PCR using the following primers, all expressed in the 5′–3′ direction: RMND1-for: gcgcttccttcttctcttcc, RMND1-rev: cttcggcactgatgtgctt; cytB-for: caatggcgcctcaatattct, cytB-rev: gccgatgtttcaggtttctg; ND6-for: cctgacccctctccttcataa, ND6-rev: ggtgctgtgggtgaaagagt; ND5-for: ccaagcctcaccccactac, ND5-rev: caggggtggagacctaattg; ND4-for: ccaccttggctatcatcacc, ND4-rev: gaagtatgtgcctgcgttca; ND3-for: tcaacaccctcctagccttactac, ND3-rev: atatagggtcgaagccgcactcgtaa; ND2-for: tccttaacctctacttctacctacgc, ND2-rev: acgttgttagatatggggagtagtg; ND1-for: ccacccttatcacaacacaaga, ND1-rev: tcatattatggccaagggtca; COX1-for: ccctcccttagcagggaac, COX1-rev: tgaaattgatggcccctaag; COX2-for: tccctcccttaccatcaaatc, COX2-rev: gccgtagtcggtgtactcgt; COX3-for: caatgatggcgcgatgta, COX3-rev: gtatcgaaggcctttttggac; ATP6-for: tttattgccacaactaacctcct, ATP6-rev: ttgggtggttggtgtaaatg; 12S rRNA-for: taaccccagggttggtca, 12S rRNA-rev: ctttacgccggcttctattg and 16S rRNA-for: aatcttaccccgcctgtttac, 16S rRNA: acctttgcacggttagggta. Levels were normalized to ACTB and GAPDH using the following primers: ACTB-for: attggcaatgagcggttc, ACTB-rev: tgaaggtagtttcgtggatgc and GAPDH-for: agccacatcgctcagac, GAPDH-rev: gcccaatacgaccaaatcc.
Analysis of the splicing variant
The mRNA decay machinery (nonsensense-mediated decay pathway; NMD) was inhibited by treating patient cells with anisomycin (100 μg/ml) for 18 h. RNA was isolated using the RNeasy mini kit (Qiagen) and RT-PCR was performed with the One-Step RT-PCR kit (Qiagen). The following primers were used: RMND1-for(exon 2): 5′-aggcatttctcatctgtatca-3′ and RMND1-rev(exon 6): 5′-gtagttaagttcttcattttc-3′. The PCR product was TA cloned using the TOPO-TA cloning kit (Invitrogen, Burlington, ON, Canada) and the resulting clones were analyzed by Sanger sequencing.
Immunoblot analysis
SDS-PAGE was used to separate denatured whole-cell extracts prepared with 1.5% lauryl maltoside/PBS solution. Twenty microgram of protein was run on 12% polyacrylamide gels. Separated proteins were transferred to a nitrocellulose membrane, and immunoblot analysis was performed with the indicated antibodies: RMND1 (Sigma, St Louis, MO, USA), SDHA (Abcam, Cambridge, UK), MRPL11 (Sigma), MRPL12 (Sigma).
Immunofluorescence experiments
Cells plated on glass coverslips were fixed for 20 min with 4% paraformaldehyde in PBS. They were permeabilized for 15 min with a solution of PBS-Triton 0.1% and blocked for nonspecific sites for 30 min in PBS-BSA 3%. Cells were then incubated for 1 h with the primary antibody in blocking solution and 30 min with appropriate anti-species secondary antibodies coupled with Alexa fluorochromes (Invitrogen) in blocking solution, where DAPI was added for nuclear staining. Coverslips were finally mounted in Fluoromount (Sigma). For BrU labeling, cells were incubated with 2.5 mm BrU for 30 min before fixation. An Anti-BrdU antibody (Becton Dickinson, Mississauga, ON, Canada) was used to detect BrU-labeled RNA. Confocal analysis was performed with an Olympus (Richmond Hill, ON, Canada) IX83 microscope coupled with a CSU-X7 spinning disc system. We used a 100 × Olympus objective (NA1.4).
BN-PAGE and mitochondrial translation
BN-PAGE analyses of the OXPHOS complexes and the RMND1 complex were carried out as previously described.4 Mitochondrial translation experiments were carried out as in ref. 10.
Results
We investigated a boy who passed away at 4 years 3 months. He was born to healthy, non-consanguineous Caucasian parents at gestational age 37+4 weeks via spontaneous vaginal delivery with a birth weight of 2.86 kg. Pregnancy was complicated by oligohydramnios of unknown cause. Family history was unremarkable; his two older siblings are healthy with normal psychomotor development. This non-dysmorphic infant did not pass the newborn hearing screening and was diagnosed with moderate sensorineural hearing loss. During the first months of life, there was failure to thrive with diarrhea and abdominal pains. At the age of 8 weeks, he was hospitalized with epileptic encephalopathy and lactic acidosis (lactate 15.6 mmol/l (reference range 0.5–2.2 mmol; pH 7.09 (reference range 7.35–7.45), dehydration, hyperkalemia, hyponatremia, and hypertension. Kidney function and renal ultrasound appeared normal at the time; urinalysis was unremarkable except for a consistently low specific gravity. Throughout the initial 2-month admission it was difficult to stabilize his electrolyte status. A working diagnosis of aldosterone insensitivity was established, and he was treated with fludrocortisone for 12 months. His normocytic anemia, present since infancy, was treated with darbopoietin, folic acid, and ferrofumerate. Echocardiogram showed mild left ventricle hypertrophy secondary to high blood pressure, the latter requiring up to four antihypertensive agents. He suffered severe feeding intolerance, requiring continuous GJ-tube feeding with elemental feeds since infancy with reduction of vomiting on oral ondansetron and pyridostigmine. He experienced several episodes of pancreatitis with dehydration, as well as line infections requiring nephrotoxic antibiotics. Related to these events there were many episodes of acute kidney injury. Nuclear medicine glomerular filtration rate declined to 23 ml/ml/1.73 m2 at 2.5 years. In hindsight, his anemia (bone marrow morphological exam was normal), as well as the initial electrolyte abnormalities were likely renal related. Since age 3 years, he exhibited end-stage renal disease characterized by a tubulopathy with hyperkalemia, hyponatremia, and hypomagnesemia, as well as a urine concentrating defect, managed with addition of decanted sodium polystyrene to feeds, sodium and magnesium supplements, and extra free water as tolerated. Lactate levels were normalized.
He developed seizures during the first year of life, which remained well controlled with two anti-epileptic medications. He suffered sleep disturbances and intermittent mottling/cutis marmorata likely secondary to dysautonomia. In terms of psychomotor development, the patient was delayed in all areas, and was severely hypotonic and myopathic. There was no speech/language development but he was quite sociable. Formal testing (Merrill-Palmer Revised Scales of Development, Vineland II Adaptive Behavior Scale) at age 3.7 years placed his estimated developmental functioning at age 12.5 months. MRI of brain (1.5 T) at age 2 months revealed increased T2 signals throughout the white matter and unremarkable spectroscopy (no lactate peak, data not shown; Figure 1a). Repeat of scan at 2 years of age showed delayed myelination with abnormal posterior periventricular white matter signals (Figure 1b). The ophthalmologic exam remained normal.
Figure 1.

Brain imaging (MRI) of the patient. This analysis shows significantly delayed myelination and abnormal posterior periventricular white matter signal. White matter signal is abnormally increased on T2 and reduced on T1. Myelination of the internal capsules is relatively preserved. There is thinning of the corpus callosum. (a) Axial MRI, age 2 months. ai, aii: Axial T1-weighted MRI without gadolinium (TR/TE 6100/ 12 ms, 4 mm); aiii, aiv: Axial T2-weighted MRI (TR/TE 4000/ 101 ms, 4 mm). (b) Axial MRI, age 2 years. bi, bii: Axial T1 MRI without gadolinium (TR/TE 1990/3.35 ms, 1 mm); biii, biv: Axial T2-weighted MRI (TR/TE 4000/ 101 ms, 5 mm).
Newborn screening, urine organic acids, acylcarnitine, and plasma amino-acid profiles during first year of life were in keeping with hyperlactatemia. Respiratory chain enzyme assay on frozen muscle showed significant reduction of complex I:citrate synthase 0.071 (reference range 0.161–0.438; 28% of mean) and complex IV:citrate synthase 0.005 (reference range 0.20–0.049; 20% of mean). Extensive investigations according to the TIDE protocol11 included plasma amino-acid profile showing elevated alanine of 798 μmol/l (reference range 134–416) consistent with markedly elevated lactates; elevated lactate/pyruvate ratio (42.3; reference range 10–20) and pyruvate (0.237 mmol/l (reference range 0.067–0.130); urine organic acids showing mild lactic aciduria and mild ketonuria; acylcarnitine profile showed mildly elevations of a number of hydroxylated carnitine species in a nonspecific pattern. Ammonia, glucose, serum biotinidase enzyme assay, plasma very long chain fatty acids, transferrins isoelectric focusing, and urine purines/pyrimidines profile were all within normal limits. Fibroblast PEP-CK, pyruvate dehydrogenase complex, and pyruvate carboxylase enzymatic testing showed normal activity. Chromosome microarray (Affymetrix; Genome-Wide Human SNP Array 6.0, Santa Clara, CA, USA) was unremarkable, as were whole-mitochondrial DNA genome sequencing and a nuclear ‘All mtDNA depletion diseases NextGen Mitochondrial Diseases panel' using DNA extracted from muscle. Autopsy showed hypoplastic kidneys with cysts and focal medullary calcification; enlarged liver with microvesicular steatosis and absent glycogen; multiple small bowel intussusceptions; and nodular adrenal glands.
Of the 64 candidates identified by whole-exome sequencing only RMND1 on chromosome 6 (NC_000006.12 (151404762..151452181, complement); GRCh38) was considered a probable candidate, based on its recently described phenotype and function,4, 12 harboring the following compound heterozygous variants: NM_017909.3:c.[613G>T][713A>G]. Sanger sequencing confirmed that the patient was compound heterozygous for both variants (Figure 2a) while the unaffected father, sister, and brother are heterozygous for NM_017909.3:c.[713A>G], and the unaffected mother for NM_017909.3:c.[613G>T].
Figure 2.

Analysis of RMND1 variants. (a) Schematic representation of RMND1 gene (NG_0333031.1) and protein (NP_060379.2) showing the position of the variants in the patient (MLS is the mitochondrial localization signal). (b) Quantitative RT-PCR measurements of RMND1 mRNA levels in the patient compared with control (n=4). Expressed as mean±SEM. (c) Alignment of the amino-acid sequences of RMND1 homologs in different species. The asparagine at position 238 is shown in the black rectangle.
The NM_017909.3:c.[613G>T] variant occurs at the conserved splice site position −1 of the 5′ donor site of intron 3. qRT-PCR analysis of mRNA from patient fibroblast revealed that RMND1 transcript levels were 50% of control (Figure 2b) and the sequencing of the RMND1 cDNA detected only the NM_017909.3:c.[713A>G] variant, demonstrating that the NM_017909.3:c.[613G>T] variant was subjected to NMD. To identify the altered splice site position, the NMD pathway was inhibited by treating patient fibroblasts with anisomycin, and resultant RNA was analyzed. A PCR product comprising exon 2 through exon 6 was amplified, TOPO-TA cloned, and individual clones were analyzed by Sanger sequencing. Clones containing the NM_017909.3:c.[613G>T] variant showed an insertion of 71 nucleotides from intron 3: r.[613g>t;613_614+1_614+71], which would result in a premature stop codon at position 209 (p.Asp205CysfsTer4), thus directing this mRNA to the NMD pathway. This analysis also confirmed that the NM_017909.3:c.[613G>T][713A>G] variants are present on two different alleles in the patient. NM_017909.3:c.[713A>G] variant results in p.Asn238Ser change at a highly conserved position through evolution (Figure 2c).
Immunoblot analysis of a frozen muscle biopsy sample revealed a severe decrease (<5% of control) in the level of the RMND1 protein (Figure 3a), suggesting that the translated protein is unstable. As a consequence, the functional 240 kDa homopolymeric RMND1 complex4 is absent in patient muscle (Figure 3b). Consistent with the enzyme activity assays, BN-PAGE analysis showed marked defects in the assembly of complexes I, III, IV, and V of the OXPHOS system (Figure 3c), suggesting a global mitochondrial translation defect.
Figure 3.
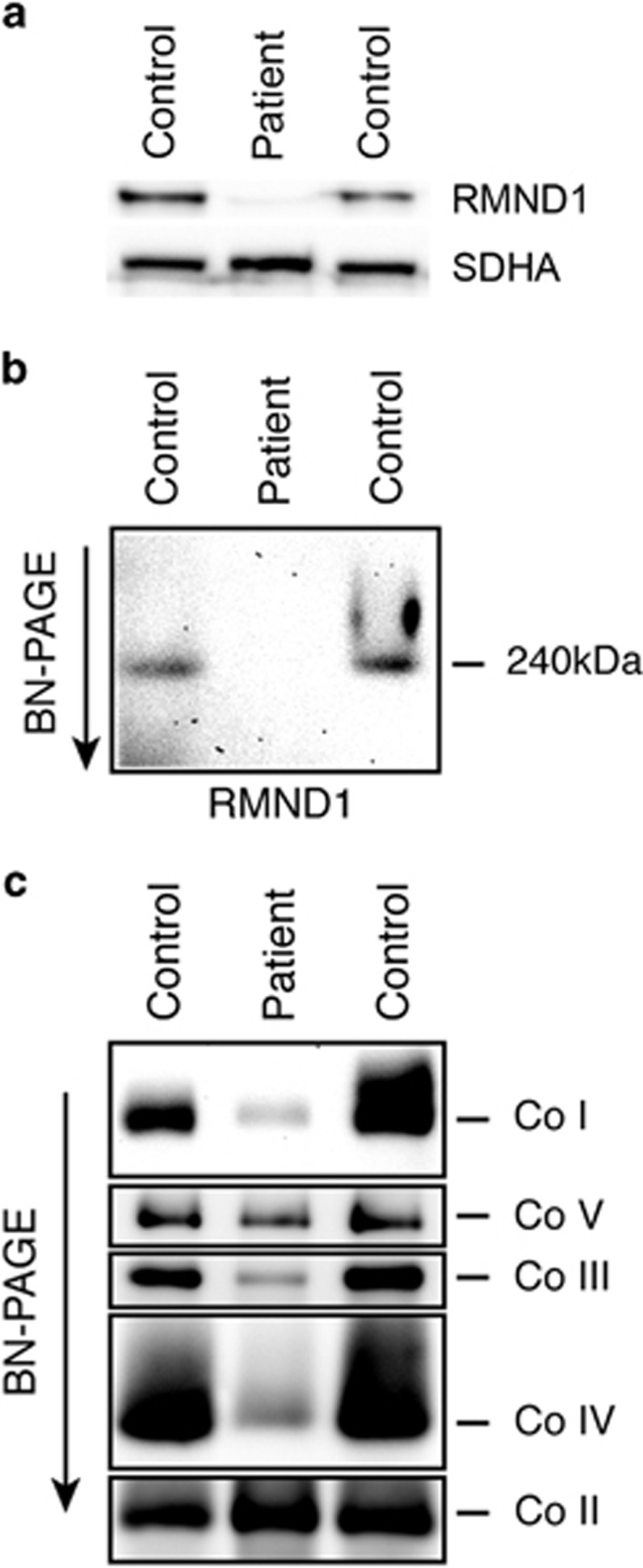
Characterization of the molecular defects in patient muscle. (a) Immunoblot analysis of control and patient muscle tissues showing very low levels of RMND1 in the patient. The 70 kDa subunit of the complex II (SDHA) was used as a loading control. (b) BN-PAGE analysis of control and patient muscle tissues showing that the 240 kDa RMND1 complex does not assemble in the patient. (c) BN-PAGE analysis of the OXPHOS complexes in control and patient muscle tissues. Each of the five OXPHOS complexes (I–V) was visualized with a subunit specific antibody that recognizes the native complexes as follows: Co I (NDUFA9), Co II (SDHA), Co III (UQCRC1), Co IV (COX4), and Co V (ATP5A1). Co II is the loading control.
To further investigate the nature of the biochemical defect, we carried out an immunoblot analysis on immortalized patient fibroblasts, which also showed a marked reduction in the steady-state level of RMND1 (<5% of control). This contrasts with our previous analysis of RMND1 levels in patient fibroblast with a homozygous NM_017909.3:p.Arg417Gln variant (herein named RMND1(R417Q); OMIM#614922),4 which is present at control levels (Figure 4a); however, in both the patients, BN-PAGE analysis showed that the 240 kDa RMND1 homopolymeric complex was absent (Figure 4b). Both the patients also exhibited a significant decrease in the levels of mitochondrial ribosomal proteins MRPL11 and MRPL12 (more substantial in the RMND1(R417Q) patient, Figure 4a), suggesting an impaired assembly or maintenance of the mitochondrial ribosome. In contrast with the results in skeletal muscle, BN-PAGE showed a milder OXPHOS assembly defect in fibroblasts (Figure 4c).
Figure 4.

Characterization of molecular defects in patient fibroblasts and rescue of the phenotype by expression of wild-type RMND1. (a) Immunoblot analysis of fibroblasts from control, the previously described RMND1(R417Q) patient, and the patient, with antibodies against RMND1 and the mitochondrial ribosomal proteins MRPL11 and MRPL12. The 70 kDa subunit of the complex II (SDHA) was used as a loading control. (b) BN-PAGE analysis of the RMND1 complex in control, RMND1(R417Q), and the patient. The 240 kDa RMND1 complex does not assemble either in RMND1(R417Q) or in the patient. (c) BN-PAGE analysis of the OXPHOS complexes in fibroblasts from control, RMND1(R417Q), and the patient. (d) Quantitative RT-PCR measurements of mitochondrial transcript levels in the patient compared with control. mRNAs coding for five subunits of the complex I (ND), one subunit of the complex III (cyt b), three subunits of the complex IV (COX), one subunit of the complex V (ATP), and mitochondrial ribosomal RNAs are indicated. (e) Analysis of mitochondrial translation by pulse labeling of mtDNA-encoded polypeptides in control, RMND1(R417Q), and the patient showing a generalized protein synthesis defect that is rescued by expression of the wild-type RMND1 cDNA. The seven subunits of the complex I (ND), one subunit of the complex III (cyt b), three subunits of the complex IV (COX), and two subunits of the complex V (ATP) are indicated. (f) Immunoblot analysis of RMND1 protein levels in fibroblasts from control, RMND1(R417Q), and the patient before and after overexpression of the wild-type RMND1 cDNA. The 70 kDa subunit of the complex II (SDHA) was used as a loading control.
To dissect the pathophysiological consequences of the RMND1 variants, we next measured the levels of the mitochondrial transcripts, which although slightly lower in the patient, were within the normal range (Figure 4d). On the contrary, the synthesis of the mtDNA-encoded polypeptides, investigated by pulse labeling the mitochondrial translation products with a mixture of [35S]methionine/cysteine, was uniformly decreased in the patient, but much less than in RMND1(R417Q) patient fibroblasts (Figure 4e). The magnitude of the mitochondrial translation defect correlated with the steady-state levels of the mitoribosome proteins (Figure 4a) and the severity of the clinical presentation. To confirm the pathogenicity of the RMND1 variant, we used a retroviral vector to express the wild-type RMND1 cDNA in patient fibroblasts (Figure 4f), and showed that the mitochondrial protein synthesis was completely restored by retroviral expression of RMND1 in subject fibroblasts (Figure 4e).
Immunofluorescence experiments showed an apparently normal mitochondrial network in both RMND1 patients (Figure 5a). RMND1 immunoreactivity was punctate throughout the mitochondrial network in controls and in the RMND1(R417Q) patient cells, but was undetectable in the current patient, consistent with the immunoblot analysis (Figure 4a). The punctate distribution of RMND1 is similar to that of the RNA-binding protein GRSF1, present in mitochondrial RNA granules,13 which are centers of mitochondrial RNA metabolism and ribosome biogenesis. To investigate the relationship between RMND1 and the RNA granules, we pulse-labeled fibroblasts for 20 min with BrU and visualized the newly synthesized mtDNA transcripts, which we previously showed appearing immediately in GRSF1-positive RNA granules.13 Co-localization experiments showed that RMND1 was immediately juxtaposed to the BrU-positive foci (Figure 5b).
Figure 5.

Sub-mitochondrial localization of RMND1. (a) Immunofluorescence analysis of fibroblasts from control, RMND1(R417Q), and the patient. Endogenous RMND1 staining is shown in green, mitochondria are stained in red (HSP60), and the nucleus is counterstained in blue with DAPI. (b) Spatial organization of RMND1 in relation to newly synthesized mitochondrial RNA. Endogenous RMND1 staining is shown in green, BrU staining in red, and the nucleus is counterstained in blue with DAPI.
Discussion
The patient we investigated here significantly expands the phenotype of the first two families reported with RMND1 variants. While we were preparing this manuscript, six other RMND1 families (five of the same ethnic origin with the same homozygous truncating founder mutation) were reported, which share some common clinical features with our patient.14 In general, RMND1 patients present with a severe condition in the neonatal period (oldest is 18 months at presentation), with a fatal course in 5/9 (age at death varies from 5 months to 10 years).4, 12, 14 Common features of RMND1 deficiency include lactic acidosis, deafness, renal dysfunction, and severe muscle involvement. Although renal dysfunction has been reported in a variety of OXPHOS disorders,15 it is not a particularly common presenting symptom. Further, CNS involvement in RMND1 patients varies from developmental delay, mild to intractable epilepsy, to white matter abnormalities and cerebral atrophy on neuroimaging. The pedigree described by Garcia-Diaz et al12 seems the most severe neurologic phenotype (including a stillborn), in that all affected cases presented with profound hypotonia, contractures and minimal movements, and all passed away during the first months of life.
We conclude that variants in RMND1 are associated with a unique constellation of clinical phenotypes that vary with the severity of the mitochondrial translation defect, all resulting from the inability to assemble a functional high-molecular-weight RMND1 complex. Although the data are still limited, it seems clear that there are marked tissue-specific biochemical phenotypes. The defect in skeletal muscle is much more profound than that in fibroblasts. Furthermore, the almost complete lack of the protein seems less severe, at least in fibroblasts, than a substitution (R417Q) in the coiled-coil domain.
Most of the core components of the mitochondrial translation system are homologous to those in bacteria. Mitochondrial and bacterial ribosomes share common drug sensitivities, but they are remarkably different structures; only about 40% of the mammalian ribosomal subunits have clear bacterial orthologues, and the RNA to protein ratio, which is about 2:1 in bacteria, is completely reversed (1:2) in mammals.16 Mammalian mitochondrial ribosomes translate only 13 polypeptides, all of which are hydrophobic inner membrane proteins. These proteins are thought to be co-translationally inserted into the inner mitochondrial membrane as they emerge from the exit tunnel of the ribosome, but exactly how mitochondrial ribosomes are targeted and stabilized near the inner membrane is not known. The presence of RMND1, which forms an integral inner membrane protein complex adjacent to RNA granules, and the decreases in the steady-state level of mitochondrial ribosomal subunits in RMND1 patient cells, all suggest that RMND1 acts to stabilize or anchor the mitochondrial ribosome adjacent to the subdomains of mitochondria where the primary transcripts are processed, thus spatially coupling post-transcriptional handling of mitochondrial mRNAs with their subsequent translation.
Acknowledgments
We are indebted to the patient and his family for participation in this study; Dr C Mammen, Dr J Hukin, Dr G Ward, Dr G Horvath, Dr Y Lillquist, Dr R Salvarinova, Mrs K Withers and Mrs B Cheng for their contributions to clinical management of the patient; Mrs M Higginson for DNA extraction and sample handling; Mrs X Han for Sanger sequencing; Dr M Thomas for consenting, Mr B Sayson for data management; and Mrs D Pak for research management support (University of British Columbia). We gratefully acknowledge Dr Robert Taylor (Newcastle, UK) for scientific communications regarding this patient. This work was supported by funding from the BC Children's Hospital Foundation (first Collaborative Area of Innovation grant; www.tidebc.org), Genome BC (SOF-195 grant), and the Michael Smith Foundation for Health Research (Scholar award) to CvK, and from the Canadian Institutes for Health Research and (MT-15460) to EAS. Informed consent was obtained, and the institutional review boards at the Montreal Neurological Institute and University of British Columbia approved the research studies.
The authors declare no conflict of interest.
References
- 1Coenen MJ, Antonicka H, Ugalde C et al: Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med 2004; 351: 2080–2086. [DOI] [PubMed] [Google Scholar]
- 2Hallberg BM, Larsson NG: Making proteins in the powerhouse. Cell Metab 2014; 20: 226–240. [DOI] [PubMed] [Google Scholar]
- 3Weraarpachai W, Antonicka H, Sasarman F et al: Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet 2009; 41: 833–837. [DOI] [PubMed] [Google Scholar]
- 4Janer A, Antonicka H, Lalonde E et al: An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet 2012; 91: 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5Antonicka H, Ostergaard E, Sasarman F et al: Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet 2010; 87: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6Riley LG, Cooper S, Hickey P et al: Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am J Hum Genet 2010; 87: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N: Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 2004; 74: 1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8Zeharia A, Shaag A, Pappo O et al: Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet 2009; 85: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9Frazier AE, Thorburn DR: Biochemical analyses of the electron transport chain complexes by spectrophotometry. Methods Mol Biol 2012; 837: 49–62. [DOI] [PubMed] [Google Scholar]
- 10Sasarman F, Shoubridge EA: Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol Biol 2012; 837: 207–217. [DOI] [PubMed] [Google Scholar]
- 11van Karnebeek CD, Shevell M, Zschocke J, Moeschler JB, Stockler S: The metabolic evaluation of the child with an intellectual developmental disorder: diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab 2014; 111: 428–438. [DOI] [PubMed] [Google Scholar]
- 12Garcia-Diaz B, Barros MH, Sanna-Cherchi S et al: Infantile encephaloneuromyopathy and defective mitochondrial translation are due to a homozygous RMND1 mutation. Am J Hum Genet 2012; 91: 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EA: The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab 2013; 17: 386–398. [DOI] [PubMed] [Google Scholar]
- 14Taylor RW, Pyle A, Griffin H et al: Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014; 312: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15Che R, Yuan Y, Huang S, Zhang A: Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol 2014; 306: F367–F378. [DOI] [PubMed] [Google Scholar]
- 16Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK: Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 2003; 115: 97–108. [DOI] [PubMed] [Google Scholar]