

Abstract
Background
Shewanella oneidensis MR-1 uses several electron acceptors to support anaerobic respiration including insoluble species such as iron(III) and manganese(IV) oxides, and soluble species such as nitrate, fumarate, dimethylsulfoxide and many others. MR-1 has complex branched electron transport chains that include components in the cytoplasmic membrane, periplasm, and outer membrane (OM). Previous studies have implicated a role for anaerobically upregulated OM electron transport components in the use of insoluble electron acceptors, and have suggested that other OM components may also contribute to insoluble electron acceptor use. In this study, the role for an anaerobically upregulated 35-kDa OM protein (Omp35) in the use of anaerobic electron acceptors was explored.
Results
Omp35 was purified from the OM of anaerobically grown cells, the gene encoding Omp35 was identified, and an omp35 null mutant (OMP35-1) was isolated and characterized. Although OMP35-1 grew on all electron acceptors tested, a significant lag was seen when grown on fumarate, nitrate, and Fe(III). Complementation studies confirmed that the phenotype of OMP35-1 was due to the loss of Omp35. Despite its requirement for wild-type rates of electron acceptor use, analysis of Omp35 protein and predicted sequence did not identify any electron transport moieties or predicted motifs. OMP35-1 had normal levels and distribution of known electron transport components including quinones, cytochromes, and fumarate reductase. Omp35 is related to putative porins from MR-1 and S. frigidimarina as well as to the PorA porin from Neisseria meningitidis. Subcellular fraction analysis confirmed that Omp35 is an OM protein. The seven-fold anaerobic upregulation of Omp35 is mediated post-transcriptionally.
Conclusion
Omp35 is a putative porin in the OM of MR-1 that is markedly upregulated anaerobically by a post-transcriptional mechanism. Omp35 is required for normal rates of growth on Fe(III), fumarate, and nitrate, but its absence has no effect on the use of other electron acceptors. Omp35 does not contain obvious electron transport moieties, and its absence does not alter the amounts or distribution of other known electron transport components including quinones and cytochromes. The effects of Omp35 on anaerobic electron acceptor use are therefore likely indirect. The results demonstrate the ability of non-electron transport proteins to influence anaerobic respiratory phenotypes.
Background
Shewanella oneidensis (formerly putrefaciens) MR-1 is a Gram-negative bacterium that can use a wide variety of terminal electron acceptors for anaerobic respiration including insoluble manganese (Mn) and iron (Fe) oxides [1-4]. Some electron transport components are common to the use of multiple electron acceptors. For example, both menaquinone and a 21-kDa tetraheme cytochrome c (CymA) are required for the use of fumarate, nitrate, Mn(IV), and Fe(III) [4-6]. Other components are specific for individual electron acceptors, including a periplasmic cytochrome that serves as the fumarate reductase [7] and the cytochrome OmcA which has a role in Mn(IV) reduction [8]. However, many components that influence or participate in electron acceptor use in MR-1 remain unidentified.
Outer membrane (OM) components could be important for the ability of MR-1 to reduce insoluble metal oxides. For example, the OM cytochromes OmcA and OmcB play a role in Mn(IV) reduction but are not essential for Fe(III) reduction [8,9]. The Mn(IV) and Fe(III) reductases in MR-1 may be distinct [9,10], and various studies suggest the presence of Fe(III) reductase in the OM [11-13]. Insights are needed into other OM components that could have roles in metal reduction.
Since metal reduction is predominant under anaerobic conditions [2,13,14], components which are upregulated under anaerobic conditions could represent candidates with potential roles in these reductive processes. In this manuscript, a 35-kDa OM protein (Omp35) that is upregulated under anaerobic conditions was identified and characterized. Although Omp35 lacks obvious electron transport moieties, a null mutant lacking Omp35 exhibited significant lags during growth on three anaerobic electron acceptors: fumarate, nitrate, and Fe(III).
Results
Purification of Omp35
While numerous procedures were used to attempt purification of Omp35, the two protocols described in the methods section resulted in a significant purification with Omp35 being the predominant protein (data not shown). The N-terminal sequence of Omp35 (EGPSFYGRLELALTNTETGA) aligned perfectly to an open reading frame (ORF) in the MR-1 genomic sequence at locus SO3896 at the coordinates 4042055-4043161 (The Institute for Genomic Research [TIGR]). This 1158-bp ORF encoded a protein with a predicted molecular mass of 39,900 Da. However, the first 23 amino acids resemble a hydrophobic leader sequence. Starting with the N-terminal protein sequence, the predicted mass of the mature protein is 37,600 kDa, which is consistent with the size seen on SDS-PAGE (35.0 ± 0.5 kDa). BLAST analysis identified the strongest alignment of Omp35 to a hypothetical OM porin (IfcO) from S. frigidimarina (emb CAB37061.1; smallest sum probability (P) value of 1.9 × 10-85; 46% identity and 61% similarity). The ifcO gene in S. frigidimarina is located on the opposite strand upstream of an iron-induced flavocytochrome Ifc3 [15].
Expression and in vivo role of Omp35
Identification of the omp35 gene allowed for the development of an Omp35-specific antibody and the generation of an insertional mutant in which part of omp35 was replaced by a kanamycin-resistance (Kmr) casette. This strain, OMP35-1, met all criteria expected for an omp35 null mutant: (i) it lacked the expected 1.7-kb wild-type PCR product for omp35; (ii) it was positive for a 3.4-kb PCR product consistent with Kmr-interrupted omp35 (Fig. 1); (iii) it was negative for a PCR product using primers specific to the cat gene of pEP185.2 (Fig. 1); and (iv) it grew in the presence of kanamycin but not in the presence of chloramphenicol. The lack of the cat gene and sensitivity to chloramphenicol are consistent with a double-crossover gene replacement.
Figure 1.
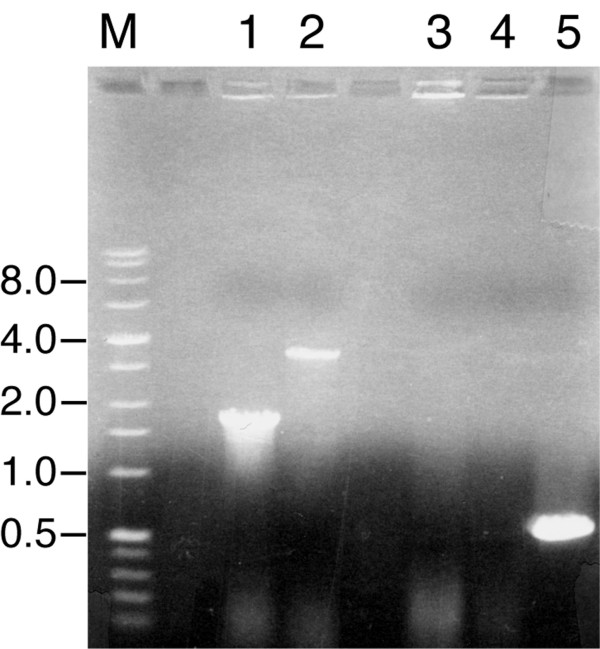
Colony PCR reactions with primers specific for the omp35 gene or the chloramphenicol acetyltransferase gene (cat) from pEP185.2. Lanes 1 and 2 are reactions with omp35 primers O1 and O2 (see Table 2) and lanes 3–5 are reactions with cat primers C1 and C2 (see Table 2). The templates for PCR were as follows: lanes 1 and 3, MR-1; lanes 2 and 4, OMP35-1; lane 5, pDSEPomp35. The sizes of the DNA markers (lane M) in kilobases are indicated on the left.
Western blots confirmed the absence of Omp35 in all subcellular fractions of OMP35-1, whereas Omp35 was readily detected in the OM and intermediate density membrane (IM) fractions of MR-1 (Fig. 2). The IM closely resembles the OM, except for a buoyant density between that of the cytoplasmic membrane (CM) and OM [16]. Omp35 was not detected in CM or soluble fractions of MR-1 (Fig. 2). This subcellular localization is consistent with its purification from the OM. The levels of Omp35 in OM fractions from the OM cytochrome mutants OMCA1 (ΔomcA) and OMCB1 (ΔomcB) [8] were the same as those for MR-1 (data not shown).
Figure 2.
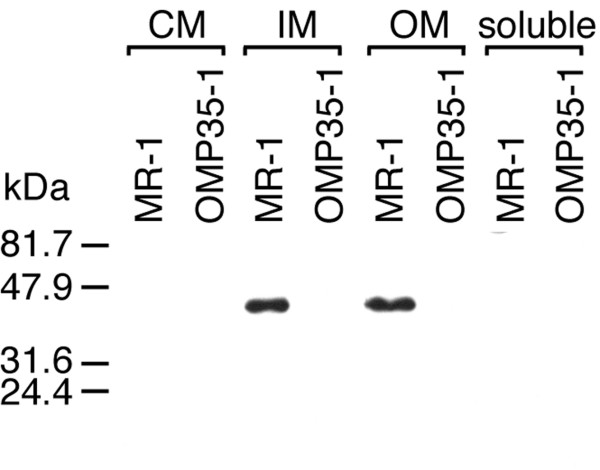
Western blot of subcellular fractions of MR-1 and OMP35-1 with an antibody specific for Omp35. The lanes were loaded with 20 ng protein from each subcellular fraction; cytoplasmic membrane (CM), intermediate membrane (IM), outer membrane (OM), and soluble fraction (soluble). Fractions were isolated in duplicate from cells grown anaerobically with fumarate as the electron acceptor. The results shown are representative.
Western blots confirmed that Omp35 is significantly upregulated under anaerobic conditions, with levels more than 7-fold higher in fumarate-grown cells compared to aerobically-grown cells (Fig. 3A,3B). This is not the result of transcriptional regulation because the levels of omp35 transcript were statistically similar in aerobically- and fumarate-grown MR-1 (Fig. 3C,3D). Levels of Omp35 protein in the OM of the etrA mutant ETRA-153 [17] were similar to the levels found in the OM of MR-1 suggesting that EtrA does not significantly regulate Omp35 (data not shown).
Figure 3.
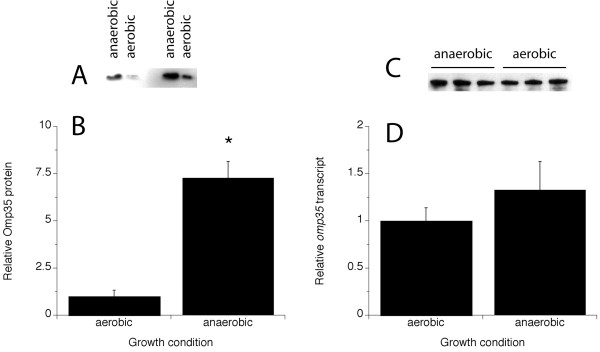
Relative levels of Omp35 protein (A, B) and omp35 transcript (C, D) in aerobically-grown versus fumarate-grown MR-1. A, B: Omp35 protein was detected by western blot of whole cells using an antibody specific for Omp35. An example of two dilutions of a representative experiment are shown in panel A, and the quantitative results from densitometric analysis of western blots from three independent experiments (mean ± S.D.) are shown in panel B. *, statistically different from aerobic to P ≤ 0.001. C, D: Transcript was determined by RNase protection using an antisense probe specific for the omp35 transcript. The data for three independent experiments are shown in panel C, and the quantitative results from densitometric analysis of the RPA blots (mean ± S.D.) are shown in panel D. For each experiment, dilutions of each sample were analyzed to ensure linearity of signal intensity.
The ability of wild-type omp35 to complement OMP35-1 was examined. Two constructs (pBComp218 and pBComp411) containing omp35 plus 218 and 411 bp of upstream DNA, respectively, in the vector pBCSK were introduced into OMP35-1. Each insert was tested in both orientations; the forward (F) is in frame with the lacZ promoter of the vector, whereas the reverse (R) is not. Western blots showed that all four constructs (pBComp218F, pBComp218R, pBComp411F, pBComp411R) restored Omp35 to OMP35-1 at levels that were greater than those of wild-type (Fig. 4).
Figure 4.
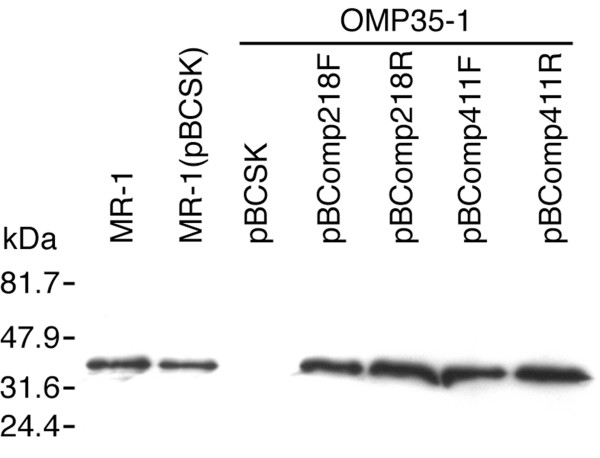
Western blot of lysed whole cells with an antibody specific for Omp35. Each lane was loaded with equivalent wet cell pellet weight (30 μg). The strains carrying the various plasmids are indicated above each lane. This blot is representative of duplicate experiments. The bars and numbers at the right indicate the migration and mass of the protein standards. The results shown are for aerobically grown cells. Analogous results were obtained with fumarate-grown cells (not shown).
The potential role of Omp35 in anaerobic respiration was assessed by a comparison of the relative abilities of MR-1 and OMP35-1 to grow on and reduce various electron acceptors. The maximal growth yields of OMP35-1 were essentially the same as those for MR-1, with no apparent growth lags on 20 mM TMAO, 5 mM DMSO, 10 mM thiosulfate, or O2 as terminal electron acceptors (data not shown). OMP35-1 also reduced 5 mM δMnO2 and AQDS at rates similar to those of MR-1 (data not shown). However, there was a distinctive lag in the onset of growth of OMP35-1 on 20 mM fumarate, 2 mM nitrate, and 10 mM Fe(III) citrate (Figs. 5,6), and in the reduction of Fe(III) citrate and 2 mM αFeOOH (Figs. 6,7). The rates of reduction of nitrate and nitrite by OMP35-1 were also slower than those of MR-1 (not shown), corresponding to the delayed growth on nitrate. The lag on fumarate was the most pronounced with MR-1(pBCSK) reaching maximal growth at 1 day, while OMP35-1(pBCSK) showed no growth until day 3 (Fig. 5A). On nitrate, OMP35-1 took one day longer than MR-1 to attain maximal growth (Fig. 5B). The growth of OMP35-1(pBCSK) on Fe(III) citrate lagged behind that of MR-1(pBCSK) for the first 12 hrs (Fig. 6B).
Figure 5.
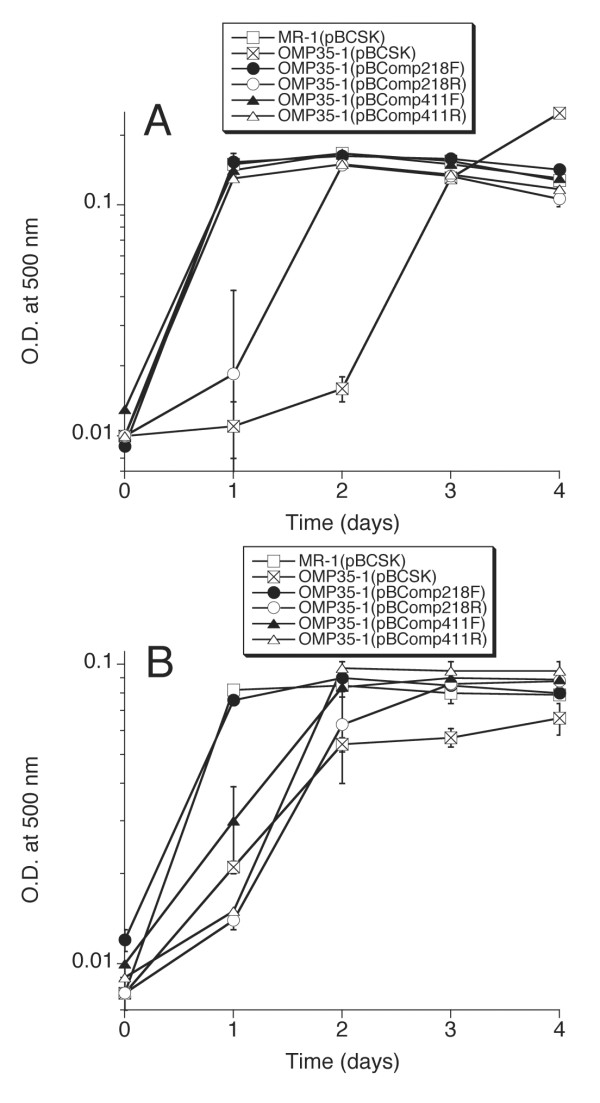
Anaerobic growth of various strains on fumarate (A) and nitrate (B). Values represent mean ± high/low for two parallel but independent experiments for each strain.
Figure 6.
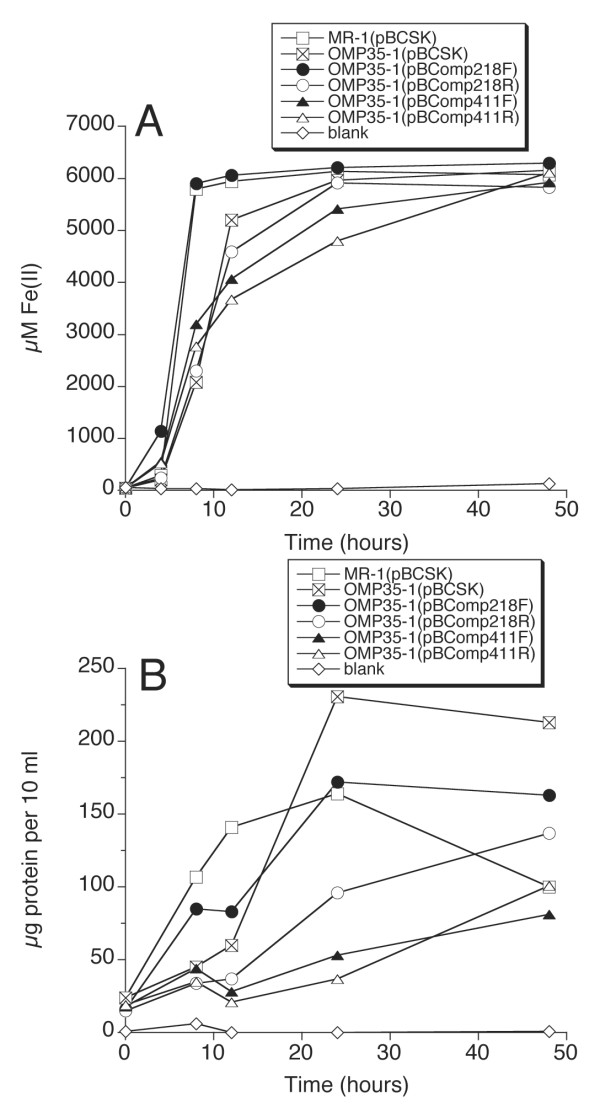
Anaerobic reduction (A) and growth (B) on Fe(III) citrate by various strains. One representative experiment from two independent experiments is shown.
Figure 7.
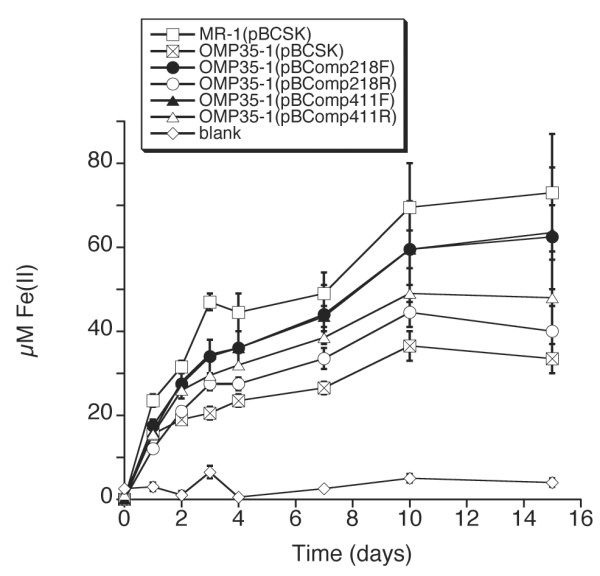
Anaerobic reduction of αFeOOH by various strains. Values represent mean ± high/low for two parallel but independent experiments for each strain.
Three of the four complementing omp35 plasmids restored the growth of OMP35-1 on fumarate to rates that were indistinguishable from those of wild-type (Fig. 5A). The growth rate of OMP35-1(pBComp218R) was less than that of MR-1, but was much closer to MR-1 than to OMP35-1 (Fig. 5A). One of the constructs (pBComp218F) restored nitrate reduction and growth to wild-type levels, whereas the other three constructs exhibited a lag similar to OMP35-1 (Fig. 5B). However, the final growth yields on nitrate were greater for the various complements than for OMP35-1 (Fig. 5B). Similar to the nitrate results, growth on and reduction of Fe(III) citrate was near wild-type for OMP35-1(pBComp218F) (Fig. 6), whereas the others lagged behind wild-type at 12 hrs (Fig. 6B). OMP35-1(pBCSK) could reduce insoluble αFeOOH, but it was significantly slower than MR-1(pBCSK) (Fig. 7). All four constructs partially restored αFeOOH reduction to levels that were between those of wild-type and OMP35-1 (Fig. 7). Both R constructs exhibited less Fe(II) accumulation than the corresponding F constructs (Fig. 7), which could indicate some control of expression of the F constructs by the lacZ promoter of pBCSK.
Cell surface exposure of Omp35
To determine the potential cell surface exposure of Omp35, proteinase K susceptibility experiments were conducted with anaerobically grown MR-1 cells and compared to control cells that were exposed to buffer. The proteinase K-to-control ratio for the periplasmic fumarate reductase was near unity, i.e. the same in proteinase K and buffer-treated cells (Fig. 8), indicating that proteinase K did not compromise the integrity of the OM. The ratio for Omp35 was also near unity (Fig. 8) indicating that Omp35 is not significantly exposed to proteinase K at the cell surface. However, proteinase K treatment of either purified OM (Fig. 8) or OM solubilized with 0.2% Z3-12 (data not shown) from fumarate-grown MR-1 resulted in complete degradation of Omp35. Thus, proteinase K can completely degrade Omp35 if given sufficient access. To confirm that the proteinase K was active in the whole cell experiments, the surface-exposed outer membrane cytochrome OmcA was substantially degraded by proteinase K (Fig. 8) in agreement with previous findings [18].
Figure 8.
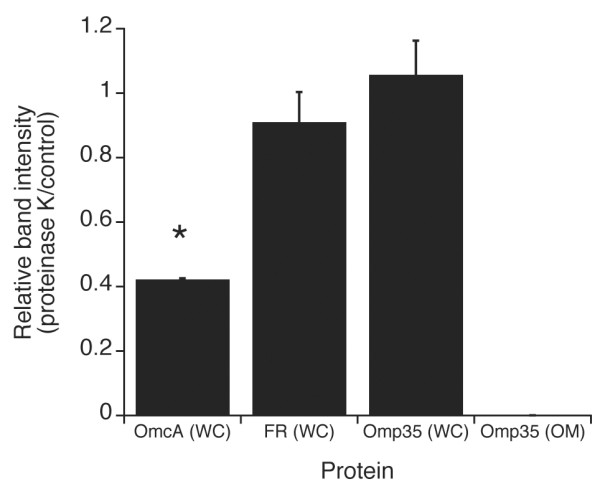
The effect of proteinase K on MR-1 proteins OmcA, fumarate reductase (FR), and Omp35. Either fumarate-grown whole cells (WC) or purified outer membrane (OM) fractions isolated from fumarate-grown MR-1 were treated with proteinase K. Results of duplicate experiments are expressed as the ratio of band intensity with proteinase K vs. protease-free control. *, statistically significant from FR control to P ≤ 0.006.
Possible roles of Omp35 in cells
Since the absence of Omp35 caused significant lags in the use of some, but not all, electron acceptors, Omp35 might have either direct or indirect roles in electron transport. While its OM location could make sense for a direct role in Fe(III) reduction, it is not clear why this would impact the use of soluble electron acceptors such as fumarate and nitrate whose terminal reductases are periplasmic (fumarate) [7,19] or likely periplasmic (nitrate). The effects on soluble electron acceptors might imply indirect effects which alter the synthesis or distribution of other electron transport components.
To examine possible indirect effects, the content and distribution of electron transport components in fumarate-grown OMP35-1 was compared to that of MR-1. Protein- and heme-stained SDS-PAGE patterns of subcellular fractions were similar for the two strains (data not shown). Cytochrome spectra of the subcellular fractions indicated that the cytochrome content and distribution of OMP35-1 were similar to MR-1 (data not shown). Western blotting of membrane and soluble fractions showed no significant differences between MR-1 and OMP35-1 in the content or distribution of the OM cytochromes OmcA and OmcB, the OM protein MtrB, and the periplasmic fumarate reductase (Fcc3) (data not shown).
Menaquinone is an important electron transport component required for the reduction of fumarate, nitrate, Fe(III), and Mn(IV) [6]. OMP35-1 displayed a delayed phenotype on three of these. However, the levels of menaquinone, methylmenaquinone, and ubiquinones were similar in MR-1, MR-1(pBCSK), OMP35-1, and OMP35-1(pBCSK) (data not shown). Analysis of Omp35 protein in the OM of the menaquinone-minus mutant CMA-1 [6] indicated that the levels were similar to those found in the OM of MR-1 (data not shown). Therefore, OMP35-1 contains a normal complement of quinones, and the absence of menaquinone and methylmenaquinone does not affect the level of Omp35 protein in CMA-1.
It was noted during the purification of Omp35 that fractions containing Omp35 had a yellow color. Because flavins (FAD and FMN) have a yellow color and are electron transfer cofactors of some proteins, the possibility that Omp35 is a flavoprotein was explored. To detect the possible presence of either FAD or FMN in partially purified or purified fractions containing Omp35, several techniques were used including fluorescence spectroscopy, thin layer chromatography (TLC), and UV-visible spectral analysis. All three detected the flavin standards (FAD and FMN) but all three failed to detect any flavin moiety in the yellow fractions containing Omp35. The lower limits of detection of the standards were 0.1 nmol and 2.5–5 pmol for TLC and fluorescence spectroscopy, respectively. Attempts to remove non-covalently bound flavin (boiling, trichloroacetic acid precipitation, and chloroform extraction) and UV-visible spectra of non-treated proteins also failed to detect any flavin. The sum total of procedures should have been able to detect either covalently or noncovalently bound flavin. Spectral analysis of Omp35 fractions was also negative for heme as were heme-stained SDS-PAGE gels. Motif searching of the Omp35 sequence resulted in no matches for heme- or flavin-binding motifs or other motifs suggestive of electron transport proteins.
A small nonproteinaceous compound is apparently excreted from MR-1 that has the ability to restore AQDS reduction to a menaquinone-minus mutant [20]. This compound is not menaquinone, but is apparently redox-active and has a yellow-orange color [20]. Studies were done to explore the possibility that Omp35 may have a role in the secretion of this compound. If true, the yellow color associated with Omp35 could conceivably be due to an affinity between this compound and Omp35. On separate plates, MR-1 and OMP35-1 were streaked adjacent to the menaquinone-minus mutant CMA-1 in a triangular pattern as done by Newman and Kolter [20]. Both MR-1 and OMP35-1 restored the ability of CMA-1 to reduce AQDS and Fe(III) (not shown). This indicates that OMP35-1 retained the ability to excrete this compound and that Omp35 is not necessary for its secretion.
Discussion
The 7-fold upregulation of Omp35 in fumarate-grown versus aerobically-grown MR-1 (Fig. 3A) is similar to the 7-fold anaerobic upregulation of the OM cytochrome OmcA [16]. This upregulation suggests an important function under anaerobic conditions. A role for OmcA is limited to Mn(IV) reduction [8], whereas the absence of Omp35 resulted in significant lags in growth on fumarate, nitrate, and Fe(III). Several other proteins in MR-1 are also increased under anaerobic conditions including the OM cytochrome OmcB, and the activities for Fe(III), nitrate and fumarate reductases, and formate dehydrogenase [8,13,19]. The mechanisms responsible for these anaerobic upregulations have not been identified in MR-1. While the transcriptional regulator EtrA has a partial role in upregulating fumarate and nitrate reductase [17], it has no effect on levels of OM cytochromes or Omp35. This coincides with the observation that the upregulation of Omp35 is not transcriptional (Fig. 3). The anaerobic induction of OM proteins with resulting effects on anaerobic electron transport have been reported in other species. For example, a major OM protein (AniA) which resembles copper-containing nitrite reductase is anaerobically induced in Neisseria gonorrhoeae [21]. However, the aniA mRNA in N. gonorrhoeae is only expressed under anaerobic conditions, with a role for Fnr and NarP as transcriptional regulators [21].
Regarding the post-transcriptional upregulation of Omp35 under anaerobic conditions (Fig. 3), possible control mechanisms include factors that affect translation efficiency, such as mRNA binding proteins or mRNA secondary structure elements that repress/activate translation [22-24]. Translational regulation of proteins that are induced anaerobically has been reported. The level of ethanol dehydrogenase (AdhE) activity is 10-fold higher in fermentatively-grown versus aerobically-grown E. coli K-12, and the translation of adhE mRNA is regulated by RNA secondary structure changes that block the RNA polymerase binding site [25]. Pseudoazurin, a periplasmic shuttle protein in Thiosphaera pantotropha, is expressed anaerobically during nitrification and is regulated by changes in RNA secondary structure [26].
The proteinase K experiments indicated that Omp35 is not significantly exposed on the cell surface. Proteinase K was chosen for these studies because it cleaves on the carboxyl side of a variety of amino acids including aliphatic, aromatic, and hydrophobic residues, and because it completely degraded Omp35 when given adequate access to the protein in purified OM fragments (Fig. 8). This is consistent with the Omp35 sequence, which indicates that 68% of the Omp35 residues are possible proteinase K cleavage sites. While proteinase K-resistant residues of Omp35 could be exposed on the cell surface in small loops, sizable extracellular loops are not likely because the longest stretch of proteinase K-resistant residues in Omp35 is four. Since Omp35 is not significantly exposed on the cell surface, it is also hard to envision a direct role in cell attachment or adhesion. Even so, while proteins that influence adhesion to insoluble Fe(III) oxides could be important [12], it is not clear why this would affect growth on nitrate and fumarate.
The significant growth lags observed with OMP35-1 on fumarate, nitrate, and Fe(III) indicate that Omp35 is required for optimal growth rates under these conditions. While complementation of OMP35-1 with pBComp218F restored wild-type growth on fumarate, nitrate, and Fe(III), the other omp35 constructs varied in their ability to restore electron acceptor use (Figs. 5,6,7), even though all restored Omp35 protein (Fig. 4). While expression of the forward constructs could be influenced by the lacZ promoter of the vector, it is unclear why OMP35-1(pBComp411F) was slower on nitrate and Fe(III) than was OMP35-1(pBComp218F) despite ~200 bp of additional upstream DNA. One possibility is that the extra upstream DNA in pBComp411F might have reduced control of the lacZ promoter on omp35 expression. If RNA secondary structure controls translation of omp35, non-optimal levels of transcript could influence protein levels. However, Omp35 levels in all four constructs were quite similar and about two-fold higher than those in MR-1 based on NIH image analysis of the western blots (Fig. 4). Since the pBComp218F construct was able to fully complement OMP35-1, elevation of Omp35 levels above those found in MR-1 is not necessarily problematic. However, closer examination indicated that OMP35-1(pBComp218F) contained 20–30% less Omp35 than the other three complements. It is unclear if such minor differences influence the phenotypes, but it is possible that the cells are sensitive to small variations in Omp35 content.
The mechanism by which Omp35 affects growth rates on only some electron acceptors is not clear. Omp35 does not contain detectable heme or flavin, and sequence analysis does not reveal any sites suggestive of iron-sulfur centers, heme- or flavin-binding sites, molybdopterin-binding sites, or other redox-active moieties. Omp35 is therefore not likely to be an electron transport protein. However, motif searching does have limitations. Some motifs can be quite variable among organisms and the motifs are based on most likely consensus domains derived from a limited number of proteins. Therefore, while Omp35 is likely not an electron transport protein, this possibility cannot be completely dismissed. However, such a role would not make sense for fumarate and nitrate given that these terminal reductases are periplasmic in MR-1. However, the size of Omp35 is consistent with the size range typical for porins [27], and sequence alignments predict that Omp35 is likely a porin (Fig. 9). Omp35 aligned most prominently to a hypothetical protein (IfcO) in S. frigidimarina NCIMB400 (46.3% identity, 61.2% homology) (Fig. 9). IfcO is located upstream of ifcA (a gene encoding an iron-induced flavocytochrome Ifc3) [15]. IfcO displays 20% identity to a cation-selective porin from Neisseria meningitidis [15]. The genes surrounding omp35 in MR-1 and ifcO in S. frigidimarina are quite different, suggesting that there could be differences in regulation and/or function. Omp35 also aligned to two putative porins at loci SO1420 and SO1557 in the MR-1 genome (31.4 and 23.0% identity, and 45.0 and 39.3% homology, respectively) (Fig. 9) and to the porin PorA [28] from N. meningitidis (20.6% identity, 35.5% homology) (Fig. 9). The sequence homologies are consistent with the presence of conserved membrane core domains and variable loops typical of porins [29,30]. The extent of sequence identity and homology is also typical for that seen with porins across other species.
Figure 9.

Amino acid sequence similarities between Omp35 of MR-1 and the top matches as identified by BLAST. Alignments were done using the ClustalW function of MacVector software. Identical residues are indicated by uppercase letters, analogous residues by lowercase letters, and unmatched residues by dots. Alignments were facilitated by introducing gaps (-). The numbers on the right indicate the relative numbering of residues within each immature protein; the total number of residues in each protein is shown in parentheses at the end of each sequence. Comparative sequences and their accession numbers are as follows: PorA N men (Neisseria meningitidis PorA, OM porin precursor, GenBank AF226349_1); SO1557 MR-1 (S. oneidensis MR-1, putative OM porin, TIGR genome locus SO1557); SO1420 MR-1 (S. oneidensis MR-1, putative OM porin, TIGR genome locus SO1420); IfcO S fri (S. frigidimarina, putative OM porin, GenBank AJ236923).
It is, however, not clear how a porin role for Omp35 relates to the observed electron acceptor phenotype of OMP35-1. If Omp35 is needed for diffusion of electron acceptors across the OM, one might have expected its absence to slow the rate of entry of all soluble electron acceptors including fumarate, nitrate, TMAO, DMSO, nitrite, and thiosulfate. All of these have molecular weights that range from 50–120 Da, well below the exclusion limit for many porins (<600 Da) [31,32]. While some porins can discriminate between substrates based on their charge [31,33], the electron acceptors affected in OMP35-1 have different charge characteristics: [fumarate2- at pH 7], nitrate [NO3-], and Fe(III) citrate or αFeOOH [neutral overall]. The reduction of thiosulfate [S2O32-] and TMAO and DMSO [neutral] were not affected in OMP35-1. Since αFeOOH is insoluble and too large to cross the OM, a porin would have no role in αFeOOH diffusion. While the loss of a porin might impair entry of general nutrients required by MR-1 (e.g. arginine, serine, glutamate, lactate, formate, ammonium sulfate, phosphate, trace metals, etc.), this should have affected growth under all conditions. Interestingly, a porA mutant of N. meningitidis displayed normal growth rates, whereas a porB mutant was somewhat retarded [34]. While N. meningitidis does not have the respiratory versatility of MR-1, these phenotypes are consistent with that of OMP35-1 and suggest that other porins may substitute for the absence of Omp35.
While many porins exhibit heat modifiability [33,35], we observed no such behavior for Omp35. In addition, OMP35-1 still contained another OM protein that migrated at 35 kDa. Another putative MR-1 porin encoded by locus SO1420 is of similar size to Omp35.
Additional OM channel proteins, often called protein transport pumps, allow the energy-dependent uptake or secretion/efflux of porin-excluded substances. Transport pumps are different than type II porins; they bind a specific substrate with a much higher affinity and their channels are not continuously open [36]. Several putative OM uptake pumps have been identified in the genome of MR-1. These include possible metal ion transporters and peptide and/or amino acid uptake systems [37]. Omp35 is not likely to represent one of these systems since non-metal electron acceptors (fumarate and nitrate) were affected in OMP35-1. Defects in amino acid uptake would likely have affected growth under a variety of conditions.
In addition to using Fe(III) as an electron acceptor, MR-1 must assimilate iron for cellular constituents such as cytochromes. Siderophores are low molecular weight Fe(III) chelators that scavenge iron from the environment [38,39]. MR-1 synthesizes siderophores that are negatively regulated by Fur (ferric uptake regulator) [40]. However, Omp35 is not likely involved in siderophore transport for several reasons: (i) Omp35 has no homology to known siderophore proteins; (ii) the specific cytochrome content of OMP35-1 resembled that of wild-type; (iii) the use of some electron acceptors (e.g. TMAO, Mn(IV)) that require iron-containing components such as cytochromes was not affected in OMP35-1; and (iv) the growth on 10 mM Fe(III) citrate should not have invoked a need for siderophore transport, because siderophores are only important under low iron conditions.
A variety of genes encoding putative efflux pump proteins have also been identified in the MR-1 genome. The membrane fusion protein (MFP) family transports larger molecules, such as peptides, proteins, and carbohydrates across the OM [41]. The MR-1 genome encodes members of this family including nine MexB-like efflux transporters [37]. Another member of this family from E. coli is the AcrAB efflux system [42]. A direct link to the OM by the AcrAB efflux system occurs through TolC, an OM protein channel that has been implicated in the secretion of proteins and the efflux of toxins [43,44]. A tolC homolog has been found to protect MR-1 from cell death due to the accumulation of AQDS through the TolC-mediated efflux of AQDS [45]. Although Omp35 did not match any known OM transport pumps, the possibility of a role as an efflux pump for the removal of toxic metabolites or the excretion of unidentified compounds remains a possibility. A small unidentified compound that is released from MR-1 restores the ability of a menaquinone mutant to reduce AQDS [20]. Our results indicate that Omp35 is not required for the secretion of this compound. Even so, a potential role for such a compound in nitrate and fumarate reduction seems improbable.
The effects of Omp35 on electron acceptor use are therefore likely indirect. This is also the case for MtrB, an OM protein of MR-1 that is required for Mn(IV) and Fe(III) reduction but that lacks obvious electron transport moieties [46,47]. MtrB is required for the proper localization and insertion of the OM cytochromes OmcA and OmcB into the OM [47]. While the localization of OM cytochromes is normal in OMP35-1, Omp35 could conceivably have a role in the localization or arrangement of OM components required for Fe(III) reduction. How this would also impact nitrate and fumarate reduction, however, remains unclear.
Conclusions
A 35-kDa probable porin (Omp35) was isolated from the OM of MR-1. Omp35 levels are markedly upregulated anaerobically by a post-transcriptional mechanism. To our knowledge, this is the first report of a porin that is upregulated anaerobically in this manner. An omp35 null mutant exhibited significant lags in anaerobic growth on fumarate, nitrate, and Fe(III). The absence of Omp35 did not affect the quinone content or the levels or distribution of various cytochromes in MR-1. Omp35 does not contain obvious electron transport moieties, so its effects on the use of electron acceptors are likely indirect. The results highlight the possibility for non-electron transport proteins to influence anaerobic respiratory phenotypes, and the importance of considering such indirect effects when characterizing electron acceptor deficiencies.
Methods
Bacterial strains, plasmids, media, and growth conditions
All materials were from sources previously described [4,5]. A list of the bacteria and strains used in this study is presented in Table 1. For molecular biology purposes, S. oneidensis strains were grown aerobically at room temperature (23–25°C) or at 30°C on Luria-Bertani (LB) medium, pH 7.4 [48]. E. coli strains were grown aerobically at 37°C on LB medium. Growth media were supplemented with appropriate antibiotics when required, including ampicillin (Ap), 50 μg mL-1; chloramphenicol (Cm), 34 μg mL-1; and kanamycin (Km), 50 μg mL-1.
Table 1.
Bacterial strains and plasmids used in this studya
| Bacterial strain or plasmid | Description | Reference, Source |
| S. oneidensis strains | ||
| MR-1 | Manganese-reducing strain from Lake Oneida, N.Y. sediments | [1] |
| OMP35-1 | omp35 mutant derived from MR-1; omp35::Kmr | This work |
| OMCA-1 | omcA mutant derived from MR-1; omcA::Kmr | [8] |
| OMCB-1 | omcB mutant derived from MR-1; omcB::Kmr | [8] |
| ETRA-153 | etrA mutant derived from MR-1; etrA::Kmr | [17] |
| CMA-1 | menaquinone-minus mutant derived from MR-1 | [6] |
| E. coli strains | ||
| JM109 | recA1 F' traD36 lacIq Δ(lacZ)M15 proA+B+/ e14-(McrA-) Δ(lac-proAB) thi gyrA96 (Nalr) endA1 hsdR17 (rK- mK+) relA1 supE44 | [61] |
| S17-1λpir | λ(pir) hsdR pro thi; chromosomally integrated RP4-2 Tc::Mu Km::Tn7; donor strain to mate pEP185.2-derived plasmids into MR-1 | [62] |
| S17-1 | C600::RP-4 2-Tc::Mu-Km::Tn7 hsdR hsdM recA thi pro; donor strain to mate pVK100- derived plasmids into MR-1 | [62] |
| TOP10 | F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15ΔlacX74 deoR recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Smr) endA1nupG; used as host for plasmids derived from pCR2.1-TOPO | Invitrogen |
| Plasmidsb | ||
| pBCSK | 3.4-kb vector; pUC ori; Cmr; used for complementation | Stratagene |
| pBComp218F & R | pBCSK with the MR-1 omp35 open reading frame plus associated 5' and 3' regions; 218 bp upstream sequence in the forward (F) and reverse (R) orientation | This work |
| pBComp411F & R | pBCSK with the MR-1 omp35 open reading frame plus associated 5' and 3' regions; 411 bp upstream sequence in F and R orientation | This work |
| pCR2.1-TOPO | 3.9-kb vector for cloning PCR products; Kmr Apr | Invitrogen |
| pUT/mini-Tn5Km | Apr; Tn5-based delivery plasmid; used as source of Kmr gene | [63] |
| pTOPO/omp35 | pCR2.1-TOPO with the MR-1 omp35 open reading frame plus associated 5' and 3' regions; 5.6 kb | This work |
| pTOPO/omp35:Km | pTOPO/omp35 with the Kmr gene replacing bases 458–850 of omp35; 7.3 kb | This work |
| pEP185.2 | 4.28-kb mobilizable suicide vector derived from pEP184; oriR6K mobRP4 Cmr | [64] |
| pDSEPomp35 | Kmr-interrupted omp35 gene cloned into the XhoI site of pEP185.2; Cmr Kmr; 7.7 kb; used for gene replacement to generate strain OMP35-1 | This work |
aNalr, Smr, Kmr, Apr, Tcr, and Cmr, resistance to nalidixic acid, streptomycin, kanamycin, ampicillin, tetracycline, and chloramphenicol, respectively. bThe vector pBCSK freely replicates in MR-1 while pEP185.2 does not.
For other applications, S. oneidensis was grown at room temperature either aerobically or anaerobically as previously described [49] in M1 defined medium [2] supplemented with 15 mM lactate and vitamin-free Casamino Acids (0.1 g L-1). For testing the growth on or reduction of electron acceptors under anaerobic conditions, 15 mM formate was also included. Anaerobic studies were conducted in an anaerobic chamber (Coy Laboratory Products, Ann Arbor, MI) with an atmosphere of 4 to 6% H2 (balance N2). For anaerobic growth or analysis of electron acceptor use, the medium was supplemented with one of the following electron acceptors: 20–30 mM disodium fumarate, 10 mM sodium thiosulfate, 20 mM trimethylamine N-oxide (TMAO), 5 mM dimethylsulfoxide (DMSO), or 2 mM sodium nitrate. For growth on TMAO, the medium was also supplemented with 30 mM HEPES to buffer against alkalinization by the product trimethylamine. Studies with Fe(III) or Mn(IV) were conducted in LM medium [3] supplemented with 15 mM lactate, 2 mM sodium bicarbonate, and one of the following electron acceptors: 10 mM Fe(III) citrate, 2 mM Fe(III) oxyhydroxide (αFeOOH), or 5 mM vernadite (δMnO2).
For electron acceptor characterization of strains, inocula were prepared from cells grown aerobically for 1–2 days on LB medium supplemented with the appropriate antibiotics. Cells were suspended in sterile distilled water and the inoculum densities were adjusted to equalize turbidity (adjustments were made in the inoculum optical density and/or volume).
Purification of Omp35
OM was isolated from MR-1 cells grown anaerobically with fumarate as the electron acceptor. Loosely associated proteins were removed by treatment of the OM at room temperature with 23 mM sodium cholate in buffer A (20 mM K2HPO4 (pH 7.4), 1 mM EDTA, 0.02% sodium azide, and 5% glycerol) containing 10 mM DTT (dithiothreitol) and 0.2 M NaCl with a cholate/protein ratio of 9:1 (wt/wt) and the protein at 1 mg mL-1. The suspension was stirred, sonicated four times (30 sec each) with 1–2 min periods of cooling, stirred for an additional 10 min, and then centrifuged for 97 min at 50,000 rpm (302,000 × g) at 4°C in a Beckman 50.2 Ti rotor. The pellets were resuspended in 3 mL buffer A and treated at room temperature with lysozyme (5 × 104 U per 7–8 mg protein) for 1 hr and with mutanolysin (6 U per mg protein) for an additional hr to digest any remaining cell wall material. The sample was diluted with buffer A and centrifuged as above. The pellet was solubilized with one of the following: (i) 1.66% Z3-12 [3-dodecyldimethylammoniopropane-1-sulfonate] in buffer A containing 0.2 M NaCl and 10 mM DTT; (ii) 1% SDS in 50 mM Tris [hydroxymethyl]aminomethane-HCl (pH 6.9) buffer containing 1 mM EDTA, 0.02% sodium azide, 0.2 M NaCl, and 10 mM DTT; or (iii) 3.65 M urea plus 1.66% Z3-12 in 50 mM Tris-HCl (pH 6.9), 1 mM EDTA, 0.02% azide, 0.2 M NaCl, and 10 mM DTT. After stirring at room temperature for 10 min, the solubilized OM was sonicated twice (30 sec each with 1 min cooling) and centrifuged for 28 min at 90,000 rpm (438,000 × g) in a Beckman TLA-100.3 rotor at 4°C (or at room temperature when 3.65 M urea was included).
Two methods were performed to separate Omp35 from other OM proteins including the OM cytochromes. In the first, Z3-12 solubilized OM was concentrated by ultrafiltration to a volume of < 2 mL using a Millipore Ultra-Free filter (30,000 MWCO), and then applied to a Sephacryl S-200 HR (Pharmacia Biotech) gel filtration column (1.6 × 86 cm) at 4°C. Proteins were eluted with 0.5% Z3-12 in buffer A containing 0.2 M NaCl and 10 mM DTT, and fractions were screened by heme- and silver-stained SDS-PAGE. Those containing a 35-kDa band were pooled and dialyzed overnight in 10 mM K2HPO4 (pH 7.4) with 5% glycerol, 1 mM EDTA, 0.02% sodium azide, 0.5% Z3-12 and 0.1 mM DTT (buffer B). The dialyzed sample was applied to a Bio-Gel hydroxylapatite column (0.5 × 2.5 cm); after equilibration and removal of nonbinding proteins with buffer B, a step gradient of increasing concentrations of K2HPO4 buffer was applied (25 mM, 50 mM, 100 mM, 200 mM, and 400 mM). A 35-kDa band was prominent in the fractions which eluted at 50–100 mM K2HPO4; this band was heme negative (i.e. not a cytochrome).
In the second method, differential ultrafiltration was used on OM that had been solubilized with each of the three detergent protocols. A Pall Filtron 50,000 MWCO filter retained the red cytochromes while the 35-kDa protein was in the filtrate. This filtrate was concentrated using a 30,000 MWCO Millipore Ultra-Free filter, and then applied to a GCL-300 gel filtration column (Isco, Inc; 1.6 × 35 cm). Fractions were obtained in which a 35-kDa protein was in high concentration relative to other minor proteins.
Fractions enriched in Omp35 were subject to SDS-PAGE and transferred to a PVDF membrane. The Omp35 band was excised from the membrane and the N-terminal sequence was determined by the Protein/Nucleic Acid Shared Facility of the Medical College of Wisconsin, using a Beckman Coulter model 2CF 3000 pulsed liquid phase protein sequencer.
DNA manipulations
A list of synthetic oligonucleotides used is presented in Table 2. Restriction enzyme digests, cloning, subcloning, and DNA electrophoresis were done according to standard techniques [48] following manufacturers' recommendations as appropriate. The following procedures were done as previously reported [8,17]: DNA ligation, isolation of plasmid and cosmid DNA, colony PCR, DNA sequencing, and determination of sizes of DNA fragments, RNA, and proteins. Electroporation and preparation of cells for electroporation was performed as previously described for either E. coli [5] or MR-1 [17]. Computer-assisted sequence analysis and comparisons were done with MacVector software (Accelrys, San Diego, CA). Oligonucleotide primers were designed by using OLIGO software (version 6.15; Molecular Biology Insights, Cascade, CO).
Table 2.
Synthetic oligonucleotides used in this study.
| Name | Oligonucleotide sequencea |
| Oligonucleotides based on omp35 | |
| O1 | 5'-TGCTCGAGGGGCGGGTAAACGTAAACTCA-3' |
| O2 | 5'-TGCTCGAGCATGGCGCCTCACCTACCT-3' |
| O3 | 5'-ATTGCGATCGATATAACCGTTGGCGATCTT-3' |
| O4 | 5'-ATTGCGATCGATTAACAAAGGTATTGCGGGTAT-3' |
| O5 | 5'-TTTAGAGAAGCGGTAGTCAGC-3' |
| O6 | 5'-ATCGCCAACGGTTATGAT-3' |
| O7 | 5'-ACGCCATATCGAGTTTACAT-3' |
| Oligonucleotide based on Km r gene | |
| K1 | 5'-ATTGCGATCGATTTATGCTTGTAAACCGTT-3' |
| Oligonucleotides based on cat gene | |
| C1 | 5'-CAGACGGCATGATGAACCTGAATC-3' |
| C2 | 5'-CCACCGTTGATATATCCCAATGGC-3' |
aThe underlined regions indicate the following restriction endonuclease sites engineered into the oligonucleotides: XhoI sites in O1 and O2 and ClaI sites in O3, O4, and K1.
Antibody specific for Omp35
Recombinant technology was used to generate a protein fusion of thioredoxin (TR) to an internal 250-residue fragment of Omp35. Specifically, a 750-bp fragment of omp35 was generated by PCR of MR-1 genomic DNA using primers O5 and O6 (Table 2). The PCR product was cloned into pBAD/Thio-TOPO (Invitrogen, Carlsbad, CA), and transformed into E. coli TOP10. After identifying a clone containing the omp35 fragment in the proper orientation, expression of the fusion protein was induced with 0.02% arabinose for 2 hr at 37°C. The cells were harvested by centrifugation, and lysed using Bugbuster Protein Extraction Reagent (Novagen, Madison, WI). The resulting fusion protein, a 250-residue fragment of Omp35 with thioredoxin (TR) at the N-terminus and a 6x histidine tag at the C-terminus, was localized primarily in inclusion bodies. After solubilization with 6 M urea, the TR-Omp35 fusion was purified using His•Bind Quick resin (Novagen) according to manufacturer's instructions. The purified fusion was dialyzed at 4°C against 20 mM Tris-HCl (pH7.5)/0.1 M glycine/5% (w/v) glycerol/1% (w/v) NaCl, and then concentrated by ultrafiltration. The purified concentrated TR-Omp35 fusion protein was used as an antigen to generate polyclonal antisera in New Zealand white rabbits using Titermax (CytRx Corp., Norcross, GA) as an adjuvant. A purified immunoglobulin G (IgG) fraction was obtained from the immune and preimmune sera using ammonium sulfate fractionation and ion exchange chromatography.
Construction of an Omp35 gene replacement mutant
An omp35 gene replacement mutant (OMP35-1) was constructed from MR-1 using a strategy analogous to that described previously [4]. A 1729-bp fragment containing the entire omp35 gene plus 5' and 3' flanking sequences was generated by PCR of MR-1 genomic DNA using custom primers O1 and O2 (Table 2). This PCR product was cloned into pCR2.1-TOPO generating pTOPO/omp35. Inverse PCR [50] of pTOPO/omp35 using custom primers O3 and O4 (Table 2) generated TOPO/omp35(Δ393), a 5.2-kb fragment that is missing 393 bp of internal omp35 sequence. The 2.1-kb Kmr gene from pUT/mini-Tn5Km was generated by PCR with the custom primer K1 (Table 2). Following digestion with ClaI, the Kmr gene was ligated to the TOPO/omp35(Δ393) fragment, generating pTOPO/omp35:Km. A 3.4-kb DNA fragment containing the Kmr-interrupted omp35 gene was cut from pTOPO/omp35:Km with XhoI and ligated into the XhoI site of the suicide vector pEP185.2, generating pDSEPomp35, which was then electroporated into the donor strain E. coli S17-1λpir. E. coli S17-1λpir(pDSEPomp35) was mated with MR-1 and MR-1 exconjugants were selected using kanamycin under aerobic conditions on defined medium with 15 mM lactate as the electron donor. Colonies were screened by colony PCR [51]; those lacking the expected wild-type 1.7 kb PCR product were pursued as putative insertional mutants. Throughout, appropriate analyses (restriction digests, PCR, DNA sequencing) were done to verify that the expected constructs were obtained.
Constructs to complement OMP35-1
Wild-type omp35 plus 218 and 411 bp of upstream DNA were amplified from MR-1 genomic DNA with custom primers O1 and O2 (Table 2) and O1 and O7 (Table 2), respectively, using the Expand High Fidelity PCR System (Roche). The products were cloned in pCR2.1-TOPO, from which the inserts were excised by either BamHI/NotI (reverse constructs) or NotI/SacI (forward constructs) and cloned in pBCSK generating pBComp218F and R and pBComp411F and R. The forward construct (F) is in frame with the lacZ promoter of the vector, whereas the reverse (R) is in the opposite orientation. These plasmids were electroporated into JM109, and the identity and orientation of the inserts was verified. They were then electroporated into OMP35-1.
Ribonuclease protection
Total RNA was isolated from either fumarate-grown or aerobically-grown (shaken at 200 rpm) cultures after 1 day of growth using a hot phenol method followed by treatment with RNase-free DNase as previously described [5,52]. To generate a probe, a 342-bp internal fragment of omp35 was cloned into pCR2.1-TOPO; the desired orientation was verified by PCR. Using M13 primers, a 585-bp fragment containing omp35, with flanking 5' and 3' vector DNA, was generated by PCR; using this fragment as a template, the biotin-labeled antisense omp35 RNA probe was generated using the MAXIscript™ Kit (Ambion) and biotin-14-CTP. The probe was gel purified on a Tris-borate-EDTA-urea polyacrylamide gel. RNase protection assays were done using 10 μg total RNA, 400 pg of probe, and the RPA III™ Kit (Ambion, Austin, TX).
Cell surface exposure studies: Proteinase K treatment of whole cells
MR-1 cells grown on fumarate (~40 hr) were harvested by centrifugation (Beckman JA-14 rotor, 10 min at 10,000 rpm, 22°C) and resuspended in 10 mM HEPES, pH 7.5 (buffer H). Cell pellets were washed twice in buffer H with 50 μM MgCl2 and suspended to a final density of 45 mg wet cell weight per mL. The cells were incubated for 15 min with proteinase K (27.5 U mL-1) or buffer as a negative control; previous time course experiments indicated 15 min was optimal for the degradation of surface-exposed proteins. Phenylmethylsulfonyl fluoride (PMSF) was then added (5 mM final concentration) to inhibit the protease. The cells were pelleted (2 min at 12,000 rpm) and washed four times in buffer containing 5 mM PMSF (7 volumes per wash relative to the original cell suspension volume in each tube). Final pellets were resuspended in buffer H with 50 μM MgCl2 and 5 mM PMSF, mixed with two volumes of SDS solubilization mix [53] containing 4 M urea and 5 mM PMSF, and boiled for 10 min. The samples were cooled to room temperature and PMSF was added to a final concentration of 5 mM. Appropriate dilutions were made in SDS solubilization mix containing urea and PMSF, and then samples were immediately run on SDS-PAGE gels. Proteinase K treatment of OM fragments was done in a similar manner except no washing steps were performed. The gels were analyzed by western blot.
Flavin analysis: Fluorescence spectroscopy, thin layer chromatography, and UV/visible spectra
Standards of FAD and FMN were made in 0.1 M K2HPO4 (pH 7.7) buffer with 0.1 mM EDTA. Riboflavin standard was dissolved in dimethylsulfoxide (DMSO).
For thin layer chromatography (TLC), samples were boiled for 3 min before use. TLC was performed on Silica GHL (Analtech) with a preabsorbant zone and developed with 5% K2HPO4. Flavins were visualized under UV at 254 nm.
Fluorescence spectroscopy was done using a Perkin Elmer Luminescence-LS-50B spectrometer. Samples and standards were prepared by a method modified from Faeder and Siegel [54]. 50 μl of sample were added to 0.95 mL of 0.1 M K2HPO4 pH 7.7 with 0.1 mM EDTA (buffer S), boiled for 3 min, and cooled rapidly to room temperature. Alternative methods to remove non-covalent flavin from protein samples were performed as well: (i) treatment with 100% trichloroacetic acid (final concentration of 20%) on ice for 20 minutes before neutralization with 5 N NaOH; and (ii) chloroform extraction. Standards of FAD and FMN (10 μM, 100 nM, 50 nM, 25 nM, 10 nM) were made in buffer S. Fluorescence measurements were done in 1-cm pathlength fluorometer cuvettes (excitation at 450 nm; emission at 535 nm or a 500–580 nm scan). Standards and samples were tested at both pH 7.7 and pH 2.6 (acidified with 0.1 mL of 1.0 N HCl). FAD and FMN have higher fluorescence under acidic and basic conditions respectively. Background fluorescence from buffer S was subtracted from the values.
Flavin content was also analyzed by comparison of reduced and oxidized spectra using an Aminco DW-2000 spectrophotometer operated in the split-beam mode. Samples were used directly without boiling or other treatments. Room temperature spectra were recorded in a quartz sub-micro sample cuvette with sample and reference slit masks, using a slit width of 2.0 nm and a scan speed of 2.0 nm s-1. To obtain reduced spectra, samples were treated with dithionite. Scans were performed in both the visible (400–700 nm) and UV (200–325 nm) ranges.
The ability of OMP35-1 to restore electron acceptor use to a menaquinone-minus mutant
Agar plates of M1 defined medium were prepared containing lactate and formate and either anthraquinone-2,6-disulfonic acid (AQDS, 5 mM final concentration) or 10 mM Fe(III) citrate. For the former, an AQDS suspension was prepared aseptically (0.206 g in 5 mL sterile distilled water) and dispensed into 100 mL medium.
Inocula were grown aerobically on LB agar. Cells were suspended in sterile distilled water and applied to the appropriate position on the agar plate using a sterile loop. Strains were inoculated in a triangular pattern as described by Newman and Kolter [20] with CMA-1 on 2 sides of the triangle and either MR-1 or OMP35-1 on the third side. Plates were placed in the anaerobic chamber immediately after inoculation. Fe(III) citrate reduction was visualized on day 2. Agar plates were covered with a ferrozine agarose solution (0.01 g ferrozine, 1.2% agarose in 10 ml 50 mM HEPES, pH 7). Magenta color corresponding to Fe(II)-ferrozine developed immediately. AQDS reduction was visualized on day 3 by examining the orange halo that formed around the streaks on the plates; this orange color was due to the reduced product 2,6-anthrahydroquinone disulphonic acid [AHDS].
Miscellaneous procedures
For most electron acceptors, growth was assessed by measuring culture turbidity at 500 nm using a Beckman DU-64 spectrophotometer. Because Fe(III) citrate interferes with the turbidity measurement, growth on Fe(III) was assessed by measuring total cellular protein as previously described [3]. Nitrate [55] and nitrite [56] were determined colorimetrically in cell-free filtrates. Fe(II) was determined by a ferrozine extraction procedure [14,57]. When Fe(III) citrate was used, Fe(II) analysis was performed in unfiltered samples. When αFeOOH was used, Fe(II) was determined in samples that were filtered through 0.2 μm filters or that were centrifuged after ferrozine was added. Mn(II) was determined in filtrates by a formaldoxime method [58,59]. δMnO2 [1] and αFeOOH [57] were prepared as described previously.
Cytoplasmic membrane (CM), intermediate membrane (IM), outer membrane (OM), and soluble fractions (cytoplasm plus periplasm) were purified by an EDTA-lysozyme-Brij 58 (polyoxyethylene cetyl ether) protocol as previously described [49]. The separation and purity of these subcellular fractions were assessed by spectral cytochrome content [49], membrane buoyant density [49], and SDS-PAGE gels [53] stained for protein with Pro-Blue (Owl Separation Systems, Woburn, MA) or for heme [60]. Protein was determined by a modified Lowry method, with bovine serum albumin as the standard [5].
Quinones were extracted from cells and were resolved by thin-layer chromatography (TLC) as previously described [4] on Merck Kieselgel 60 F254 plates.
Western blotting was performed as previously described and developed using either the ImmunoPure NBT/BCIP Substrate Kit (Pierce, Inc., Rockford, IL) [16] or the SuperSignal West Pico Kit (Pierce) [47]. Polyclonal antibodies specific for the OM cytochromes OmcA and OmcB [9], the OM protein MtrB [47], and the periplasmic fumarate reductase (Fcc3) [17] have been described previously. The antibody specific for Omp35 (above) was diluted to a final purified IgG concentration of 0.4 μg mL-1.
Statistical analysis was performed using single-factor ANOVA (analysis of variance). Relative band densities of the RNA or protein blots were determined using NIH image software http://rsb.info.nih.gov/nih-image/.
Authors' contributions
TMM conducted most of the experiments, analyzed and organized the data, and drafted most of the manuscript. CRM conducted some of the expression studies, conceived the overall project, provided experimental guidance, and drafted portions of the manuscript. Both authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
The authors are grateful to J. M. Myers for providing expertise and advice in the laboratory. This work was supported by National Institute of Health grant R01GM50786 to CRM. The graduate fellowship for TMM was provided in part by the Robert G. Zach Family Foundation.
The MR-1 genomic sequence was obtained from the Institute for Genomic Research (TIGR) through the website at http://www.tigr.org. Sequencing of the MR-1 genome by TIGR was accomplished with support from the Dept. of Energy.
Contributor Information
Tamara M Maier, Email: tmaier@mcw.edu.
Charles R Myers, Email: cmyers@mcw.edu.
References
- Myers CR, Nealson KH. Bacterial manganese reduction and growth with manganese oxide as the sole electron acceptor. Science. 1988;240:1319–1321. doi: 10.1126/science.240.4857.1319. [DOI] [PubMed] [Google Scholar]
- Myers CR, Nealson KH. Respiration-linked proton translocation coupled to anaerobic reduction of manganese(IV) and iron(III) in Shewanella putrefaciens MR-1. J Bacteriol. 1990;172:6232–6238. doi: 10.1128/jb.172.11.6232-6238.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Ferric iron reduction-linked growth yields of Shewanella putrefaciens MR-1. J Appl Bacteriol. 1994;76:253–258. doi: 10.1111/j.1365-2672.1994.tb01624.x. [DOI] [PubMed] [Google Scholar]
- Myers JM, Myers CR. Role of the tetraheme cytochrome CymA in anaerobic electron transport in cells of Shewanella putrefaciens MR-1 with normal levels of menaquinone. J Bacteriol. 2000;182:67–75. doi: 10.1128/jb.182.1.67-75.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Cloning and sequence of cymA, a gene encoding a tetraheme cytochrome c required for reduction of iron(III), fumarate, and nitrate by Shewanella putrefaciens MR-1. J Bacteriol. 1997;179:1143–1152. doi: 10.1128/jb.179.4.1143-1152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Role of menaquinone in the reduction of fumarate, nitrate, iron(III) and manganese(IV) by Shewanella putrefaciens MR-1. FEMS Microbiol Lett. 1993;114:215–222. doi: 10.1016/0378-1097(93)90522-4. [DOI] [Google Scholar]
- Maier TM, Myers JM, Myers CR. Identification of the gene encoding the sole physiologocal fumarate reductase of Shewanella oneidensis MR-1. J Basic Microbiol. 2003;43:312–327. doi: 10.1002/jobm.200390034. [DOI] [PubMed] [Google Scholar]
- Myers JM, Myers CR. Role for outer membrane cytochromes OmcA and OmcB of Shewanella putrefaciens MR-1 in reduction of manganese dioxide. Appl Environ Microbiol. 2001;67:260–269. doi: 10.1128/AEM.67.1.260-269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JM, Myers CR. Genetic complementation of an outer membrane cytochrome omcB mutant of Shewanella putrefaciens MR-1 requires omcB plus downstream DNA. Appl Environ Microbiol. 2002;68:2781–2793. doi: 10.1128/AEM.68.6.2781-2793.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Nealson KH. Iron mineralization by bacteria: metabolic coupling of iron reduction to cell metabolism in Alteromonas putrefaciens strain MR-1. In: Frankel R B and Blakemore R P, editor. Iron Biominerals. New York, N.Y., Plenum Press; 1990. p. 131–149. [Google Scholar]
- Dobbin PS, Requena Burmesiter LM, Heath SL, Powell AK, McEwan AG, Richardson DJ. The influence of chelating agents upon the dissimilatory reduction of Fe(III) by Shewanella putrefaciens. Part 2. Oxo- and hydroxo-bridged polynuclear Fe(III) complexes. BioMetals. 1996;9:291–301. [Google Scholar]
- Lower SK, Hochella M.F. Jr., Beveridge TJ. Bacterial recognition of mineral surfaces: naoscale interactions between Shewanella and a-FeOOH. Science. 2001;292:1360–1363. doi: 10.1126/science.1059567. [DOI] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Ferric reductase is associated with the membranes of anaerobically grown Shewanella putrefaciens MR-1. FEMS Microbiol Lett. 1993;108:15–22. doi: 10.1016/0378-1097(93)90480-P. [DOI] [Google Scholar]
- Myers CR, Nealson KH. Microbial reduction of manganese oxides: interactions with iron and sulfur. Geochim Cosmochim Acta. 1988;52:2727–2732. doi: 10.1016/0016-7037(88)90041-5. [DOI] [Google Scholar]
- Dobbin PS, Butt JN, Powell AK, Reid GA, Richardson DJ. Characterization of a flavocytochrome that is induced during the anaerobic respiration of Fe3+ by Shewanella frigidimarina NCIMB400. Biochem J. 1999;342:439–448. doi: 10.1042/0264-6021:3420439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Outer membrane cytochromes of Shewanella putrefaciens MR-1: spectral analysis, and purification of the 83-kDa c-type cytochrome. Biochim Biophys Acta. 1997;1326:307–318. doi: 10.1016/s0005-2736(97)00034-5. [DOI] [PubMed] [Google Scholar]
- Maier TM, Myers CR. Isolation and characterization of a Shewanella putrefaciens MR-1 electron transport regulator etrA mutant: reassessment of the role of EtrA. J Bacteriol. 2001;183:4918–4926. doi: 10.1128/JB.183.16.4918-4926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Cell surface exposure of the outer membrane cytochromes of Shewanella oneidensis MR-1. Lett Appl Microbiol. 2003;37:254–258. doi: 10.1046/j.1472-765x.2003.01389.x. [DOI] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Fumarate reductase is a soluble enzyme in anaerobically grown Shewanella putrefaciens MR-1. FEMS Microbiol Lett. 1992;98:13–20. doi: 10.1016/0378-1097(92)90125-8. [DOI] [Google Scholar]
- Newman DK, Kolter R. A role for excreted quinones in extracellular electron transfer. Nature. 2000;405:94–97. doi: 10.1038/35011098. [DOI] [PubMed] [Google Scholar]
- Householder TC, Belli WA, Lissenden S, Cole JA, Clark VL. cis- and trans-acting elements involved in regulation of aniA, the gene encoding the major anaerobically induced outer membrane protein in Neisseria gonorrhoeae. J Bacteriol. 1999;181:541–551. doi: 10.1128/jb.181.2.541-551.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedding G, Draper DE. Allosteric mechanism for translational repression in the Escherichia coli alpha operon. Proc Natl Acad Sci USA. 1993;90:4399–4403. doi: 10.1073/pnas.90.10.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett PS. Translational attenuation as the regulator of inducible cat genes. J Bacteriol. 1990;172:1–6. doi: 10.1128/jb.172.1.1-6.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykxhoorn DM, St Pierre R, Van Ham O, Linn T. An efficient protocol for linker scanning mutagenesis: analysis of the translational regulation of an Escherichia coli RNA polymerase subunit gene. Nucl Acids Res. 1997;25:4209–4218. doi: 10.1093/nar/25.21.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aristarkhov A, Mikulskis A, Belasco JG, Lin EC. Translation of the adhE transcript to produce ethanol dehydrogenase requires RNase III cleavage in Escherichia coli. J Bacteriol. 1996;178:4327–4332. doi: 10.1128/jb.178.14.4327-4332.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung YC, Chan C, Reader JS, Willis AC, van Spanning RJ, Ferguson SJ, Radford SE. The pseudoazurin gene from Thiosphaera pantotropha: analysis of upstream putative regulatory sequences and overexpression in Escherichia coli. Biochem J. 1997;321:699–705. doi: 10.1042/bj3210699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte W, Nestel U, Wacker T, Diederichs K. Structure and function of the porin channel. Kidney Int. 1995;48:930–940. doi: 10.1038/ki.1995.374. [DOI] [PubMed] [Google Scholar]
- Pizza M, Scarlato V, Masignani V, Giuliani MM, Arico B, Comanducci M, Jennings GT, Baldi L, Bartolini E, Capecchi B, Galeotti CL, Luzzi E, Manetti R, Marchetti E, Mora M, Nuti S, Ratti G, Santini L, Savino S, Scarselli M, Storni E, Zuo P, Broeker M, Hundt E, Knapp B, Blair E, Mason T, Tettelin H, Hood DW, Jeffries AC, Saunders NJ, Granoff DM, Venter JC, Moxon ER, Grandi G, Rappuoli R. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science. 2000;287:1816–1820. doi: 10.1126/science.287.5459.1816. [DOI] [PubMed] [Google Scholar]
- van der Ley P, Heckels JE, Virji M, Hoogerhout P, Poolman JT. Topology of outer membrane porins in pathogenic Neisseria spp. Infect Immun. 1991;59:2963–2971. doi: 10.1128/iai.59.9.2963-2971.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart A, Dankert J, van der Ende A. Antigenic variation of the class I outer membrane protein in hyperendemic Neisseria meningitidis strains in the Netherlands. Infect Immun. 1999;67:3842–3846. doi: 10.1128/iai.67.8.3842-3846.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebnik R, Locher KP, Van Gelder P. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol. 2000;37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- Decad GM, Nikaido H. Outer membrane of gram-negative bacteria. XII. Molecular-sieving function of cell wall. J Bacteriol. 1976;128:325–336. doi: 10.1128/jb.128.1.325-336.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler HM. Pore-forming molecules in gram-negative anaerobic bacteria. Clin Infect Dis. 1997;25 Suppl 2:S284–286. doi: 10.1086/516225. [DOI] [PubMed] [Google Scholar]
- Tommassen J, Vermeij P, Struyve M, Benz R, Poolman JT. Isolation of Neisseria meningitidis mutants deficient in class 1 (PorA) and class 3 (PorB) outer membrane proteins. Infect Immun. 1990;58:1355–1359. doi: 10.1128/iai.58.5.1355-1359.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H, Nakae T. The outer membrane of Gram-negative bacteria. Adv Microb Physiol. 1979;20:163–250. doi: 10.1016/s0065-2911(08)60208-8. [DOI] [PubMed] [Google Scholar]
- Nikaido H, Saier MHJ. Transport proteins in bacteria: common themes in their design. Science. 1992;258:936–942. doi: 10.1126/science.1279804. [DOI] [PubMed] [Google Scholar]
- Heidelberg JF, Paulsen IT, Nelson KE, Gaidos EJ, Nelson WC, Read TD, Eisen JA, Seshadri R, Ward N, Methe B, Clayton RA, Meyer T, Tsapin A, Scott J, Beanan M, Brinkac L, Daugherty S, DeBoy RT, Dodson RJ, Durkin AS, Haft DH, Kolonay JF, Madupu R, Peterson JD, Umayam LA, White O, Wolf AM, Vamathevan J, Weidman J, Impraim M, Lee K, Berry K, Lee C, Mueller J, Khouri H, Gill J, Utterback TR, McDonald LA, Feldblyum TV, Smith HO, Venter JC, Nealson KH, Fraser CM. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nature Biotechnol. 2002;20:1118–1123. doi: 10.1038/nbt749. [DOI] [PubMed] [Google Scholar]
- Neilands JB. Siderophores: structure and function of microbial iron transport compounds. J Biol Chem. 1995;270:26723–26726. doi: 10.1074/jbc.270.45.26723. [DOI] [PubMed] [Google Scholar]
- Braun V, Killmann H. Bacterial solutions to the iron-supply problem. Trends Biochem Sci. 1999;24:104–109. doi: 10.1016/S0968-0004(99)01359-6. [DOI] [PubMed] [Google Scholar]
- Thompson DK, Beliaev AS, Giometti CS, Tollaksen SL, Khare T, Lies DP, Nealson KH, Lim H, Yates J., 3rd, Brandt CC, Tiedje JM, Zhou J. Transcriptional and proteomic analysis of a ferric uptake regulator (fur) mutant of Shewanella oneidensis: possible involvement of fur in energy metabolism, transcriptional regulation, and oxidative stress. Appl Environ Microbiol. 2002;68:881–892. doi: 10.1128/AEM.68.2.881-892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh T, Paulsen IT, Saier M. H., Jr. A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. J Bacteriol. 1994;176:3825–3831. doi: 10.1128/jb.176.13.3825-3831.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol. 1995;16:45–55. doi: 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- Postle K, Vakharia H. TolC, a macromolecular periplasmic 'chunnel'. Nature Struct Biol. 2000;7:527–530. doi: 10.1038/76726. [DOI] [PubMed] [Google Scholar]
- Fralick JA. Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. J Bacteriol. 1996;178:5803–5805. doi: 10.1128/jb.178.19.5803-5805.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu JBH, Lies DP, Newman DK. Protective role of tolC in efflux of the electron shuttle anthraquinone-2,6-disulfonate. J Bacteriol. 2002;184:1806–1810. doi: 10.1128/JB.184.6.1806-1810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliaev AS, Saffarini DA. Shewanella putrefaciens mtrB encodes an outer membrane protein required for Fe(III) and Mn(IV) reduction. J Bacteriol. 1998;180:6292–6297. doi: 10.1128/jb.180.23.6292-6297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CR, Myers JM. MtrB is required for proper incorporation of the cytochromes OmcA and OmcB into the outer membrane of Shewanella putrefaciens MR-1. Appl Environ Microbiol. 2002;68:5585–5594. doi: 10.1128/AEM.68.6.2781-2793.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd. 1–3. Cold Spring Harbor, N.Y., Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Myers CR, Myers JM. Localization of cytochromes to the outer membrane of anaerobically grown Shewanella putrefaciens MR-1. J Bacteriol. 1992;174:3429–3438. doi: 10.1128/jb.174.11.3429-3438.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend KM, Frost AJ, Lee CW, Papadimitriou JM, Dawkins HJS. Development of PCR assays for species- and type-specific identification of Pasteurella multocida isolates. J Clin Microbiol. 1998;36:1096–1100. doi: 10.1128/jcm.36.4.1096-1100.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HR, Archer CD, Liu J, Turnbough C.L., Jr. Translational control of pyrC expression mediated by nucleotide-sensitive selection of transcriptional start sites in Escherichia coli. J Bacteriol. 1992;174:514–524. doi: 10.1128/jb.174.2.514-524.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JM, Myers CR. Isolation and sequence of omcA, a gene encoding a decaheme outer membrane cytochrome c of Shewanella putrefaciens MR-1, and detection of omcA homologs in other strains of S. putrefaciens. Biochim Biophys Acta. 1998;1373:237–251. doi: 10.1016/s0005-2736(98)00111-4. [DOI] [PubMed] [Google Scholar]
- Faeder EJ, Siegel LM. A rapid micromethod for determination of FMN and FAD in mixtures. Anal Biochem. 1973;53:332–336. doi: 10.1016/0003-2697(73)90442-9. [DOI] [PubMed] [Google Scholar]
- Cataldo DA, Haroon M, Schrader LE, Youngs VL. Rapid colorimetric determination of nitrate in plant tissue by nitration of salicylic acid. Commun Soil Sci Plant Anal. 1975;6:71–80. [Google Scholar]
- van'T Riet J, Stouthamer AH, Planta RJ. Regulation of nitrate assimilation and nitrate respiration in Aerobacter aerogenes. J Bacteriol. 1968;96:1455–1464. doi: 10.1128/jb.96.5.1455-1464.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovley DR, Phillips EJP. Organic matter mineralization with reduction of ferric iron in anaerobic sediments. Appl Environ Microbiol. 1986;51:683–689. doi: 10.1128/aem.51.4.683-689.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong PB, Lyons WB, Gaudette HE. Application of formaldoxime colorimetric method for the determination of manganese in the pore water of anoxic estuarine sediments. Estuaries. 1979;2:198–201. [Google Scholar]
- Brewer PG, Spencer DW. Colorimetric determination of manganese in anoxic waters. Limnol Oceanogr. 1971;16:107–110. [Google Scholar]
- Thomas PE, Ryan D, Levin W. An improved staining procedure for the detection of the peroxidase activity of cytochrome P-450 on sodium dodecyl sulfate polyacrylamide gels. Anal Biochem. 1976;75:168–176. doi: 10.1016/0003-2697(76)90067-1. [DOI] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Simon R, Priefer U, Pühler A. A broad host-range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Bio/Technology. 1983;1:784–791. [Google Scholar]
- de Lorenzo V, Herrero M, Jakubzik U, Timmis KN. Mini-Tn5 transposon derivatives for insertional mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol. 1990;172:6568–6572. doi: 10.1128/jb.172.11.6568-6572.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinder SA, Badger JL, Bryant GO, Pepe JC, Miller VL. Cloning of the YenI restriction endonuclease and methyltransferase from Yersinia enterocolitica serotype O8 and construction of a transformable R-M+ mutant. Gene. 1993;136:271–275. doi: 10.1016/0378-1119(93)90478-L. [DOI] [PubMed] [Google Scholar]