

Abstract
Background
Huntington’s disease is an inherited neurodegenerative disorder characterised by motor, cognitive and psychiatric disturbances. Patients exhibit other symptoms including sleep and mood disturbances, muscle atrophy and weight loss which may be linked to hypothalamic pathology and dysfunction of hypothalamo-pituitary axes.
Methods
We studied neuroendocrine profiles of corticotropic, somatotropic and gonadotropic hypothalamo-pituitary axes hormones over a 24-hour period in controlled environment in 15 healthy controls, 14 premanifest and 13 stage II/III Huntington’s disease subjects. We also quantified fasting levels of vasopressin, oestradiol, testosterone, dehydroepiandrosterone sulphate, thyroid stimulating hormone, free triiodothyronine, free total thyroxine, prolactin, adrenaline and noradrenaline. Somatotropic axis hormones, growth hormone releasing hormone, insulin-like growth factor-1 and insulin-like factor binding protein-3 were quantified at 06:00 (fasting), 15:00 and 23:00. A battery of clinical tests, including neurological rating and function scales were performed.
Results
24-hour concentrations of adrenocorticotropic hormone, cortisol, luteinizing hormone and follicle-stimulating hormone did not differ significantly between the Huntington’s disease group and controls. Daytime growth hormone secretion was similar in control and Huntington’s disease subjects. Stage II/III Huntington’s disease subjects had lower concentration of post-sleep growth hormone pulse and higher insulin-like growth factor-1:growth hormone ratio which did not reach significance. In Huntington’s disease subjects, baseline levels of hypothalamo-pituitary axis hormones measured did not significantly differ from those of healthy controls.
Conclusions
The relatively small subject group means that the study may not detect subtle perturbations in hormone concentrations. A targeted study of the somatotropic axis in larger cohorts may be warranted. However, the lack of significant results despite many variables being tested does imply that the majority of them do not differ substantially between HD and controls.
Introduction
Huntington´s disease (HD) is an inherited neurodegenerative disease, caused by a CAG triplet repeat expansion in the gene encoding huntingtin [1]. Classical features of HD include motor manifestations, cognitive and psychiatric symptoms [2]. However, these are not the sole manifestations in HD, and disruption of circadian rhythms [3–6], alterations in sleep patterns [7–9], altered glucose homeostasis [10–13], muscle atrophy [14] and weight loss [15–17] may also impact on the quality of life of the patients and can precede motor symptoms by many years. Extensive research in animal models of HD [18,19] and HD patients [20–26] suggests that these symptoms could be linked to progressive hypothalamic pathology and changes in the neuroendocrine systems.
The hypothalamus exerts control over many bodily functions via three major outputs: autonomic, endocrine and behavioural systems. It acts as the coordinating centre for the neuroendocrine system. Hypothalamic endocrine efferent output is mediated through the hypothalamic pituitary axes [27,28], regulating the function of the thyroid gland, the adrenal gland, and the gonads, and, thereby, the circulating levels of growth hormone, thyroid hormones, cortisol, testosterone and oestrogens [29,30].
Alterations of the hypothalamic-pituitary-adrenal (HPA) axis have been shown in HD patients [24,26,31,32] and in HD mouse models [19]. Interestingly, increased cortisol levels [22–24,26] can cause symptoms that are common in HD patients such as depression [33–36], skeletal muscle atrophy, altered glucose tolerance and memory impairment [37].
Since the thyrotropic axis is involved in the regulation of body weight and metabolism [38], which are affected in HD [15], several studies have evaluated hypothalamic-pituitary-thyroid (HPT) axis function in patients with HD, with conflicting results [24,39–41].
The hypothalamic-pituitary-gonadal (HPG) axis has also been shown altered in HD mice [42,43] and in men with HD, where reduced testosterone levels have been shown to be linked to disease severity [24,44]. Gonadotropic axis hormones have not been carefully investigated in female HD patients.
We conducted a study to analyse the corticotropic, thyrotropic, gonadotropic, somatotropic and lactotropic axes in detail over a 24-hour period in a controlled environment, using cohorts of premanifest and moderate HD subjects and healthy controls.
Methods
Participants
Patients were eligible for enrolment if they were 18 years of age or older, had completed either a predictive or diagnostic genetic test for HD (CAG repeat ≥40). Patients were excluded from the study if they had: pre-existent endocrine disease, central nervous system disorder other than HD, history of alcohol or drug abuse, treatment with corticosteroids, phenothiazine anti-emetics, antipsychotic medication (including neuroleptics, SSRI drugs), or hypnotic drugs for preceding 6 months, night shift working and weight change in the preceding 6 months. Controls were recruited principally from the partners, spouses, or carers of the HD group with no clinical evidence or family history of HD and the same exclusion criteria applied. Fifteen healthy controls, 14 gene carriers (premanifest HD) and 13 stage II/III HD patients were enrolled into a study analysing neuroendocrine factors. The study was conducted at the Royal Free London NHS Foundation Trust. Participants were recruited through the HD Multidisciplinary Clinic at the National Hospital for Neurology and Neurosurgery, London, UK. Written informed consent was obtained from all subjects. The study protocol was approved by the joint UCL/UCLH ethics committee and was conducted in accordance with the Declaration of Helsinki.
Clinical protocol
Study subjects underwent to a 24-hour inpatient stay in the hospital. Subjects were advised to observe their usual bedtime, wake time, and mealtime patterns in the week preceding the study. During the study, subjects could walk freely or watch television, but not fall asleep or snack outside scheduled mealtimes. A cannula was inserted 60 minutes before the start of blood sampling at 14:00 hours. Sampling was performed through a long line to minimise sleep disruption. The aim for female subjects was to be sampled, where possible, at the midpoint of their menstrual cycle in order to standardise hormone analysis.
The following pituitary axis hormones were assayed: vasopressin, corticotropic-axis hormones (adrenocorticotropic hormone [ACTH] and cortisol), somatotropic-axis hormones (growth hormone releasing hormone [GHRH], growth hormone [GH], insulin-like growth factor-1 [IGF-1], and insulin-like factor binding protein-3 [IGF-BP3]), gonadotropic axis hormones (luteinizing hormone [LH], follicle-stimulating hormone [FSH], oestradiol and testosterone in female and male), thyrotropic axis hormones (thyroid stimulating hormone [TSH], free triiodothyronine [fT3], and free total thyroxine [fT4]) and lactotropic axis hormone (prolactin). Samples for ACTH, cortisol, GH, LH and FSH analysis were taken hourly from 14:00 on day 1 until 13:00 on day 2. Plasma for catecholamines (adrenaline, noradrenaline), vasopressin, fT3, fT4 and TSH, prolactin, total testosterone, oestradiol and dehydroepiandrosterone sulphate (DHEAS) analysis was drawn at 06:00 on day 2 after an overnight fast. Plasma for GHRH, IGF-1and IGF-BP3 was taken at 06:00, 15:00 and 23:00. During the study, urine was collected over 24 hours for the determination of cortisol concentration.
Clinical assessment and HD rating scales were performed by neurologist with expertise in HD. The Unified Huntington’s Disease Rating Scale (UHDRS), motor section (MS), Total Functional Capacity (TFC), and Functional activity (FA), were assessed. Cognitive function was assessed by Stroop Test Evaluation Colour Naming (STECN), Stroop Test Evaluation Word Reading (STEWR), Stroop Test Evaluation Interference (STEI), Symbol Digit Test (SDT) and Verbal Fluency Test (VFT). Problem behaviours assessment (PBA) was also used and behavioural score was calculated [45–47]. For HD gene carriers, subjects who were scored as 4 on the diagnostic confidence score of the Motor scale (motor abnormalities >99% likely to be due to HD) were classed as affected and those who scored 0 or 1 were classed as premanifest.
Sample collection and analysis
Assays were performed according to the manufacturer’s instructions for the relevant kit. Blood for the analysis of serum cortisol, GH, LH, FSH, oestradiol, DHEAS, testosterone, prolactin and thyroid hormones were collected in BD Vacutainer Serum Separation Tubes while blood for ACTH analysis was collected into EDTA containing tubes. Plasma samples for catecholamines and vasopressin were collected in BD Vacutainer Lithium Heparin tubes. Specimens for ACTH analysis were immediately placed on ice and were centrifuged at 2500 rotations per minute (rpm), at 4°C, for 5 minutes, soon after venesection. Specimens for catecholamines were also spun immediately after collection while all the other specimens were kept at room temperature and centrifuged within 60 minutes of sampling at 2500 rpm, at 4°C, for 5 minutes. Plasma and serum samples were immediately frozen on dry ice and stored at ‐80°C prior to analysis.
24 hour urine samples for free cortisol measurement were collected into plain containers, the bottles were weighed and the total urine volume recorded. 600 μL aliquots of urine underwent liquid-liquid extraction with 3 mL dichloromethane. 1.5 mL of the non-polar layer was transferred into glass boiling tubes and heated at 50°C until dry. The samples were reconstituted in 300 μL Elecsys Universal Diluent (Roche) and cortisol measured using the assay described below. The 24 hour excretion values were calculated by multiplying the cortisol concentration by the 24 hour urine volume.
Serum IGF-1, IGF-BP3 and GHRH were measured in duplicate by enzyme-linked immunosorbent assays (R&D Systems Europe, Ltd. Abingdon, UK). Concentrations of cortisol (measuring range (MR): 0.5–1750 nmol/L), FSH (MR: 0.100–200 mIU/mL), LH (MR: 0.100–200 mIU/mL), FT3 (MR: 0.40–50.00 pmol/L), FT4 (MR: 0.3–100 pmol/L), TSH (MR: 0.005–100 μIU/mL), prolactin (MR: 1.00–10000 μIU/mL), total testosterone (MR: 2.50–1500 ng/dL), DHEAS (MR: 0.003–27.0 μmol/L) and oestradiol (MR: 0.003–27.0 μmol/L) were determined by electrochemiluminescent immunoassay on a Roche Modular E170 autoanalyser. Serum GH and ACTH were quantified using chemiluminescent immunoassays on a Siemens Immulite Analyser (GH MR: 0.01–40.00 μg/L, ACTH MR: 5–1250 pg/mL). Plasma catecholamines were quantified by reverse phase partition high performance liquid chromatography HPLC.
Statistical analysis
Results are expressed as mean ± standard deviation (SD) or median (minimum-maximum). Disease burden score, which correlates with and is an indicator of the severity of neuropathology of HD, was calculated for each HD gene carrier using the formula [CAG repeat length – 35.5} x age [48]. Inter-group differences were assessed by linear regression of the relevant variable (or its log if necessary for normality) on disease group and adjusting for age and gender, or (in the absence of normality or log-normality) by using a permutation test after Freedman and Lane [49]. This latter test is based on permutations of the residuals calculated under the reduced model of the full linear regression one, with the same predictor variables [50]. For relevant sex hormones, where there are marked differences by gender, analyses were performed separately by gender. All tests were two-tailed and significance level was set at p<0.05. There was no formal adjustment for multiple testing, since all hypotheses tested are interesting in their own right, although the number of comparisons made is borne in mind in the interpretation of any significant results [51]. Statistical analyses were performed using SPSS for Windows (release 16.0, SPSS, Inc., Chicago, IL) and using Stata (release 13: StataCorp Stata Statistical Software: Release 13. College Station, TX: StataCorp LP).
Fourier Transformation (FT) analysis was utilised to measure spectral power of ACTH, cortisol, GH and LH oscillations, enabling analysis of the strength/power of hormonal oscillations at different frequencies [52,53] (http://ora.ox.ac.uk/objects/ora:2356). Signal–noise ratio was improved using a 3-point moving average and any trend in the data was removed by a difference + mean regression. Data were analysed using the EASY-TSA program (Oxford University, UK: Nathan.hill@phc.ox.ac.uk) [53]. Comparisons between waveforms of time series analysis were performed by probability of SEM at any discrete time-point overlapping the SEM of the comparative group.
Results
The demographic data and clinical characteristics of the healthy controls, premanifest, and stage II/III HD patients are reported in Table 1. This shows some differences between groups, particularly in age, where median age is lower in the premanifest group, and older in the moderate/ severe Huntington group, compared to controls. For clinical ratings there is some overlap in range for premanifest and manifest HD groups. This reflects the fact that HD pathology is a continuum and although the diagnostic confidence score identifies the groups there is not a strict dichotomy for the two groups. For further discussion of the diagnosis and classification of HD see review by Ross et al [54]. Three patients with stage II/III HD were excluded on screening as they were taking neuroleptic medication. Table 2 presents hormone concentrations.
Table 1. Demographic characteristics and clinical features of controls, pre-manifest and stage II/III HD cohorts.
Data presented as median (range).
| Disease stage | Controls | Pre-manifest HD | Stage II/III HD |
|---|---|---|---|
| Number of subjects | 15 | 14 | 13 |
| Age | 54 (29–69) | 45 (31–58) | 58 (42–70) |
| Female: Male | 6:9 | 9:5 | 5:8 |
| BMI | 25 (20–37) | 27 (23–39) | 26 (20–33) |
| CAG | - | 42 (40–47) | 42 (42–47) |
| Disease burden score | - | 299 (207–434) | 410 (273–702) |
| Functional Assessment | 25 | 25 (21–25) | 21 (11–24) |
| UHDRS Total Functional Capacity | 13 | 13 (12–13) | 9 (5–12) |
| UHDRS Motor Score | 0 | 0 (0–11) | 38 (10–65) |
| STECN | 82 (60–93) | 71 (47–91) | 45 (30–81) |
| STEWR | 100 (83–102) | 96 (64–110) | 57 (37–100) |
| STEI | 45 (29–68) | 42 (34–52) | 24 (20–44) |
| SDT | 51 (42–65) | 52 (37–62) | 31 (17–50) |
| VFT | 48 (30–66) | 41 (26–70) | 27 (12–58) |
| CS | 322 (276–359) | 298 (225–369) | 182 (133–308) |
| PBA | 2 (0–10) | 8 (2–31) | 21 (5–55) |
Table 2. Hormone levels in control, premanifest and stage II/III HD cohorts.
Data are presented as Mean ±SD for normally distributed data and as median [minimum–maximum] for skewed data.
| Axis | Control | Premanifest HD | Stage II/III HD | p |
|---|---|---|---|---|
| Corticotropic axis | ||||
| ACTH, 24 h, ng/L | 11.4 [7.4–23.5] | 13.0 [7.7–28.0] | 13.4 [7.1–31.4] | 0.63L |
| Cortisol, 24 h, nmol/L | 232 [165–636] | 224 [100–498] | 212 [97–363] | 0.54L |
| Cortisol, urine, nmol/24 h | 179 [58–758] | 136 [35–434] | 121 [50–295] | 0.63L |
| Cortisol: ACTH ratio, 24 h | 79200 [7800–802000] | 63300 [9100–489300] | 65100 [10600–317200] | 0.35L |
| Cortisol: ACTH ratio, evening | 93600 [37000–705200] | 70800 [25300–226900] | 60800 [22900–142500] | 0.06L |
| Vasopressin, pg/ml | 0.84 [0.56–1.92] | 0.79 [0.59–1.49] | 0.77 [0.53–1.68] | 0.89LP |
| Somatotropic axis | ||||
| GH, μg/L | ||||
| 24 h | 0.7 [0.1–5.7] | 0.7 [0.1–1.6] | 0.4 [0.1–1.7] | 0.48LP |
| Post-sleep | 2.1 [0.2–8.4] | 1.1 [0.2–3.5] | 0.3 [0.1–2.2] | 0.09L |
| GHRF, ng/ml | ||||
| 06:00 | 0.77 ± 0.24 | 0.76 ± 0.16 | 0.71 ± 0.23 | 0.7 |
| 15:00 | 0.76 ± 0.18 | 0.80 ± 0.38 | 0.82 ± 0.19 | 0.83P |
| 23:00 | 0.78 ± 0.17 | 0.79 ± 0.16 | 0.72 ± 0.18 | 0.29 |
| IGF-1, ng/ml | ||||
| 06:00 | 106 [76–154] | 121 [69–218] | 90 [66–232] | 0.88L |
| 15:00 | 128 [77–154] | 110 [65–230] | 109 [69–250] | 0.70L |
| 23:00 | 107 [75–161] | 113 [64–257] | 106 [76–207] | 0.997L |
| IGF-BP3, ng/ml | ||||
| 06:00 | 2590 ± 400 | 2481 ± 584 | 2308 ± 531 | 0.38P |
| 15:00 | 2823 ± 575 | 2444 ± 407 | 2625 ± 553 | 0.08P |
| 23:00 | 2501 ± 557 | 2499 ± 358 | 2254 ± 494 | 0.50P |
| IGF-1:GH ratio | ||||
| 06:00 | 801 [77–2194] | 352 [11–2211] | 987 [49–1642] | 0.80LP |
| 15:00 | 744 [31–3070] | 613 [20–2451] | 1389 [17–3043] | 0.96LP |
| 23:00 | 271 [15–1505] | 300 [20–5132] | 1149 [26–2630] | 0.23L |
| Gonadotropic axis | ||||
| LH, U/L | ||||
| Female, 24 h | 14.0 [1.6–46.5] | 7.2 [0.1–30.9] | 21.8 [3.2–32.1] | 0.87L |
| Male, 24 h | 5.2 [2.5–7.8] | 5.0 [3.1–6.1] | 4.9 [3.4–6.0] | 0.98L |
| FSH, U/L | ||||
| Female, 24 h | 46.1 [1.5–140.1] | 5.0 [0.6–278.9] | 32.9 [7.3–65.3] | 0.88L |
| Male, 24 h | 5.9 [2.4–12.0] | 5.5 [3.1–6.0] | 5.3 [1.8–7.4] | 0.53L |
| Testosterone, nmol/L | ||||
| Females | 0.73 ± 0.27 | 0.84 ± 0.38 | 0.50 ± 0.27 | 0.6 |
| Males | 15.5 ± 7.7 | 15.5 ± 5.3 | 16.8 ± 4.6 | 0.59 |
| Oestradiol, pmol/L | ||||
| Females | 50 [50–1432] | 171 [50–791] | 84 [56–218] | 0.89LP |
| Males | 109 [50–156] | 100 [71–334] | 121 [50–221] | 0.57L |
| DHEAS, umol/L | 4.81 ± 2.21 | 4.12 ± 1.92 | 2.77 ± 2.31 | 0.04 |
| Females | 3.7 ± 1.2 | 3.3 ± 1.3 | 2.1 ± 1.4 | 0.24 |
| Males | 5.6 ± 2.4 | 6.0 ± 1.8 | 3.2 ± 2.7 | 0.17 |
| Lactotropic axis | ||||
| Prolactin, mU/L | ||||
| Females | 360 ± 125 | 432 ± 195 | 461 ± 257 | 0.33P |
| Males | 245 ± 98 | 247 ± 47 | 235 ± 60 | 0.98 |
| Thyrotropic axis | ||||
| TSH, mU/L | 2.61 ± 1.34 | 3.23 ± 3.01 | 1.98 ± 1.10 | 0.81 |
| fT3, pmol/L | 4.48 ± 0.70 | 5.04 ± 0.64 | 4.91 ± 0.59 | 0.08 |
| fT4, pmol/L | 14.7 ± 2.19 | 15.4 ± 2.31 | 15.5 ± 2.2 | 0.62 |
| fT4:fT3 ratio | 3.32 ± 0.42 | 3.09 ± 0.41 | 3.21 ± 0.54 | 0.52 |
| Catecholamines | ||||
| Adrenaline, nmol/L | 0.46 ± 0.17 | 0.36 ± 0.17 | 0.42 ± 0.21 | 0.29 |
| Noradrenaline, nmol/L | 1.86 ± 0.71 | 2.16 ± 0.63 | 1.89 ± 0.80 | 0.64 |
P-value = p-value from linear regression of specified variable on age and gender (or else by gender as indicated), L indicates that logs were taken of the specified variable prior to linear regression; P indicates Freedman and Lane permutation tests were performed to account to non-normality, LP indicates Freedman and Lane permutation tests were performed after taking logs of the specified variable. In all cases, age and gender were adjusted for.
Corticotropic axis
In all three groups the 24-hour profiles of ACTH and cortisol display a typical pattern with early morning peak concentrations and declining levels throughout daytime, with lowest values around midnight (Fig 1A and 1B). There was an elevation of ACTH (Fig 1A) and cortisol (Fig 1B) levels during late sleep in all three groups.
Fig 1. Analysis of ACTH and cortisol in control, premanifest and stage II/III HD cohorts.

A: Mean ACTH concentrations over 24 hour sampling period in the three groups. B: Mean ACTH concentrations over 24 hour sampling period in the three groups. C: FT analysis of ACTH plotting strength/power (%) against frequency (minutes) of ACTH oscillations for the three groups. D: FT analysis of cortisol plotting strength/power (%) against frequency (minutes) of cortisol oscillations for the three groups. E: Mean molar cortisol:ACTH ratio over 24 hour sampling period in the three groups.
Relative FT analysis of ACTH levels showed an initial pulsatility at 132 minutes for all groups (Fig 1C) (oscillatory power: control 0.57, SEM 0.10; premanifest HD 0.50, SEM 0.10; stage II/III HD 0.57, SEM 0.09) with a further periodicity peak at 440 minutes (oscillatory power: control 1.0, SEM 0.17; premanifest HD 0.76, SEM 0.17; stage II/III HD 0.85, SEM 0.23). Overall between-group comparison at both 132 and 440 minutes did not reach statistical significance. For cortisol (Fig 1D), initial dominant pulsatility was at 132 minutes (oscillatory power: control 7.5, SEM 0.88; premanifest HD 6.31, SEM 1.16; stage II/III HD 7.04, SEM 1.17) with a further periodicity peak at 440 minutes (oscillatory power: control 16.42, SEM 3.22; premanifest HD 13.94, SEM 2.02; stage II/III HD 13.64, SEM 1.98) and again there was no significant difference between groups.
Mean levels of ACTH and cortisol over 24 hours (Table 2) did not differ significantly between groups, nor did 24-hour urinary cortisol. A comparison of cortisol to ACTH ratio for the three groups (Fig 1E) also did not demonstrate any significant differences.
Vasopressin
We measured baseline plasma levels of vasopressin. There was no significant difference in plasma vasopressin levels between the groups.
Somatotropic axis
24-hour profile of GH, and baseline levels of GHRH (a hormone that stimulates GH secretion from the pituitary), IGF-1 and IGF-BP3 are shown in Fig 2A and Table 2. In all three groups, the 24-hour profile of GH plasma levels consisted of low levels interrupted by bursts of secretion (Fig 2A). It has been reported that GH is released in a pulsatile manner with the largest and most reproducible pulse occurring shortly after sleep onset [55]. Since the subjects in this study retired to bed at 22:00, we calculated the mean GH concentration during the first half of the night (22:00–01:00). Mean concentrations of 24 hour and post-sleep GH were lower in the stage II/III HD patients but analysis corrected for age and gender did not demonstrate a significant difference (Table 2).
Fig 2. Analysis of GH in control, premanifest and stage II/III HD cohorts.
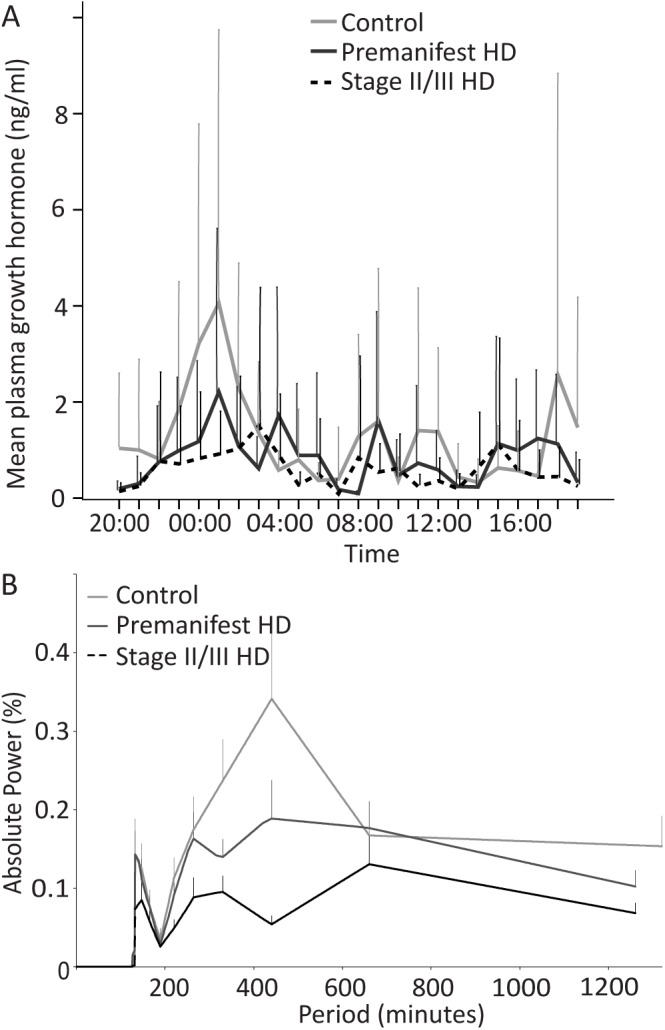
A: Mean GH concentrations over 24 hour sampling period in the three groups. B: FT analysis of GH plotting strength/power (%) against frequency (minutes) of GH oscillations for the three groups.
Relative FT of GH levels showed an initial pulsatility at 132 minutes for all groups (Fig 2B) (oscillatory power: control 0.14, SEM 0.04; premanifest HD 0.07, SEM 0.01; stage II/III HD 0.14, SEM 0.03) with a further periodicity peak at 440 minutes (oscillatory power: control 0.34, SEM 0.11; premanifest HD 0.05, SEM 0.01; stage II/III HD 0.18, SEM 0.05). Overall between-group comparison at 132 minutes did not reach statistical significance.
We also evaluated other variables of the somatotropic axis hormones, including GHRH, IGF-1 and IGF-BP3 at 06:00 (fasting), 15:00 and 23:00. There was no significant difference in plasma GHRH, IGF-1, IGF-BP3 or the IGF-1:GH ratio among the three groups at the three time-points we sampled (Table 2).
Gonadotropic axis
We assessed the 24-hour profiles of gonadotropic axis hormones LH and FSH, as well as fasting levels of testosterone, oestradiol and DHEAS in serum from control, premanifest and stage II/III HD subjects. Since gender has a major effect on the levels of gonadotropic axis hormones, the data for all gonadotropic hormones was analysed separately for males and females.
In males, the pattern of LH release in all three groups exhibited episodic pulses with large inter-individual variability and no obvious diurnal variation (Fig 3B). The 24-hour FSH profile in males was similar in all three groups with very low levels throughout the day, without any diurnal variation (Fig 4B).
Fig 3. Analysis of LH in control, premanifest and stage II/III HD cohorts.
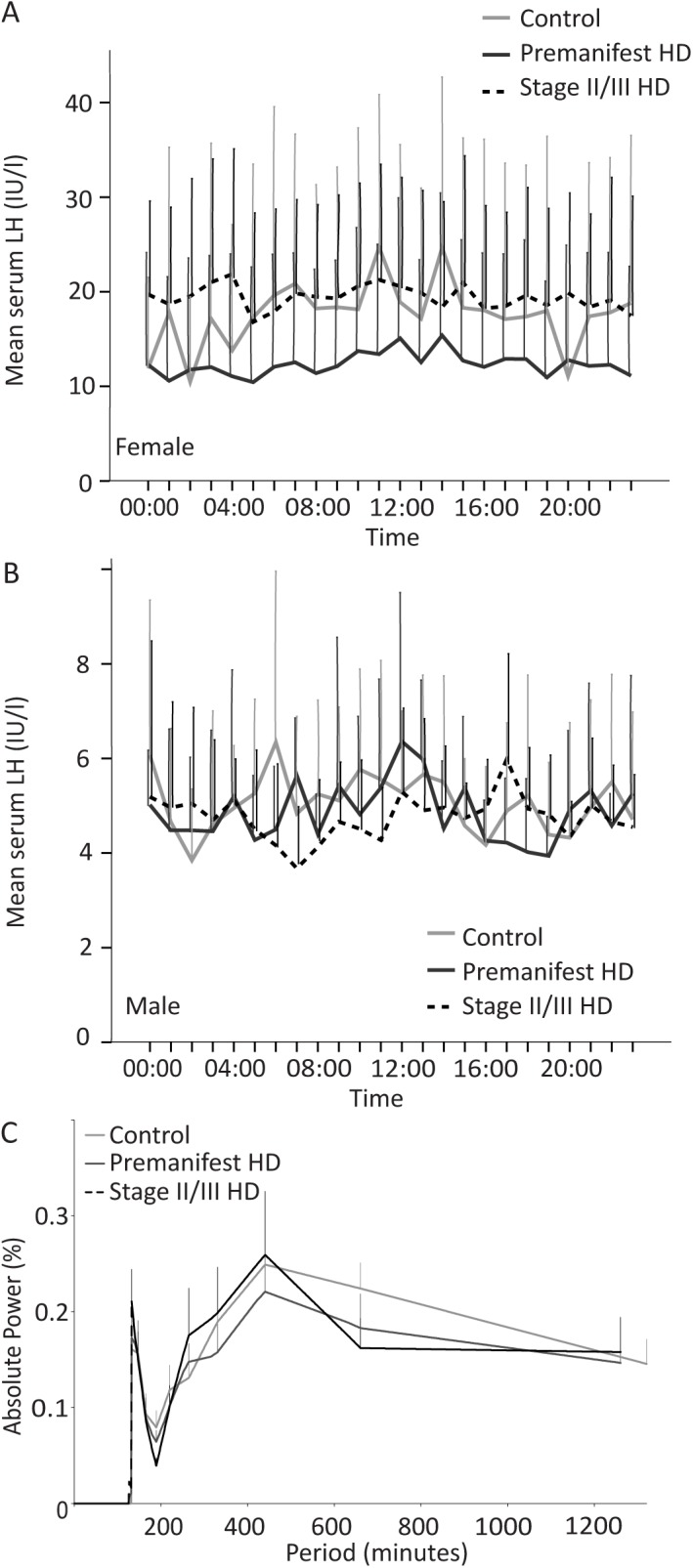
A: Mean LH concentrations over 24 hour sampling period for female subjects in the three groups. B: Mean LH concentrations over 24 hour sampling period for male subjects in the three groups. C: FT analysis of LH plotting strength/power (%) against frequency (minutes) of LH oscillations for the three groups.
Fig 4. Analysis of FSH in control, premanifest and stage II/III HD cohorts.
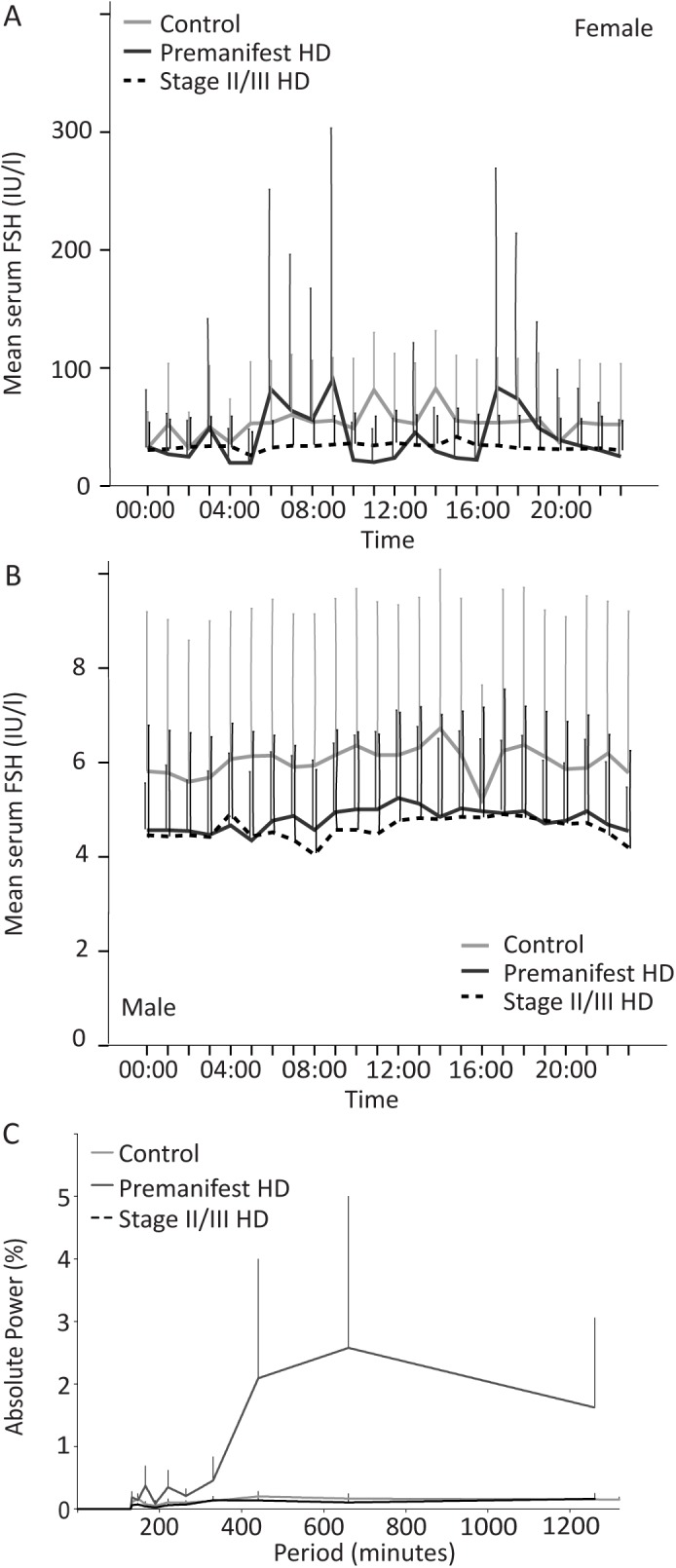
A: Mean FSH concentrations over 24 hour sampling period for female subjects in the three groups. B: Mean FSH concentrations over 24 hour sampling period for male subjects in the three groups. C: FT analysis of FSH plotting strength/power (%) against frequency (minutes) of FSH oscillations for the three groups.
Although the aim for female subjects was to be sampled, where possible, at the midpoint of their menstrual cycle in order to standardise hormone analysis, this was not always the case. The young female participants were using different contraceptive methods (pill, implant or injections). Other female participants were either at a premenopausal stage with irregular menstrual cycle or menopausal. Therefore, the 24-hour LH and FSH profiles for female subjects may not be very informative. However, it can be observed that the LH and the FSH 24-hour profiles do not differ between control, premanifest HD and stage II/III HD females (Figs 3A and 4A, respectively).
Relative FT analysis of LH release showed an initial pulsatility at 132 minutes (Fig 3C) (oscillatory power: control 0.16, SEM 0.02; premanifest HD 0.21, SEM 0.03; stage II/III HD 0.17, SEM 0.05) with a further periodicity peak at 440 minutes (oscillatory power: control 0.25, SEM 0.04; premanifest HD 0.26, SEM 0.07; stage II/III HD 0.22, SEM 0.05). Overall between-group comparison at 132 and 440 minutes did not reach statistical significance.
Relative FT analysis of FSH release showed oscillatory power was supressed in both the control and stage II/III HD group in comparison to the premanifest HD group although this did not reach statistical significance. There was an initial pulsatility at 147 minutes for all groups (Fig 4C) (oscillatory power: control 0.17, SEM 0.09; premanifest HD 0.14, SEM 0.07; stage II/III HD 0.07 SEM 0.03) with a further periodicity peak in the premanifest HD group only at 660 minutes (oscillatory power: 2.58, SEM 2.43). Overall between-group comparison at 147 and 660 minutes periodicity did not reach statistical significance.
There was no significant difference in oestradiol or testosterone levels between the three groups in males or females (Table 2). Linear regression of disease group adjusting for age and sex suggested a significant difference in fasting DHEAS levels (males and females combined, F2,36 = 3.51, p 0.04); the means observed in the three groups shows that means were substantially lower in stage II/III HD, than in controls (with mean in premanifest HD nearer to those in controls). Analysis of DHEAS concentrations in male and female subjects separately showed lower levels in stage II/III disease groups but no significant difference in comparison with controls/premanifest HD groups (Table 2).
Lactotropic axis
Fasting serum levels of prolactin were not significantly different between the three study cohorts (Table 2).
Thyrotropic axis
Fasting concentrations of thyrotropic axis hormones TSH, fT3 and fT4 and ratio of fT4:fT3 in control, premanifest HD and stage II/III HD serum showed that TSH and fT4 were similar across the three groups (Table 2), and linear regression analysis adjusting for age and gender showed was no significant between group difference.
Catecholamines
Analysis of fasting plasma concentrations of catecholamines (adrenaline and noradrenaline) did not demonstrated significant differences between the groups (Table 2).
Discussion
Huntington’s disease (HD) is a devastating neurodegenerative disorder for which the underlying pathophysiologic mechanisms affecting the brain and other organs remain unclear. Analyses of peripheral tissues from HD patients suggest that the molecular mechanisms by which mutated huntingtin leads to cell dysfunction may be similar with those in the central nervous system [56].
The hypothalamus is an important site for pathology in HD. MRI studies have reported hypothalamic atrophy in early stage HD patients as well as in transgenic HD mice [57–61]. Grey matter alterations in the hypothalamus are among the earliest features detected in HD [62]. Microglial activation and dopamine D2 receptor reduction occur in the hypothalamic region before onset of motor symptoms in HD gene carriers [63]. Furthermore, reduction in orexin and loss of orexigenic neurons in the hypothalamus [64] and alterations in the HPA axis have been shown in mouse models and patients with HD from an early disease stage [15,19,24,26,64]. In bacterial artificial chromosome-mediated transgenic HD mice, a metabolic phenotype including impaired glucose metabolism, insulin and leptin resistance was identified that could be reversed by inactivation of mutant huntingtin in the hypothalamus [65].
A number of studies describe the effect of HD on the function of the hypothalamic-pituitary axes. However, many of these report conflicting data and are difficult to compare due to methodological issues such as different sampling times, analysis of patients with different disease stages, inclusion of patients on medication that can affect hormone levels and fasting status. In addition, with the exception of two studies that quantified vasopressin [66] and prolactin [67], previous studies did not include premanifest HD gene carriers, and thus do not inform on the state of the hypothalamic-pituitary axis early on in the disease course.
The strength of our study is that we included both premanifest and manifest HD gene carriers, and have measured all the hormones in the same cohorts during a standardised day allowing a direct comparison of hormone levels. Most previous studies applied a single or a few baseline measurements, which is inadequate to assess either the pulsatile or circadian nature of hormonal secretion, which are essential for normal hormone function [68]. We therefore performed a detailed study of hormone secretory dynamics for the main hypothalamo-pituitary axes in premanifest HD gene carriers, moderate HD patients and controls, to identify potential HD state markers and inform on neuroendocrine abnormalities associated with HD. Due to the intensive nature of this study on participants, cohort sizes were relatively small which meant that small but biologically relevant changes could be overlooked.
Endocrine dysfunction in HD
There is evidence of abnormal function of the hypothalamus [64], pituitary gland [18], pancreatic islets [69,70] and adrenal glands [71] in animal models of HD, and to some extent in HD patients. This may lead to dysfunction of the neuroendocrine system with downstream effects on the metabolism of various cell types, including muscle cells and adipocytes, contributing to peripheral tissue abnormalities observed in HD such as weight loss, energy metabolism changes and altered glucose homeostasis [15–17].
Corticotropic axis
In the R6/2 mouse model increased adrenal cortex volume is seen from the age of 7 weeks [71] and adrenal glands in 12-week-old mice contained intranuclear huntingtin-inclusions and weighed 37% more than in WT mice [71]. This adrenal cortical hypertrophy may explain the increased serum and urine corticosterone detected in R6/2 mice [19].
Corticotropic axis hormones ACTH and cortisol are released in pulsatile fashion with circadian and ultradian rhythms governing their secretion [72–76]. Both are released approximately hourly, with ACTH pulses preceding cortisol by approximately 10 minutes [74–76].
In HD, ACTH levels have been reported to be both increased [22] and unchanged [24]. Saleh et al found no significant difference between HD patients and controls in fasting morning ACTH levels [24]. Heuser et al found significantly higher basal cortisol and ACTH concentrations in HD patients than controls (at 19:00 hours), HD subjects had a tendency towards blunted ACTH in response to CRH stimulation, but released normal amounts of cortisol, suggesting dysregulation of the HPA axis [22].
We measured 24-hour secretion of ACTH and cortisol at hourly intervals and found no significant difference between HD patients and controls in ACTH levels over 24 hours. In agreement with Saleh et al, fasting levels of ACTH in the morning were not significantly different between HD patients and controls. In contrast to Heuser et al, we did not see increased ACTH levels in the evening, although we did not include late stage HD patients, which could explain why we do not see a significant increase in ACTH levels in the evening.
Previous studies have consistently reported that morning levels of cortisol are increased in HD patients [23,24,26], whereas one study reported similar levels in the evening [21]. While we found no significant difference in cortisol levels in HD subjects compared to controls over 24 hours, we observed higher cortisol levels in HD gene carrier in the morning but this was not statistically significant. It is possible that we would need a higher number of subjects to replicate the previously reported increase in morning cortisol.
Aziz et al measured cortisol levels during a 24-hour period at 10 minutes intervals [26] and found that the total cortisol secretion rate and the amplitude of the diurnal cortisol profile were significantly higher in early HD patients compared with controls. Transient awakenings interrupting sleep consistently trigger pulses of cortisol secretion [77–79] and nocturnal arousals increase morning cortisol levels [80]. It is therefore possible that the frequent sampling regime contributed to the increased cortisol levels observed.
Studies have also shown that approximately 30% of patients with depression have increased cortisol secretion [33,35,36,76] and this may be a factor contributing to increased levels seen in other HD studies. We are not able to comment from our data as no participant had significant depression.
Vasopressin
Vasopressin is released in a circadian pattern by neurons of the hypothalamus. It plays a key role in homeostasis by the regulation of water retention through diuresis and thirst. Other roles of vasopressin include regulation of temperature, blood pressure, social behaviour and sexual motivation. Neuropathological analysis of the hypothalamus in post-mortem brain tissue has revealed a reduction in the number of vasopressin-expressing neurons [81], and interestingly was also present in a case of early stage HD who died of unrelated causes [82]. A previous study of post-mortem hypothalamic tissue showed no significant difference in the number of vasopressin neurons or vasopressin expression between controls and HD patients [83]. However, in the R6/2 mouse model the number of immunoreactive vasopressin neurons in the paraventricular nucleus of the hypothalamus were significantly decreased, suggesting that the change in drinking behaviour seen in mice may be the result of hypothalamic dysfunction [66]. In the same study patients with HD had increased thirst, unaffected urine osmolality and increased serum vasopressin, suggesting a dysregulation in the control of hypothalamic vasopressin release [66]. In another transgenic mouse model the hypothalamic expression of vasopressin was down-regulated without observing a significant loss of hypothalamic neurons [84]. In our study there was no significant difference in the fasting levels of plasma vasopressin levels between premanifest HD, stage II/III HD patients and controls. There are several differences between our study and that of Wood et al [66] that could account for this difference: 1. vasopressin levels differ in serum and plasma; 2. Wood et al included patients taking medication that may influence hormone levels; 3. Since vasopressin has a circadian rhythm [85], levels observed will depend on time of sampling: our sampling time was at 06:00 hours, whereas the sampling time in the Wood et al study was in the afternoon between 14:00–17:00 [66]; 4. Since hypoglycaemia causes the release of vasopressin [86], the fasting status may also have an effect on the levels of vasopressin.
Somatotropic axis
Studies analysing GH concentration in HD have yielded conflicting results with four studies reporting no significant difference between HD patients and controls [87–90], and three studies reporting an increase in HD [24,25,91]. GH exerts its effects by stimulating IGF-1 release from the liver. Saleh et al [24] have reported an increase in both central (GH) and peripheral (IGF-1) somatotropic hormones in early HD patients compared to healthy controls, as well as increase with disease severity. The study included a large cohort of patients (n = 217), but only a single morning sample in the morning was analysed and the cohorts were not controlled for medication (56% patients used neuroleptics, 61% antidepressants, 37% tranquillizers) [24]. This is important since neuroleptic treatment can influence GH levels by altering dopaminergic regulation [92]. Our study excluded patients using neuroleptics in the preceding 6 months.
Phillipson et al [25] also reported elevated fasting plasma GH levels in HD patients compared to controls. In our study, we found no significant difference in morning, fasting levels of GH or IGF-1, which is in agreement to several previous studies [87–90]. In a 24-hour study of plasma GH concentration in female HD patients free from centrally-active medication and matched controls, Durso et al showed increased levels of GH in HD females throughout the 24-hour period [91], which is in contrast to our findings. However, it is known in women that the amplitude of GH secretory pulses correlate with circulating levels of oestradiol [93,94]. In our study many of the female subjects had low oestradiol levels, which may lead to the low GH concentrations. In addition, in menstruating women, 24-hour GH levels are higher compared to age-matched men [93]. Thus, since in our study we had both males and females, this may account for the lower levels of GH observed compared to the Durso et al study [91]. However, when we analyse the female data separately, we still do not see an increase in GH levels in HD patients.
In this study we observed lower concentrations of 24 hour and post-sleep GH in stage II/III HD patients but these were not significant. Potentially, a study of larger cohorts may verify whether this is a biologically significant observation. Decreased concentrations of post-sleep GH in premanifest and stage II/III HD subjects may be indicate delay in sleep onset since GH secretion in early sleep is temporally and quantitatively associated with the amount of slow wave sleep [95,96]. Transient awakenings during sleep inhibit GH release [97], suggesting the decrease in nocturnal GH levels in HD subjects are secondary to fragmented sleep. GHRH inhibits sleep as well as GH secretion [98], thus if decreased GH levels in the HD subjects are associated with sleep delay, one would expect GHRH release to also be lower. We did not observe any change in GHRH levels, but this did not include measurement of post-sleep levels. The normality of GHRH levels at 06:00, 15:00 and 23:00 is in keeping with the finding of no significant difference on GH concentration at these time points.
IGF-1 decreases GH release from the pituitary by negative feedback, implying that low post-sleep GH levels in HD subjects would be associated with higher IGF-1 levels. The relationship between IGF-1 and GH may be better reflected by the ratio of the two in serum at any particular time and analysis of our data showed a higher IGF-1:GH ratio in stage II/III HD patients at 23.00 but this was not statistically significant. Further studies examining sleep quality in HD patients and measuring levels of GHRH and IGF-1, in parallel with GH in larger groups may be helpful. Another reason for possible reduction in sleep-related GH levels in HD gene carriers may be linked to a reduction in the night-time melatonin release we observed in these cohorts [99], since it has been shown that melatonin administration increases basal GH release and GH responsiveness to GHRH, possibly by inhibiting endogenous somatostatin release at the hypothalamic level [100,101].
Gonadotropic axis
In men with HD, reduced testosterone concentrations linked to disease severity have been reported [24,44]. In R6/2 mice, reduced testosterone is found and is accompanied by a reduction of gonadotropin releasing hormone (GnRH) neurons in the hypothalamus, suggesting this may be secondary to hypothalamic dysfunction [42]. In contrast, YAC128 mice develop testicular degeneration before levels of testosterone decrease and before loss of GnRH neurons in the hypothalamus can be detected, suggesting that testicular pathology results from a direct toxic effect of mutant huntingtin in the testes [43]. In contrast to previous studies, we did not observe an alteration in testosterone levels in HD males, nor in the levels of LH, FSH or oestradiol. The studies that reported lower testosterone levels in HD [24,44] included patients at different disease stages (TFC 0–13, and motor score 1–110), whereas, in our study, we had a mixture of premanifest and stage II/III HD subjects (TFC 5–13 and motor score 0–65).
To date, only two studies have analysed LH and FSH levels in male HD patients [24,44]. Both studies found no significant difference in FSH levels between control and HD males [24,44], which is in agreement with our data. Saleh et al found no significant difference in fasting, morning levels of LH [24], whereas Markianos et al reported lower LH levels in HD males in samples obtained between 10:00 and 12:00 [44]. We found no significant difference in LH levels between HD and control subjects (males or females) over 24 hours.
Gonadotropic axis hormones have not been carefully investigated in female HD patients. In our study, information on menopausal and menstrual cycle phase at the time of blood sampling for was available. We observed increased LH and FSH levels in pre- and post-menopausal women compared to other females, however, there was no significant difference between HD females and control females. More detailed study of gonadotrophic hormones at different phases of the menstrual cycle are needed to properly evaluate whether these hormones are altered in females with HD.
In the current study the only significant difference found in the gonadotropic axis was in fasting DHEAS levels, where a significant reduction could be seen in stage II/III patients with HD. This is in agreement with two previous studies that reported reduced DHEAS levels in HD patients [21,23]. Here, DHEAS was measured at only one timepoint, thus, measuring the circadian rhythm of DHEAS may be of interest. We also observed a non-significant reduction in DHEAS levels with increasing disease burden. DHEAS should ideally be analysed separately in males and females [102], however, we do not have a high number of subjects and when analysed separately for males and females, DHEAS levels are lower in HD gene carriers, however this is not statistically significant.
Lactotropic axis
Politis et al 2008 demonstrated D2 receptor loss in the hypothalamus of both early-stage HD patients and premanifest HD mutation carriers [63]. D2 receptors stimulation inhibits prolactin release [103]. Prolactin levels in patients with HD have been reported to be unchanged [24,39,41,87–89,91], increased [67,104] or even decreased [105,106]. In a 24-hour study with 6 medication-free male patients, Aziz et al (2010) demonstrated that prolactin secretion tended to be non-significantly higher in HD patients and significantly more irregular[39]. In our study we did not detect any significant difference in fasting prolactin levels between control and HD individuals. To date it remains unclear whether the lactotropic axis is indeed affected in HD.
Prolactin rhythmicity is primarily regulated by the sleep-wake cycle and maximal secretion occurs when sleep and circadian effects are superimposed [107–109]. Like GH, increased prolactin secretion is temporally associated with slow wave sleep and fragmented sleep results in decreased prolactin levels [110].
Thyrotropic axis
Progressive weight loss and muscle wasting are common in HD patients [14–16,111–114]. Since the thyrotropic axis is involved in the regulation of body weight and metabolism [38], several studies have evaluated hypothalamic-pituitary-thyroid axis function in patients with HD. Many studies have shown similar levels of TSH, total T4, T3 and free T4 in HD patients and normal controls [24,39–41,88]. However, in a retrospective study of HD patients, levothyroxine was found to be the most commonly prescribed drug for problems ‘unrelated’ to HD [115].
Although TSH synthesis and secretion are primarily controlled by the stimulatory action of thyrotropin-releasing hormone (TRH) and the negative feedback by thyroid hormones, other factors such as dopamine exert important modulatory effects [38]. Dopamine inhibits TSH synthesis and release through D2 receptor activation, whereas it stimulates TRH secretion [38].
One study showed similar TSH and T4 levels but higher T3 levels in early HD subjects which were not significantly increased compared with healthy controls [39]. Our findings do not support mild thyrotropic axis hyperactivity in HD but indicate a more focussed analysis in a larger cohort is warranted.
Since we only measured TSH at one time-point in the morning, we do not know whether its circadian rhythmicity is affected in HD subjects since TSH levels are low and relatively stable throughout the daytime and begin to increase in the early evening, with maximal levels around the beginning of the sleep period [116]. TSH levels progressively decline during the latter part of sleep, and this decrease is associated with slow wave sleep, while awakening result in increased TSH levels [117–119]. It has been suggested that the TSH evening rise shifts in concordance with the melatonin rhythm [120] and since we observed a reduction in the night-time melatonin in our HD cohorts [99], it is possible that TSH rhythm is also affected. Also, since the fT3 diurnal rhythm parallels TSH variations [121], the fT3 rhythmicity may also be altered in HD.
Correlation with clinical measures
It has previously been shown in a longitudinal study that IGF-1 correlates with cognitive measures: Stroop Word Reading, Stroop Colour Naming, Symbol Digit Modalities Test and Verbal Fluency [122]. We did not observe significant correlations between IGF-1 and any of these cognitive measures. Although the correlations in our study were not significant, they were all negative. This may reflect a true biological finding although may also reflect low number of HD subjects in our study. Thus, a longitudinal follow-up of our patients is needed to elucidate whether high IGF-1 levels correlate with poor cognition.
Conclusion
In contrast to many previous studies we describe overwhelmingly negative results, with no statistically significant differences. This may in part be down to small sample sizes and resulting low statistical power to find moderately sized differences. However, this lack of significant differences may also reflect the strict inclusion criteria, excluding patients on medication which is anticipated to alter these pathways (and perhaps, as a result, excluding some of the more severely affected HD patients). Our results are at least suggestive that with our patient inclusion criteria, there are differences of any appreciable size in not more than a small proportion of these biological markers, since even in the absence of any such associations a few significant results could readily have been found. We found only very subtle non-significant differences in hypothalamic-pituitary hormones in HD subjects, (e.g. decreased post-sleep GH and fasting DHEAS) that may warrant study in larger controlled cohorts.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The study was funded by a contract from Cure Huntington's Disease Initiative # A-2377. Scientific advisors from CHDI contributed to study design.
References
- 1. The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72: 971–983. [DOI] [PubMed] [Google Scholar]
- 2. Brandt J (1991) Cognitive impairments in Huntington's disease: Insights into the neuropsychology of the striatum In: Grafman FBaJ, editor. Handbook of neuropsychology. Amsterdam: Elsevier; pp. 241–264. [Google Scholar]
- 3. Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES (2005) Disintegration of the sleep-wake cycle and circadian timing in Huntington's disease. J Neurosci 25: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aziz NA, Anguelova GV, Marinus J, Lammers GJ, Roos RA (2010) Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington's disease. Parkinsonism Relat Disord 16: 345–350. 10.1016/j.parkreldis.2010.02.009 [DOI] [PubMed] [Google Scholar]
- 5. Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, et al. (2011) Dysfunctions in circadian behavior and physiology in mouse models of Huntington's disease. Exp Neurol 228: 80–90. 10.1016/j.expneurol.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morton AJ (2013) Circadian and sleep disorder in Huntington's disease. Exp Neurol 243: 34–44. 10.1016/j.expneurol.2012.10.014 [DOI] [PubMed] [Google Scholar]
- 7. Videnovic A, Leurgans S, Fan W, Jaglin J, Shannon KM (2009) Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord 15: 471–474. 10.1016/j.parkreldis.2008.10.002 [DOI] [PubMed] [Google Scholar]
- 8. Arnulf I, Nielsen J, Lohmann E, Schiefer J, Wild E, Jennum P, et al. (2008) Rapid eye movement sleep disturbances in Huntington disease. Arch Neurol 65: 482–488. 10.1001/archneur.65.4.482 [DOI] [PubMed] [Google Scholar]
- 9. Wiegand M, Moller AA, Lauer CJ, Stolz S, Schreiber W, Dose M, et al. (1991) Nocturnal sleep in Huntington's disease. J Neurol 238: 203–208. [DOI] [PubMed] [Google Scholar]
- 10. Farrer LA (1985) Diabetes mellitus in Huntington disease. Clin Genet 27: 62–67. [DOI] [PubMed] [Google Scholar]
- 11. Podolsky S, Leopold NA, Sax DS (1972) Increased frequency of diabetes mellitus in patients with Huntington's chorea. Lancet 1: 1356–1358. [DOI] [PubMed] [Google Scholar]
- 12. Podolsky S, Leopold NA (1977) Abnormal glucose tolerance and arginine tolerance tests in Huntington's disease. Gerontology 23: 55–63. [DOI] [PubMed] [Google Scholar]
- 13. Lalic NM, Maric J, Svetel M, Jotic A, Stefanova E, Lalic K, et al. (2008) Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol 65: 476–480. 10.1001/archneur.65.4.476 [DOI] [PubMed] [Google Scholar]
- 14. Sanberg PR, Fibiger HC, Mark RF (1981) Body weight and dietary factors in Huntington's disease patients compared with matched controls. Med J Aust 1: 407–409. [DOI] [PubMed] [Google Scholar]
- 15. Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, Roos RA (2008) Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 71: 1506–1513. 10.1212/01.wnl.0000334276.09729.0e [DOI] [PubMed] [Google Scholar]
- 16. Djousse L, Knowlton B, Cupples LA, Marder K, Shoulson I, Myers RH (2002) Weight loss in early stage of Huntington's disease. Neurology 59: 1325–1330. [DOI] [PubMed] [Google Scholar]
- 17. Hamilton JM, Wolfson T, Peavy GM, Jacobson MW, Corey-Bloom J (2004) Rate and correlates of weight change in Huntington's disease. J Neurol Neurosurg Psychiatry 75: 209–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petersen A, Bjorkqvist M (2006) Hypothalamic-endocrine aspects in Huntington's disease. Eur J Neurosci 24: 961–967. [DOI] [PubMed] [Google Scholar]
- 19. Bjorkqvist M, Petersen A, Bacos K, Isaacs J, Norlen P, Gil J, et al. (2006) Progressive alterations in the hypothalamic-pituitary-adrenal axis in the R6/2 transgenic mouse model of Huntington's disease. Hum Mol Genet 15: 1713–1721. [DOI] [PubMed] [Google Scholar]
- 20. Aziz NA, Swaab DF, Pijl H, Roos RA (2007) Hypothalamic dysfunction and neuroendocrine and metabolic alterations in Huntington's disease: clinical consequences and therapeutic implications. Rev Neurosci 18: 223–251. [DOI] [PubMed] [Google Scholar]
- 21. Bruyn GW, de Yong FH, van der Molen JH (1972) Huntington's chorea and the adrenal. Br Med J 1: 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heuser IJ, Chase TN, Mouradian MM (1991) The limbic-hypothalamic-pituitary-adrenal axis in Huntington's disease. Biol Psychiatry 30: 943–952. [DOI] [PubMed] [Google Scholar]
- 23. Leblhuber F, Peichl M, Neubauer C, Reisecker F, Steinparz FX, Windhager E, et al. (1995) Serum dehydroepiandrosterone and cortisol measurements in Huntington's chorea. J Neurol Sci 132: 76–79. [DOI] [PubMed] [Google Scholar]
- 24. Saleh N, Moutereau S, Durr A, Krystkowiak P, Azulay JP, Tranchant C, et al. (2009) Neuroendocrine disturbances in Huntington's disease. PLoS One 4: e4962 10.1371/journal.pone.0004962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phillipson OT, Bird ED (1976) Plasma growth hormone concentrations in Huntington's chorea. Clin Sci Mol Med 50: 551–554. [DOI] [PubMed] [Google Scholar]
- 26. Aziz NA, Pijl H, Frolich M, van der Graaf AW, Roelfsema F, Roos RA (2009) Increased hypothalamic-pituitary-adrenal axis activity in Huntington's disease. J Clin Endocrinol Metab 94: 1223–1228. 10.1210/jc.2008-2543 [DOI] [PubMed] [Google Scholar]
- 27. Harris GW, Reed M, Fawcett CP (1966) Hypothalamic releasing factors and the control of anterior pituitary function. Br Med Bull 22: 266–272. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimura F, Gorbman A (1986) Pars distalis of the pituitary gland: structure, function, and regulation: proceedings of the First International Symposium on the Pituitary Gland, Tokyo, Japan, November 14–17, 1984. Amsterdam; New York: Excerpta Medica, Elsevier Science Pub. Co. xiv, 549 p. p.
- 29. Lopez M, Tena-Sempere M, Dieguez C (2010) Cross-talk between orexins (hypocretins) and the neuroendocrine axes (hypothalamic-pituitary axes). Front Neuroendocrinol 31: 113–127. 10.1016/j.yfrne.2009.07.001 [DOI] [PubMed] [Google Scholar]
- 30. Tonsfeldt KJ, Chappell PE (2012) Clocks on top: the role of the circadian clock in the hypothalamic and pituitary regulation of endocrine physiology. Mol Cell Endocrinol 349: 3–12. 10.1016/j.mce.2011.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shirbin CA, Chua P, Churchyard A, Hannan AJ, Lowndes G, Stout JC (2013) The relationship between cortisol and verbal memory in the early stages of Huntington's disease. J Neurol 260: 891–902. 10.1007/s00415-012-6732-y [DOI] [PubMed] [Google Scholar]
- 32. van Duijn E, Selis MA, Giltay EJ, Zitman FG, Roos RA, van Pelt H, et al. (2010) Hypothalamic-pituitary-adrenal axis functioning in Huntington's disease mutation carriers compared with mutation-negative first-degree controls. Brain Res Bull 83: 232–237. 10.1016/j.brainresbull.2010.08.006 [DOI] [PubMed] [Google Scholar]
- 33. Rubin RT, Poland RE, Lesser IM, Winston RA, Blodgett AL (1987) Neuroendocrine aspects of primary endogenous depression. I. Cortisol secretory dynamics in patients and matched controls. Arch Gen Psychiatry 44: 328–336. [DOI] [PubMed] [Google Scholar]
- 34. Young EA, Altemus M, Parkison V, Shastry S (2001) Effects of estrogen antagonists and agonists on the ACTH response to restraint stress in female rats. Neuropsychopharmacology 25: 881–891. [DOI] [PubMed] [Google Scholar]
- 35. Carroll BJ, Curtis GC, Mendels J (1976) Neuroendocrine regulation in depression. II. Discrimination of depressed from nondepressed patients. Arch Gen Psychiatry 33: 1051–1058. [DOI] [PubMed] [Google Scholar]
- 36. Halbreich U, Asnis GM, Shindledecker R, Zumoff B, Nathan RS (1985) Cortisol secretion in endogenous depression. I. Basal plasma levels. Arch Gen Psychiatry 42: 904–908. [DOI] [PubMed] [Google Scholar]
- 37. William GH, Dluhy RG (1998) "Hyperfunction of the Adrenal Cortex." Fauci AS, editor. New York: McGraw-Hill. [Google Scholar]
- 38. Zoeller RT, Tan SW, Tyl RW (2007) General background on the hypothalamic-pituitary-thyroid (HPT) axis. Crit Rev Toxicol 37: 11–53. [DOI] [PubMed] [Google Scholar]
- 39. Aziz NA, Pijl H, Frolich M, Roelfsema F, Roos RA (2010) Altered thyrotropic and lactotropic axes regulation in Huntington's disease. Clin Endocrinol (Oxf) 73: 540–545. [DOI] [PubMed] [Google Scholar]
- 40. Goodman AO, Murgatroyd PR, Medina-Gomez G, Wood NI, Finer N, Vidal-Puig AJ, et al. (2008) The metabolic profile of early Huntington's disease—a combined human and transgenic mouse study. Exp Neurol 210: 691–698. 10.1016/j.expneurol.2007.12.026 [DOI] [PubMed] [Google Scholar]
- 41. Lavin PJ, Bone I, Sheridan P (1981) Studies of hypothalamic function in Huntington's chorea. J Neurol Neurosurg Psychiatry 44: 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papalexi E, Persson A, Bjorkqvist M, Petersen A, Woodman B, Bates GP, et al. (2005) Reduction of GnRH and infertility in the R6/2 mouse model of Huntington's disease. Eur J Neurosci 22: 1541–1546. [DOI] [PubMed] [Google Scholar]
- 43. Van Raamsdonk JM, Murphy Z, Selva DM, Hamidizadeh R, Pearson J, Petersen A, et al. (2007) Testicular degeneration in Huntington disease. Neurobiol Dis 26: 512–520. [DOI] [PubMed] [Google Scholar]
- 44. Markianos M, Panas M, Kalfakis N, Vassilopoulos D (2005) Plasma testosterone in male patients with Huntington's disease: relations to severity of illness and dementia. Ann Neurol 57: 520–525. [DOI] [PubMed] [Google Scholar]
- 45. Siesling S, van Vugt JP, Zwinderman KA, Kieburtz K, Roos RA (1998) Unified Huntington's disease rating scale: a follow up. Mov Disord 13: 915–919. [DOI] [PubMed] [Google Scholar]
- 46. Huntington Study Group (1996) Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord 11: 136–142. [DOI] [PubMed] [Google Scholar]
- 47. Shoulson I (1981) Huntington disease: functional capacities in patients treated with neuroleptic and antidepressant drugs. Neurology 31: 1333–1335. [DOI] [PubMed] [Google Scholar]
- 48. Penney JB Jr., Vonsattel JP, MacDonald ME, Gusella JF, Myers RH (1997) CAG repeat number governs the development rate of pathology in Huntington's disease. Ann Neurol 41: 689–692. [DOI] [PubMed] [Google Scholar]
- 49. Freedman D, Lane D (1983) A nonstochastic interpretation of reported significance levels. Journal of Business & Economic Statistics 1: 292–298. [Google Scholar]
- 50. Shadrokh A, 'Aubigny Gd (2010) An analytic comparison of permutation methods for tests of partial regression coefficients in the linear model. Applied Mathematical Sciences 4: 857–878. [Google Scholar]
- 51. Rothman KJ (1990) No adjustments are needed for multiple comparisons. Epidemiology 1: 43–46. [PubMed] [Google Scholar]
- 52. Matthews DR (1988) Time series analysis in endocrinology. Acta Paediatr Scand Suppl 347: 55–62. [PubMed] [Google Scholar]
- 53.Hill NR, Matthews D, University of Oxford. Medical Sciences Division. (2008) Analysis of non-steady state physiological and pathological processes [Thesis (D Phil)]: University of Oxford, 2008. 2 v. p.
- 54. Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10: 204–216. 10.1038/nrneurol.2014.24 [DOI] [PubMed] [Google Scholar]
- 55. Van Cauter E, Plat L, Copinschi G (1998) Interrelations between sleep and the somatotropic axis. Sleep 21: 553–566. [PubMed] [Google Scholar]
- 56. Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A (2009) Huntington's disease: the current state of research with peripheral tissues. Exp Neurol 219: 385–397. 10.1016/j.expneurol.2009.05.012 [DOI] [PubMed] [Google Scholar]
- 57. Douaud G, Gaura V, Ribeiro MJ, Lethimonnier F, Maroy R, Verny C, et al. (2006) Distribution of grey matter atrophy in Huntington's disease patients: a combined ROI-based and voxel-based morphometric study. Neuroimage 32: 1562–1575. [DOI] [PubMed] [Google Scholar]
- 58. Kassubek J, Juengling FD, Kioschies T, Henkel K, Karitzky J, Kramer B, et al. (2004) Topography of cerebral atrophy in early Huntington's disease: a voxel based morphometric MRI study. J Neurol Neurosurg Psychiatry 75: 213–220. [PMC free article] [PubMed] [Google Scholar]
- 59. Sawiak SJ, Wood NI, Williams GB, Morton AJ, Carpenter TA (2009) Voxel-based morphometry in the R6/2 transgenic mouse reveals differences between genotypes not seen with manual 2D morphometry. Neurobiol Dis 33: 20–27. 10.1016/j.nbd.2008.09.016 [DOI] [PubMed] [Google Scholar]
- 60. Kremer HP, Roos RA, Dingjan G, Marani E, Bots GT (1990) Atrophy of the hypothalamic lateral tuberal nucleus in Huntington's disease. J Neuropathol Exp Neurol 49: 371–382. [DOI] [PubMed] [Google Scholar]
- 61. Kremer HP, Roos RA, Dingjan GM, Bots GT, Bruyn GW, Hofman MA (1991) The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington's disease. Neurosci Lett 132: 101–104. [DOI] [PubMed] [Google Scholar]
- 62. Soneson C, Fontes M, Zhou Y, Denisov V, Paulsen JS, Kirik D, et al. (2010) Early changes in the hypothalamic region in prodromal Huntington disease revealed by MRI analysis. Neurobiol Dis 40: 531–543. 10.1016/j.nbd.2010.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Politis M, Pavese N, Tai YF, Tabrizi SJ, Barker RA, Piccini P (2008) Hypothalamic involvement in Huntington's disease: an in vivo PET study. Brain 131: 2860–2869. 10.1093/brain/awn244 [DOI] [PubMed] [Google Scholar]
- 64. Petersen A, Gil J, Maat-Schieman ML, Bjorkqvist M, Tanila H, Araujo IM, et al. (2005) Orexin loss in Huntington's disease. Hum Mol Genet 14: 39–47. [DOI] [PubMed] [Google Scholar]
- 65. Hult S, Soylu R, Bjorklund T, Belgardt BF, Mauer J, Bruning JC, et al. (2011) Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab 13: 428–439. 10.1016/j.cmet.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 66. Wood NI, Goodman AO, van der Burg JM, Gazeau V, Brundin P, Bjorkqvist M, et al. (2008) Increased thirst and drinking in Huntington's disease and the R6/2 mouse. Brain Res Bull 76: 70–79. 10.1016/j.brainresbull.2007.12.007 [DOI] [PubMed] [Google Scholar]
- 67. Markianos M, Panas M, Kalfakis N, Vassilopoulos D (2009) Plasma homovanillic acid and prolactin in Huntington's disease. Neurochem Res 34: 917–922. 10.1007/s11064-008-9851-1 [DOI] [PubMed] [Google Scholar]
- 68. Veldhuis JD, Keenan DM, Pincus SM (2008) Motivations and methods for analyzing pulsatile hormone secretion. Endocr Rev 29: 823–864. 10.1210/er.2008-0005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Andreassen OA, Dedeoglu A, Stanojevic V, Hughes DB, Browne SE, Leech CA, et al. (2002) Huntington's disease of the endocrine pancreas: insulin deficiency and diabetes mellitus due to impaired insulin gene expression. Neurobiol Dis 11: 410–424. [DOI] [PubMed] [Google Scholar]
- 70. Bjorkqvist M, Fex M, Renstrom E, Wierup N, Petersen A, Gil J, et al. (2005) The R6/2 transgenic mouse model of Huntington's disease develops diabetes due to deficient beta-cell mass and exocytosis. Hum Mol Genet 14: 565–574. [DOI] [PubMed] [Google Scholar]
- 71. Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F, et al. (1999) Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet 8: 813–822. [DOI] [PubMed] [Google Scholar]
- 72. Carnes M, Lent S, Feyzi J, Hazel D (1989) Plasma adrenocorticotropic hormone in the rat demonstrates three different rhythms within 24 h. Neuroendocrinology 50: 17–25. [DOI] [PubMed] [Google Scholar]
- 73. Follenius M, Simon C, Brandenberger G, Lenzi P (1987) Ultradian plasma corticotropin and cortisol rhythms: time-series analyses. J Endocrinol Invest 10: 261–266. [DOI] [PubMed] [Google Scholar]
- 74. Veldhuis JD, Iranmanesh A, Johnson ML, Lizarralde G (1990) Twenty-four-hour rhythms in plasma concentrations of adenohypophyseal hormones are generated by distinct amplitude and/or frequency modulation of underlying pituitary secretory bursts. J Clin Endocrinol Metab 71: 1616–1623. [DOI] [PubMed] [Google Scholar]
- 75. Veldhuis JD, Iranmanesh A, Johnson ML, Lizarralde G (1990) Amplitude, but not frequency, modulation of adrenocorticotropin secretory bursts gives rise to the nyctohemeral rhythm of the corticotropic axis in man. J Clin Endocrinol Metab 71: 452–463. [DOI] [PubMed] [Google Scholar]
- 76. Young EA, Carlson NE, Brown MB (2001) Twenty-four-hour ACTH and cortisol pulsatility in depressed women. Neuropsychopharmacology 25: 267–276. [DOI] [PubMed] [Google Scholar]
- 77. Spath-Schwalbe E, Gofferje M, Kern W, Born J, Fehm HL (1991) Sleep disruption alters nocturnal ACTH and cortisol secretory patterns. Biol Psychiatry 29: 575–584. [DOI] [PubMed] [Google Scholar]
- 78. Pruessner JC, Wolf OT, Hellhammer DH, Buske-Kirschbaum A, von Auer K, Jobst S, et al. (1997) Free cortisol levels after awakening: a reliable biological marker for the assessment of adrenocortical activity. Life Sci 61: 2539–2549. [DOI] [PubMed] [Google Scholar]
- 79. Caufriez A, Moreno-Reyes R, Leproult R, Vertongen F, Van Cauter E, Copinschi G (2002) Immediate effects of an 8-h advance shift of the rest-activity cycle on 24-h profiles of cortisol. Am J Physiol Endocrinol Metab 282: E1147–1153. [DOI] [PubMed] [Google Scholar]
- 80. Ekstedt M, Akerstedt T, Soderstrom M (2004) Microarousals during sleep are associated with increased levels of lipids, cortisol, and blood pressure. Psychosom Med 66: 925–931. [DOI] [PubMed] [Google Scholar]
- 81. Gabery S, Murphy K, Schultz K, Loy CT, McCusker E, Kirik D, et al. (2010) Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol 120: 777–788. 10.1007/s00401-010-0742-6 [DOI] [PubMed] [Google Scholar]
- 82. Gabery S, Halliday G, Kirik D, Englund E, Petersen A (2015) Selective loss of oxytocin and vasopressin in the hypothalamus in early Huntington disease: a case study. Neuropathol Appl Neurobiol. 10.1111/nan.12236 [DOI] [PubMed] [Google Scholar]
- 83. van Wamelen DJ, Aziz NA, Anink JJ, Roos RA, Swaab DF (2012) Paraventricular nucleus neuropeptide expression in Huntington's disease patients. Brain Pathol 22: 654–661. 10.1111/j.1750-3639.2012.00565.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kotliarova S, Jana NR, Sakamoto N, Kurosawa M, Miyazaki H, Nekooki M, et al. (2005) Decreased expression of hypothalamic neuropeptides in Huntington disease transgenic mice with expanded polyglutamine-EGFP fluorescent aggregates. J Neurochem 93: 641–653. [DOI] [PubMed] [Google Scholar]
- 85. Tominaga K, Shinohara K, Otori Y, Fukuhara C, Inouye ST (1992) Circadian rhythms of vasopressin content in the suprachiasmatic nucleus of the rat. Neuroreport 3: 809–812. [DOI] [PubMed] [Google Scholar]
- 86. Fisher BM, Baylis PH, Thornton S, Frier BM (1989) Arginine vasopressin and oxytocin responses to insulin-induced hypoglycemia in type 1 (insulin-dependent) diabetes. J Clin Endocrinol Metab 68: 688–692. [DOI] [PubMed] [Google Scholar]
- 87. Chalmers RJ, Johnson RH, Keogh HJ, Nanda RN (1978) Growth hormone and prolactin response to bromocriptine in patients with Huntington's chorea. J Neurol Neurosurg Psychiatry 41: 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Levy CL, Carlson HE, Sowers JR, Goodlett RE, Tourtellotte WW, Hershman JM (1979) Growth hormone and prolactin secretion in Huntington's disease. Life Sci 24: 743–749. [DOI] [PubMed] [Google Scholar]
- 89. Murri L, Iudice A, Muratorio A, Polleri A, Barreca T, Murialdo G (1980) Spontaneous nocturnal plasma prolactin and growth hormone secretion in patients with Parkinson's disease and Huntington's chorea. Eur Neurol 19: 198–206. [DOI] [PubMed] [Google Scholar]
- 90. Popovic V, Svetel M, Djurovic M, Petrovic S, Doknic M, Pekic S, et al. (2004) Circulating and cerebrospinal fluid ghrelin and leptin: potential role in altered body weight in Huntington's disease. Eur J Endocrinol 151: 451–455. [DOI] [PubMed] [Google Scholar]
- 91. Durso R, Tamminga CA, Ruggeri S, Denaro A, Kuo S, Chase TN (1983) Twenty-four hour plasma levels of growth hormone and prolactin in Huntington's disease. J Neurol Neurosurg Psychiatry 46: 1134–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Caraceni T, Panerai AE, Paratl EA, Cocchi D, Muller EE (1977) Altered growth hormone and prolactin responses to dopaminergic stimulation in Huntington's chorea. J Clin Endocrinol Metab 44: 870–875. [DOI] [PubMed] [Google Scholar]
- 93. Ho KY, Evans WS, Blizzard RM, Veldhuis JD, Merriam GR, Samojlik E, et al. (1987) Effects of sex and age on the 24-hour profile of growth hormone secretion in man: importance of endogenous estradiol concentrations. J Clin Endocrinol Metab 64: 51–58. [DOI] [PubMed] [Google Scholar]
- 94. Shah N, Evans WS, Veldhuis JD (1999) Actions of estrogen on pulsatile, nyctohemeral, and entropic modes of growth hormone secretion. Am J Physiol 276: R1351–1358. [DOI] [PubMed] [Google Scholar]
- 95. Van Cauter E, Kerkhofs M, Caufriez A, Van Onderbergen A, Thorner MO, Copinschi G (1992) A quantitative estimation of growth hormone secretion in normal man: reproducibility and relation to sleep and time of day. J Clin Endocrinol Metab 74: 1441–1450. [DOI] [PubMed] [Google Scholar]
- 96. Holl RW, Hartman ML, Veldhuis JD, Taylor WM, Thorner MO (1991) Thirty-second sampling of plasma growth hormone in man: correlation with sleep stages. J Clin Endocrinol Metab 72: 854–861. [DOI] [PubMed] [Google Scholar]
- 97. Van Cauter E, Caufriez A, Kerkhofs M, Van Onderbergen A, Thorner MO, Copinschi G (1992) Sleep, awakenings, and insulin-like growth factor-I modulate the growth hormone (GH) secretory response to GH-releasing hormone. J Clin Endocrinol Metab 74: 1451–1459. [DOI] [PubMed] [Google Scholar]
- 98. Ocampo-Lim B, Guo W, DeMott-Friberg R, Barkan AL, Jaffe CA (1996) Nocturnal growth hormone (GH) secretion is eliminated by infusion of GH-releasing hormone antagonist. J Clin Endocrinol Metab 81: 4396–4399. [DOI] [PubMed] [Google Scholar]
- 99. Kalliolia E, Silajdzic E, Nambron R, Hill NR, Doshi A, Frost C, et al. (2014) Plasma melatonin is reduced in Huntington's disease. Mov Disord. 10.1002/mds.26003 [DOI] [PubMed] [Google Scholar]
- 100. Valcavi R, Zini M, Maestroni GJ, Conti A, Portioli I (1993) Melatonin stimulates growth hormone secretion through pathways other than the growth hormone-releasing hormone. Clin Endocrinol (Oxf) 39: 193–199. [DOI] [PubMed] [Google Scholar]
- 101. Meeking DR, Wallace JD, Cuneo RC, Forsling M, Russell-Jones DL (1999) Exercise-induced GH secretion is enhanced by the oral ingestion of melatonin in healthy adult male subjects. Eur J Endocrinol 141: 22–26. [DOI] [PubMed] [Google Scholar]
- 102. Sulcova J, Hill M, Hampl R, Starka L (1997) Age and sex related differences in serum levels of unconjugated dehydroepiandrosterone and its sulphate in normal subjects. J Endocrinol 154: 57–62. [DOI] [PubMed] [Google Scholar]
- 103. Muller EE, Cocchi D, Mantegazza P, Parati EA, Caraceni T (1977) Prolactin control in Huntington's chorea. Lancet 2: 764–765. [DOI] [PubMed] [Google Scholar]
- 104. Caine E, Kartzinel R, Ebert M, Carter AC (1978) Neuroendocrine function in Huntington's disease: dopaminergic regulation of prolactin release. Life Sci 22: 911–918. [DOI] [PubMed] [Google Scholar]
- 105. Hayden MR, Vinik AI, Paul M, Beighton P (1977) Impaired prolactin release in Huntington's chorea. Evidence for dopaminergic excess. Lancet 2: 423–426. [DOI] [PubMed] [Google Scholar]
- 106. Kremer HP, Roos RA, Frolich M, Radder JK, Nieuwenhuijzen Kruseman AC, Van der Velde A, et al. (1989) Endocrine functions in Huntington's disease. A two-and-a-half years follow-up study. J Neurol Sci 90: 335–344. [DOI] [PubMed] [Google Scholar]
- 107. Spiegel K, Follenius M, Simon C, Saini J, Ehrhart J, Brandenberger G (1994) Prolactin secretion and sleep. Sleep 17: 20–27. [DOI] [PubMed] [Google Scholar]
- 108. Waldstreicher J, Duffy JF, Brown EN, Rogacz S, Allan JS, Czeisler CA (1996) Gender differences in the temporal organization of proclactin (PRL) secretion: evidence for a sleep-independent circadian rhythm of circulating PRL levels- a clinical research center study. J Clin Endocrinol Metab 81: 1483–1487. [DOI] [PubMed] [Google Scholar]
- 109. Desir D, Van Cauter E, L'Hermite M, Refetoff S, Jadot C, Caufriez A, et al. (1982) Effects of "jet lag" on hormonal patterns. III. Demonstration of an intrinsic circadian rhythmicity in plasma prolactin. J Clin Endocrinol Metab 55: 849–857. [DOI] [PubMed] [Google Scholar]
- 110. Spiegel K, Luthringer R, Follenius M, Schaltenbrand N, Macher JP, Muzet A, et al. (1995) Temporal relationship between prolactin secretion and slow-wave electroencephalic activity during sleep. Sleep 18: 543–548. [PubMed] [Google Scholar]
- 111. Farrer LA, Meaney FJ (1985) An anthropometric assessment of Huntington's disease patients and families. Am J Phys Anthropol 67: 185–194. [DOI] [PubMed] [Google Scholar]
- 112. Farrer LA, Yu PL (1985) Anthropometric discrimination among affected, at-risk, and not-at-risk individuals in families with Huntington disease. Am J Med Genet 21: 307–316. [DOI] [PubMed] [Google Scholar]
- 113. Trejo A, Tarrats RM, Alonso ME, Boll MC, Ochoa A, Velasquez L (2004) Assessment of the nutrition status of patients with Huntington's disease. Nutrition 20: 192–196. [DOI] [PubMed] [Google Scholar]
- 114. van der Burg JM, Bjorkqvist M, Brundin P (2009) Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol 8: 765–774. 10.1016/S1474-4422(09)70178-4 [DOI] [PubMed] [Google Scholar]
- 115. Nance MA, Sanders G (1996) Characteristics of individuals with Huntington disease in long-term care. Mov Disord 11: 542–548. [DOI] [PubMed] [Google Scholar]
- 116. Brabant G, Prank K, Ranft U, Schuermeyer T, Wagner TO, Hauser H, et al. (1990) Physiological regulation of circadian and pulsatile thyrotropin secretion in normal man and woman. J Clin Endocrinol Metab 70: 403–409. [DOI] [PubMed] [Google Scholar]
- 117. Hirschfeld U, Moreno-Reyes R, Akseki E, L'Hermite-Baleriaux M, Leproult R, Copinschi G, et al. (1996) Progressive elevation of plasma thyrotropin during adaptation to simulated jet lag: effects of treatment with bright light or zolpidem. J Clin Endocrinol Metab 81: 3270–3277. [DOI] [PubMed] [Google Scholar]
- 118. Goichot B, Brandenberger G, Saini J, Wittersheim G, Follenius M (1992) Nocturnal plasma thyrotropin variations are related to slow-wave sleep. J Sleep Res 1: 186–190. [DOI] [PubMed] [Google Scholar]
- 119. Gronfier C, Luthringer R, Follenius M, Schaltenbrand N, Macher JP, Muzet A, et al. (1995) Temporal link between plasma thyrotropin levels and electroencephalographic activity in man. Neurosci Lett 200: 97–100. [DOI] [PubMed] [Google Scholar]
- 120. Buxton OM, Frank SA, L'Hermite-Baleriaux M, Leproult R, Turek FW, Van Cauter E (1997) Roles of intensity and duration of nocturnal exercise in causing phase delays of human circadian rhythms. Am J Physiol 273: E536–542. [DOI] [PubMed] [Google Scholar]
- 121. Russell W, Harrison RF, Smith N, Darzy K, Shalet S, Weetman AP, et al. (2008) Free triiodothyronine has a distinct circadian rhythm that is delayed but parallels thyrotropin levels. J Clin Endocrinol Metab 93: 2300–2306. 10.1210/jc.2007-2674 [DOI] [PubMed] [Google Scholar]
- 122. Saleh N, Moutereau S, Azulay JP, Verny C, Simonin C, Tranchant C, et al. (2010) High insulinlike growth factor I is associated with cognitive decline in Huntington disease. Neurology 75: 57–63. 10.1212/WNL.0b013e3181e62076 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.