

Abstract
Histone deacetylases (HDAC) are metal-dependent enzymes and considered as important targets for cell functioning. Particularly, higher expression of class I HDACs is common in the onset of multiple malignancies which results in deregulation of many target genes involved in cell growth, differentiation and survival. Although substantial attempts have been made to control the irregular functioning of HDACs by employing various inhibitors with high sensitivity towards transformed cells, limited success has been achieved in epigenetic cancer therapy. Here in this study, we used ligand-based pharmacophore and 2-dimensional quantitative structure activity relationship (QSAR) modeling approaches for targeting class I HDAC isoforms. Pharmacophore models were generated by taking into account the known IC50 values and experimental energy scores with extensive validations. The QSAR model having an external R2 value of 0.93 was employed for virtual screening of compound libraries. 10 potential lead compounds (C1-C10) were short-listed having strong binding affinities for HDACs, out of which 2 compounds (C8 and C9) were able to interact with all members of class I HDACs. The potential binding modes of HDAC2 and HDAC8 to C8 were explored through molecular dynamics simulations. Overall, bioactivity and ligand efficiency (binding energy/non-hydrogen atoms) profiles suggested that proposed hits may be more effective inhibitors for cancer therapy.
Introduction
Histone Acetyltransferases (HATs) and HDACs regulate the acetylation and deacetylation events of small alkaline histones associated with DNA double helical structure [1,2]. Interactions of positively charged amino-terminal tails of histones with negatively charged phosphodiester backbones of DNA results in chromatin compaction [3,4]. The associated conformational changes which occur due to acetylation of lysine residues result in chromatin remodeling. Thus, HATs mediated acetylation promotes chromatin relaxation by loosening the packed histones and DNA, thereby facilitating the accession of transcription factors to bind to respective DNA templates [5–7]. In contrast to acetylation, compactness of nucleosome units and controlled gene expression as a result of deacetylation is mediated by HDACs [1]. Histones play crucial roles in stabilizing the heritable epigenetic changes in gene activity and expression [2]. Any disturbances in these functions may lead to the abnormal expression of genetic material that may cause fatal diseases like diabetes and cancer [8,9].
The critical roles of HATs and HDACs in tumor progression, cardiac and brain disorders have been analyzed in many studies [10–13]. In human, at least 18 HDACs have been identified that are grouped into four classes and comprise of two major categories: Zn+2 and NAD+ dependent [14]. This classification is based on the structural, functional and phylogenetic analysis of HDACs [15]. A detailed overview of cellular compartments and HDAC involvements in diverse biological processes is given in Table 1. Class I comprises of HDAC1-3 and 8 [16–18] which controls many functional and regulatory mechanisms [19]. Importantly, implication of class I HDACs has been monitored in hematological malignancies, resulting in differentiation and proliferation abnormalities of myeloid cells [20]. In tumor progression, most prevailing alteration is linked to abnormal expressions of HDACs [21]. Aberrant expression pattern of HDAC1 has been observed in prostate, gastric, breast and colon [22–25] cancers. Similarly, onset of gastric, cervical and colorectal carcinoma [26–28] is associated with increased expression of HDAC2. Abnormal expression patterns of class I HDAC members are also evidenced in cell proliferation and migration during ovarian and breast carcinomas [29]. In these tumor cells, down-regulation of e-cadherin is associated with HDAC3 overexpression. Unbalanced expressions of HDAC members also result in Acute Promyelocytic Leukemia (APL) such as lymphoblastic APL and non-Hodgkin’s lymphomas [28,30].
Table 1. Classification and biological roles of HDACs.
| Classes | Cofactors | HDACs | Cellular Locations | Biological Processes | References |
|---|---|---|---|---|---|
| Class I | Zn+2 dependent | 1, 2, 3, 8 | Nucleus, Cytoplasm, Transcriptional repressor complex, Spindle microtubule, Replication fork | Cell cycle regulation, Cell differentiation, DNA damage response, Epidermis development, Regulating cardiac myocyte proliferation on the course of cardiac development | [16–18] |
| Class II | Zn+2 dependent | 4, 5, 6, 7, 9, 10 | Cytoplasm, Nucleus, Neuromuscular junction, Golgi apparatus, Cytosol caveola | Regulation of transcription and cell differentiation, Regulation of cardiac muscle contraction, Inflammatory response, Nervous system development, Heart development, Protein polyubiquitination, Response to toxic and organic substances, Macroautophagy, Vasculogenesis | [1, 17] |
| Class III | NAD+ dependent | Sirtuins SIRT1-SIRT7 | Nucleus, Cytoplasm | Histone deacetylation, Regulation of phosphorylation, Regulation of double-strand break repair via homologous recombination, DNA repair mechanism | [16] |
| Class IV | Zn+2 dependent | 11 | Nucleus | Transcription, DNA-dependent chromatin modification, Histone Deacetylation | [16] |
Another mechanism of HDAC-mediated tumor onset is due to transcriptional repression of tumor suppressor genes and their aberrant recruitments to promoter regions. In recent years, many studies potentiate the ways of targeted HDAC inhibition in the context of tumor control [31,32]. Transcriptional activation of tumor suppressor genes by the inhibition of HDAC activity is considered as an ideal and innovative strategy. To date, several HDAC inhibitors (HDACi) like hydroxamic acids, benzamides, short chain carbolic acids, and cyclic tetrapeptides have been characterized in vitro and ex vivo for various cancers [33–36]. Hydroxamic acids (hydroximates) include vorinostat (SAHA), belinostat (PXD101), panobinostat (LBH589), dacinostat (LAQ824), givinostat (ITF-2357) and trichostatin A (TSA). Benzamides are the derivatives of benzoic acid which include entinostat (MS-275), p-N-acetyldinaline (CI-994), mocetinostat (MGCD0103) and SK-7041. Short chain carboxylic acids (aliphatic acids) are relatively weak inhibitors which comprise of valproic acid or VPA, butyrate and sodium phenyl butyrate. Cyclic tetrapeptides or cyclic peptides are structurally complex molecules which consist of romidepsin (depsipeptide), apicidin (OSI-2040), trapoxin A and trapoxin B (cyclic hydroxamic acid) [37,38]. Currently, these inhibitors are under investigations in clinical trials [39–44]. In cancer patients, functional activity of HDACi is mediated extrinsically as well as intrinsically due to cell cycle arrest at G1/G2 phases and up-regulation of apoptosis [31,45]. Sensitivity of actinotheraphy and chemotherapy for cancers is also enhanced by HDAC inhibition [46].
In silico drug designing strategies are extensively applied for the identification of novel inhibitors through modeled HDAC structures and their pharmacophore-based studies [47–53]. However, these inhibitors are unique in terms of HDAC targeting actions. Here, through comparative ligand-based approaches, several novel candidate hits were proposed and added in the list of HDACi which are able to target entire class I of HDACs.
Methodology
Dataset
High resolution co-crystallized structures of class I HDACs were retrieved through PDB database [54] with following PDB IDs: 4BKX (HDAC1), 4LXZ (HDAC2), 4A69 (HDAC3) and 1T64 (HDAC8) (Table A in S1 File). These structures were further refined by adding charges and missing residues through dock prep module of UCSF Chimera 1.10 [55]. Binding and catalytic site details of class I HDACs were extracted through PDBSum [56]. Structural superimposition was performed by UCSF Chimera 1.10 [55]. Additionally, comparative binding mode of class I HDACs has been reviewed through literature [47,48,53].
Pharmacophore/QSAR Model Generation
In order to generate the ligand-based quantitative structure-activity relationship (QSAR) and pharmacophore models, 16 known inhibitors (Table B in S1 File) were selected. These known HDAC inhibitors were collected through literature survey [57–73] and classified on the basis of their inhibition potential values for class I HDACs. Chemical structures and bioactivities of these inhibitors were extracted through EMBL-EBI ChEMBL database [74], while their 2D/3D molecular descriptors values were retrieved through ChemAxon’s Chemicalize [75] and Molinspiration [76]. Molecular dockings of these inhibitors were performed against class I HDACs by AutoDock VINA with default parameters i.e. exhaustiveness = 8 and energy range = 3 [77].
The selection of training set compounds was based on bioactivity and binding affinity attributes of docked protein-inhibitor complexes. The best docked conformations of inhibitors having least IC50 values and high binding abilities were filtered out for pharmacophore generation. In order to design 2D QSAR model, IC50 values of training set compounds against HDAC1, 2, 3 and 8 were converted into pIC50 values and average pIC50 values were calculated for each of the training set compound.
Test data set was created to evaluate the quality of pharmacophore hypotheses. Known selective and non-selective synthetic molecules were gathered through ChEMBL [74] which were utilized in human assays [68,69,71,73,78–109] to generate a dataset of 71 compounds (Table C in S1 File) and their experimental IC50 values were assessed. Compounds having weak inhibition potential (IC50 > 2500 nM) for class I HDACs were grouped in decoy set, whereas high inhibitory compounds (IC50 < 2500 nM) were placed in the active dataset. By implying the above criteria, 41 compounds were found to be active and 30 were declared as decoys. 3D generation and optimization of data set compounds was performed by ACD ChemSketch [110].
To generate pharmacophore models, training set hits were employed in the ligand-based module of the LigandScout 3.0 [111]. Pharmacophoric sites such as hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), hydrophobic sites, aromatic ring and positive and negative ionizable groups were carefully characterized. To incorporate the associated features of selected compounds, merge feature model generation and atom overlap scoring function of LigandScout 3.0 was applied. Subsequently, descriptor selection analysis was performed through forward selection (FS), backward elimination (BE) and stepwise selection (SS), the classical methods of variable selection [112]. All unique descriptors signifying the model were selected, while excluding the non-significant descriptors. QSAR model was created by employing multiple linear regression (MLR) technique [113]. Regression module of the IBM statistical package for social sciences (SPSS) version 22 [114] was used to create the regression model by employing Eq 1.
| (1) |
Where, y (pIC50) is the dependent variable, x is the independent variable, β is the coefficient and ε is the error value. QSAR and pharmacophore models were statistically evaluated to check their reliabilities prior to implementation.
Top five models exhibiting significant statistical values were scrutinized as 3D query for virtual screening. Validated 3D pharmacophore models were screened against a total of 48,386 compounds (2,601 of Aurora [115] and 45,785 of Princeton [116] libraries) to prey novel drug targets. The novel hits were selected for docking analysis on the basis of high query fit values relative to pharmacophore models.
Molecular docking and dynamic simulation assays
Molecular docking analyses of highly ranked compounds were performed for class I HDACs through above mentioned parameters of AutoDock VINA [77]. Subsequently, energy scores and HDAC bound poses of these compounds were carefully evaluated. To study the structural dynamics of receptor-ligand complex, molecular dynamics (MD) simulations of HDAC2 and HDAC8 were performed. Gromacs 4.5.5 [117] and GROMOS96 43a1 force field with SPC water model was used for MD simulations. Subsequently, energy minimization was performed by steepest descent method and appropriate counter ions were added to neutralize the system [118,119]. Finally, a 12 ns MD simulation run was performed at constant temperature (300 K) and pressure (1 atm). GROMACS analysis tools were used for the analysis of MD simulation trajectories (118). The stability of secondary structure elements, conformational changes and interactions were assessed by computing root mean square deviation (RMSD), root mean square fluctuation (RMSF) and hydrogen bonds obtained throughout MD trajectories. For RMSD, RMSF, gyration and hydrogen bonds g_rms, g_rmsf, g_gyrate and g_hbond modules of Gromacs were applied, respectively. RMS cluster distribution of HDAC2 and HDAC8 backbone was computed using g_cluster module with nearest neighbor method. Energy values along the MD trajectories were calculated by Eq 2.
| (2) |
All simulations were carried out through an open SUSE 11.2 system with Intel(R) core (TM) i5-2300 CPU containing Linux 2.6.31.5–0.1 operating system. The binding patterns were carefully monitored through Accelrys Discovery Studio 4 [120].
ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties were predicted through AdmetSAR [121] server to hypothetically depict the positive and negative biological effects of compounds. Bioavailability, Rule of Five (ROF), lead-likeness and other filters were assessed using Chemicalize [75]. Toxicity parameters such as mutagenic and tumorigenic effects of selected compounds were evaluated through OSIRIS [122].
Expected IC50 values of compounds were predicted through QSAR model. Physiochemical descriptors of compounds were extracted by Chemicalize [75] and Molinspiration [76] servers. To avoid any redundancy issue, predicted compounds were compared against small molecule library available in ChEMBL [74] database, based on ≥90% similarity threshold. Synthetic accessibility analysis of predicted hits was performed through SYLVIA [123] tool.
Results
Binding site analysis
Binding sites of class I HDAC family members were evaluated by structural comparisons (Fig 1). Co-crystallized structures of HDAC1, 2, 3 and 8 showed structural similarities with an RMSD value of 0.5Å (Fig 1A). Zn+2 ions were coordinated by one HIS and two ASP residues of individual HDACs (Fig 1B). These catalytic residues resided within the 4Å region of surface forming the tunnel shaped pocket.
Fig 1. Zn+2 dependent class I HDACs.

(a) Structural superimposition of HDACs. (b) Binding sites of class 1 HDACs. HDAC1, 2, 3 and 8 are shown in orange, pink, green and blue, respectively. Zn+2 is shown in brown color.
Pharmacophore-based 2D QSAR modeling
In order to select training set compounds, binding affinities, IC50 and molecular descriptors values of known class I HDAC inhibitors were monitored (Tables B, D and E in S1 File). These known hits included MS-275, LBH-589, LAQ-824, Trichostatin A, Saha, Belinostat, Oxamflatin, Pyroxamide, Mocetinostat, and Scriptaid with IC50 values ranging from 5–300 nM. 5–7 pharmacophore models were generated based on the information of docking poses of training set compounds and their pharmacophore fit values were evaluated. The ideal pharmacophore models with high query fit values were shown in Figure A in S1 File. These models exhibited good pharmacophoric features such as hydrogen bond donors, hydrogen bond acceptors, hydrophobic sites and negative ionizable groups (Zn+2 binding locations). QSAR regression model was generated using 5 descriptors of training set compounds, as shown in Eq 3. Their pIC50 values were shown in Table F in S1 File.
| (3) |
To determine the reliability and predictability of models, several statistical parameters (Table G in S1 File) were applied which showed positive results. Higher sensitivity and specificity values, percent of active yield (%Y), enrichment factor and Guner-Henry score depict the performance quality of pharmacophore model [124] (Fig 2A). Our generated models showed high sensitivity and specificity values; models 1 and 2 resulted in 80% and 75% retrieval of active compounds, respectively. Scores of other statistical parameters were comprised of 80% score of number of active percent of yields (%Y) and G.H scores of 0.7 and 0.65 for models 1 and 2, respectively. Statistical evaluation of QSAR model included goodness of fit i.e. R2 (correlation coefficient) and adjusted R2 (goodness of fit) values (coefficient of determination). The R2 value (0.93) and adjusted R2 (0.84) were close to 1 and standard error value was close to 0 (Table 2). External validation was performed by predicting pIC50 values of training set compounds through generated model and cross-validated against the given activity values (Fig 2B). Observed IC50 values of training set compounds were comparable with the predicted values and residual error values were ≤ 0.5. ANalysis of VAriance (ANoVA) test [125] also verified the model with a p-value of 0.02.
Fig 2. Statistical validation of designed pharmacophore and QSAR models.
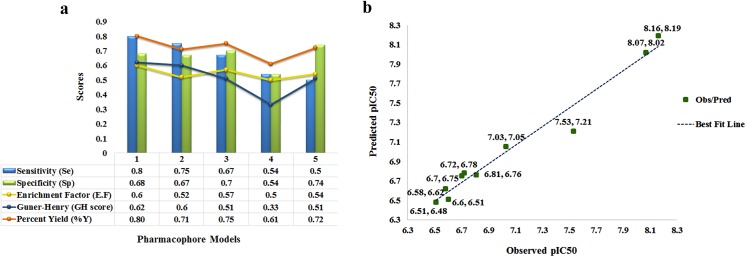
(a) Top five pharmacophore models labeled as Model (1–5). (b) Correlation analysis of QSAR regression model. Green dots depict the observed vs predicted IC50 values and correlation best fit dotted line is shown in blue.
Table 2. Statistical parameters for regression model.
| N | R | R2 | Adjusted R2 | Std. Error | Significance |
|---|---|---|---|---|---|
| 10 | 0.96 | 0.93 | 0.84 | 0.24 | 0.02 |
Virtual screening
Pharmacophore model was incorporated to the virtual screening of compound libraries and about 2000 hits were screened out with high to low query fit values. 81 top scoring hits were selected for docking and drug-like analysis. 16 novel hits (1–16 in Table H in S1 File) exhibited interactions with specified binding sites of class I HDACs. These novel compounds were further exploited for the analysis of steric and physiochemical properties (Table I in S1 File) and 10 compounds (C1-C10) were identified as potential inhibitors for class I HDACs. The 2D structures and binding profile of these compounds are listed in Fig 3 and Table J in S1 File indicates their IUPAC names and SMILE codes. These compounds (C1-C10) exhibited interactions with Zn+2 and binding pocket residues of class I HDACs. C8 and C9 showed class specific binding (Fig 4), while other hits were specific for individual HDACs (Figures B-E in S1 File). Binding pattern of new drug-like compounds coincides with the experimental known binding of existing inhibitors. Bound complexes of SAHA with HDAC2 and Trichostatin A with HDAC8 were shown in Figure F in S1 File which depicted that these inhibitors have similar interaction mode as described for the newly identified inhibitors (Figures B-E in S1 File). Interacting residues and distances of proposed compounds (C1-C10) were also in good agreement to the experimental results.
Fig 3. 2D structures and binding energies of compounds C1-C10.
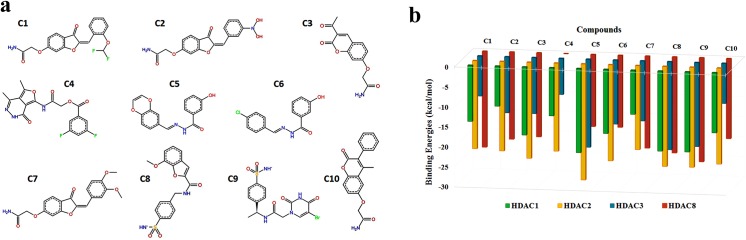
(a) 2D structures (b) Binding energy overview.
Fig 4. Binding pattern of compound C8 with class 1 HDACs.

C8 (7-methoxy-N-((4-sulfamoylphenyl)methyl)-1-benzofuran-2-carboxamide) was shown in blue, whereas hydrogen bonding and hydrophobic residues were shown in pink and yellow, respectively. (a) Hit C8 forms hydrogen bonds with HIS140, HIS148, HIS178, ASP264 and TYR303 of HDAC1; (b) HIS145, HIS146, HIS183 and TYR308 of HDAC2; (c) HIS134, HIS135, HIS172, ASP259 and (d)TYR298 of HDAC3 and HIS142, HIS143, HIS180 and TYR306 of HDAC8. C8 bonding with Zn+2 is shown in green color with an average distance of 2Å. Hydrophobic residues involved in interaction are (a) ASP99, GLY149, PHE150 and GLY301 in HDAC1; (b) PHE155, PHE210, ASP269 and GLY306 in HDAC2; (c) ASP93, PHE144, ASP170, PHE209 and GLY296 in HDAC3 and (d) PHE152, PHE208, MET274 and GLY304 in HDAC8.
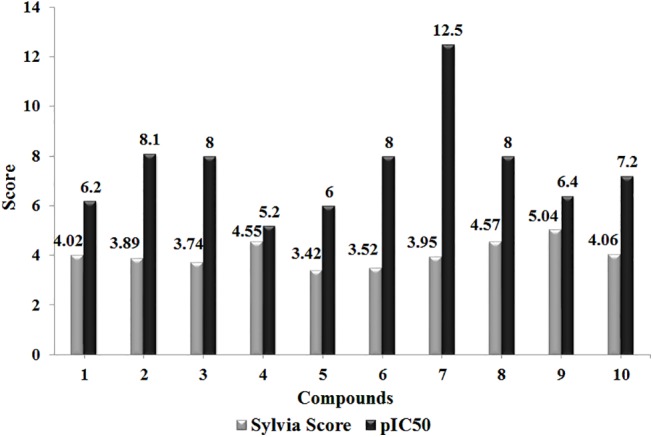
ADMET analysis, binding contributions and physiochemical properties suggested that predicted hits (C1-C10) (Table 3) may act as more potent inhibitors against class I HDACs. Synthetic accessibility scores (<5) of these hits (Fig 5) further validated these hits. The expected pIC50 values of these hits were predicted by QSAR model (Eq 1) and compared with the known pIC50 values of training set compounds. As evident, hit C7 (pIC50 = 12.5) is the most bioactive compound (Fig 5).
Table 3. ADME and Toxicity Analysis.
| Compounds | Human Intestinal Absorption | CYP450 Inhibition | Blood Brain Barrier | Bioavailability | No Mutagenic | No Tumorigenic | No Irritant |
|---|---|---|---|---|---|---|---|
| C1 | 1 | 0.72 | 0.964 | + | + | + | + |
| C2 | 0.8 | 0.6 | 0.7 | + | + | + | + |
| C3 | 0.9 | 0.8 | 0.7 | + | + | + | + |
| C4 | 0.9 | 0.6 | 0.7 | + | + | + | + |
| C5 | 0.7 | 0.6 | 0.7 | + | + | + | + |
| C6 | 0.9 | 0.8 | 0.9 | + | + | + | + |
| C7 | 1 | 0.8 | 0.7 | + | + | + | + |
| C8 | 1 | 0.8 | 0.8 | + | + | + | + |
| C9 | 0.9 | 0.8 | 0.6 | + | + | + | + |
| C10 | 1 | 0.7 | 0.6 | + | + | + | + |
Fig 5. Synthetic accessibility score and expected pIC50 values.
Recently, the properties like ligand efficiency (LE) or binding energy of ligand per atom [126–128] and lipophilic efficiency (LipE or LLE) [129, 130] have been considered essential for lead optimization [131,132], which rely on both potency and lipophilicity profiles. LE is the ratio of free energy of binding to the number of heavy atoms and was calculated by assuming the standard conditions of aqueous solution i.e. 300K, neutral pH and remaining concentrations of 1M (Eq 4) [133]. To access the lipophilicity of predicted hits, logP (activity/size) values were calculated (Table 4) using Bio-Loom version 1.5 [134] and compared with pIC50 values. LE (Eq 4) [135,133] and LipE (Eq 5) [129] profiles of inhibitors were used to identify the hits with higher activities (Table 4).
Table 4. Ligand efficiency (LE), Lipophilic Efficiency (LipE) and Fit Quality (FQ) values of C1-C10 hits.
| Compounds | pIC50 | HA | clogP | LE | LEScale | LipE | FQ |
|---|---|---|---|---|---|---|---|
| C1 | 6.2 | 25 | 2.94 | 0.34 | 0.36 | 3.26 | 0.95 |
| C2 | 8.1 | 25 | 0.81 | 0.44 | 0.36 | 7.29 | 1.22 |
| C3 | 8 | 19 | 0.27 | 0.58 | 0.43 | 7.73 | 1.35 |
| C4 | 5.2 | 27 | 1.49 | 0.26 | 0.34 | 3.71 | 0.76 |
| C5 | 6 | 22 | 4.11 | 0.37 | 0.29 | 1.89 | 1.28 |
| C6 | 8 | 19 | 3.38 | 0.58 | 0.43 | 4.62 | 1.35 |
| C7 | 12.5 | 26 | 2.23 | 0.66 | 0.35 | 10.27 | 1.89 |
| C8 | 8 | 25 | 1.7 | 0.44 | 0.36 | 6.3 | 1.22 |
| C9 | 6.4 | 25 | -0.2 | 0.35 | 0.36 | 6.6 | 0.97 |
| C10 | 7.2 | 23 | 2.5 | 0.43 | 0.38 | 4.7 | 1.13 |
| (4) |
| (5) |
Size-independent ligand efficiency values (LEScale) of these hits were calculated through fitting the top LE values with heavy atom count through a simple exponential function (Eq 6), as described [127,128]. “Fit Quality” or “FQ” scoring function (Eq 7) is the ratio of LE and LEScale which was computed to detect the optimal ligand binding properties of predicted hits.
| (6) |
| (7) |
Conventionally, clogP <3, LipE > 5 and FQ close to 1 are considered as optimal properties for highly bioactive compounds [127–129]. Clearly, clogP values of C1, C2, C3, C4, C7, C8 and C10 hits ranged in 0.2 to 2.5 (Fig 6A), while about 50% compounds exhibited LipE > 5 (Table 4). Similarly, FQ scores of these compounds were also in acceptable range (Fig 6B), indicating that proposed hits may have better in vivo performance based on their ligand binding, potency and lipophilicity profiles.
Fig 6. Ligand efficiency analysis of 10 selected compounds (C1-C10). (a) ClogP and (b) Fit Quality graph.

Molecular dynamics simulation analysis
To elucidate the dynamic behavior of class I HDACs upon binding to inhibitor and to gauge the pattern of system stability, HDAC2 and HDAC8 were subjected to molecular dynamics (MD) simulations. The stability of secondary structure elements and conformational changes were assessed by computing root mean square deviation (RMSD) and root mean square fluctuation (RMSF) plots of values obtained throughout MD trajectories. RMSD is a measure of stable interaction pattern of docked complex. In case of HDAC2, RMSD trend remained stable for both bound and unbound complexes within the range of 1.5-2Å (Fig 7A). However, In case of apo HDAC8, an increasing trend of RMSD profile (1.5Å to 2.8Å) was observed between 2–4 ns, while in its bound form with C8, system attained lower RMSD values in a range of 1.5Å- 2.2Å (Fig 7B). Our analysis indicated that backbone RMSD profiles of HDAC2 and HDAC8 systems remained stable during 12 ns MD runs.
Fig 7. RMSD plot for 12 ns MD simulation.
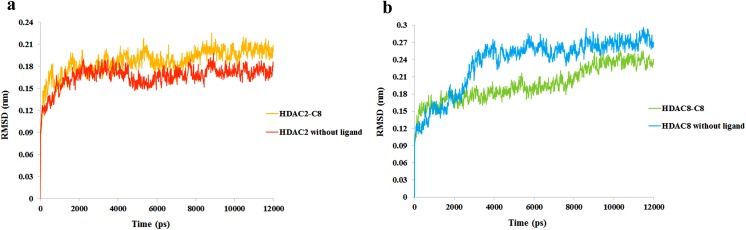
(a) RMSD plot for HDAC2-C8 complex (yellow) and HDAC2 without ligand (red). (b) RMSD plot for HDAC8-C8 complex (green) and HDAC8 without ligand (blue).
RMSF plots provided insight into the residual fluctuations upon binding to inhibitor (Fig 8). In HDAC2 bound to C8, higher RMSF values were observed for ILE40-THR43 (2–2.7Å), PHE210 (0.8-2Å) and LEU333 (1.5-2Å) residues (Fig 8A), while residues involved in inhibitor binding namely, HIS145, HIS146, ASP181, HIS183, ASP269 and TYR308 were quite stable. In HDAC8-C8 system, fluctuations were more common in ASP87, ASP88, SER193-SER199 and GLY320 residues. However, RMSF profiles of binding site and Zn+2 coordinated residues (HIS142, HIS143, ASP178, HIS180, ASP267 and TYR306) exhibited a lower trend (Fig 8B). The observed conformational changes occurring in the proximal residues of HDAC2 and HDAC8 binding sites induced more flexibility to accommodate the inhibitor.
Fig 8. RMSF plot for 12 ns MD simulation.
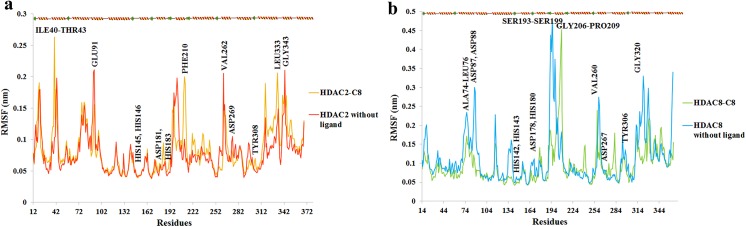
(a) RMSF plot for HDAC2-C8 complex (yellow) and HDAC2 without ligand (red). (b) RMSF plot for HDAC8-C8 complex (green) and HDAC8 without ligand (blue).
The potential energy of system is a measure of its stability. By plotting potential energy as a function of time, we observed that systems were well equilibrated and remained stable throughout MD simulations. The HDAC2-C8 (-74100 kcal/mol) system has shown lower potential energy values compared to HDAC8-C8 (-727500 kcal/mol) complex (Fig 9). The binding characteristics of HDAC2 and HDAC8 with C8 were analyzed through plotting time-dependent intermolecular hydrogen bonds. Compared to HDAC8-C8 system, the intermolecular hydrogen bonds were increased in HDAC2-C8 complex after 2 ns of simulation time, indicating higher interactions (Fig 10). Overall, hydrogen bond interactions remained stable throughout the simulation time. These results substantiated that C8 exhibited more stable binding to HDAC2 compared to HDAC8, which is in good agreement to the distribution of RMSD distances of trajectory conformation pairs. The lower mean RMS values in case of HDAC2-C8 complex displayed the stable nature of system compared to apo HDAC2, while in C8 bound and unbound HDAC8, the pattern of RMS distribution was quite similar (Fig 11). Analysis of radius of gyration (Rg) together with RMSD profile revealed a convergence of Rg values between 1.88 nm to 1.91 nm for HDAC2-C8 complex (Fig 12A). In case of apo HDAC8, Rg trend was quite different compared to its bound form (Fig 12B). These data indicated that structural transitions in HDAC2 resulted in less tight packing, while in HDAC8, binding of C8 induced more compaction in the structure.
Fig 9. Energy plot for 12 ns MD simulation.
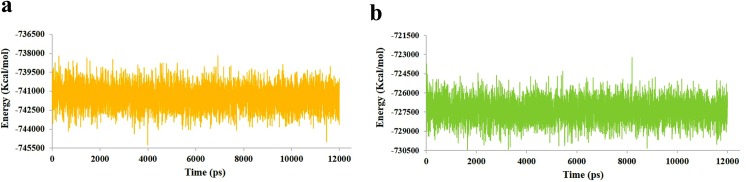
(a) Energy plot for HDAC2-C8 complex (yellow). (b) Energy plot for HDAC8-C8 complex (green).
Fig 10. Hydrogen bonds plot for 12 ns MD simulation.
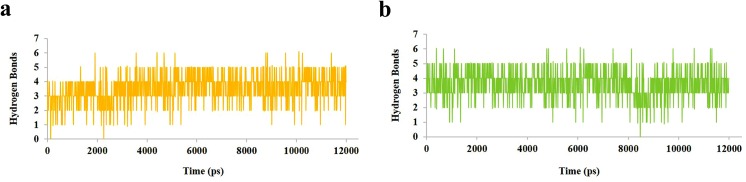
(a) Hydrogen bonds for HDAC2-C8 complex (yellow). (b) Hydrogen bonds for HDAC8-C8 complex (green).
Fig 11. RMS distribution for 12 ns MD simulation.
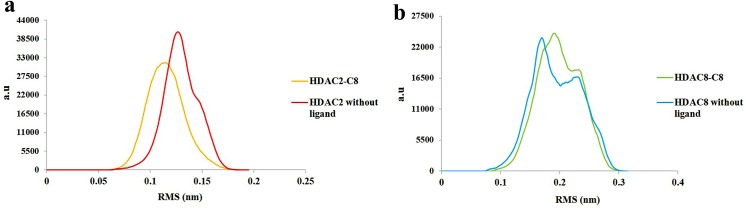
(a) RMS distribution pattern for HDAC2-C8 complex (yellow) and HDAC2 without ligand (red). (b) RMS distribution for HDAC8-C8 complex (green) and HDAC8 without ligand (blue).
Fig 12. Radius of gyration (Rg) analysis for 12 ns MD simulation.
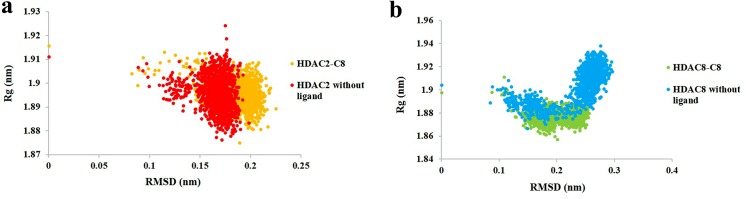
(a) Rg/RMSD plot for HDAC2-C8 complex (yellow) and HDAC2 without ligand (red). (b) Rg/RMSD for HDAC8-C8 complex (green) and HDAC8 without ligand (blue).
Discussion
Ligand-based pharmacophore modeling, based on the energetic binding values of inhibitors (e-pharmacophore) is considered as effective tool in rational drug design [53]. Here we retrieved a diverse set of inhibitors for class I HDACs by generating pharmacophore/QSAR models based on QSAR properties of MS-275, LBH-589, LAQ-824, Trichostatin A, Saha, Belinostat, Oxamflatin, Pyroxamide, Mocetinostat, and Scriptaid. The quality of our selected model (R2 value of 0.93) was quite high, compared to previously proposed pharmacophore/QSAR models for screening HDAC inhibitors [49–51]. Subsequently, 10 novel hit compounds (C1-C10) were determined through their query fit values and pharmacophoric features they possess. Our current protocol of pharmacophore/QSAR-based virtual screening was quite efficient in predicting the reliable inhibitors as it was based on a subset of selective descriptors which were based on known scaffolds.
Through docking analysis, the candidate hits with binding affinities for all members of class I HDACs were selected for detailed analysis. The structural features of predicted hits were much similar by having a capping region, a linker and a Zn+2 ion binding region. The predicted hits were derivatives of benzamides, acetamides, carboxamides and hydrazides. Out of these hits, C8 (7-methoxy-N-[(4-sulfamoylphenyl)methyl]-1-benzofuran-2-carboxamide) and C9 2-(5-bromo-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-N-[1-(4-sulfamoylphenyl)ethyl]acetamide showed ideal binding energetics for HDAC1, 2, 3 and 8 (Fig 3). In C8, carboxamide moiety was involved in hydrogen bonding with Zn+2 metal ion as well as other active site residues. Due to possessing Zn+2-binding amide moieties and stereo selectivity, carboxamides are considered as more potent inhibitors for HDACs [136,137]. Recent experimental evidences support benzofuran-2-carboxamide derivatives for their antitumor and anti-proliferative properties [138]. Similarly, various benzofuran-2-carboxylic acids bearing (chlorometyl) indoline or benzoyl nitrogen as DNA-binding group serve as structural subunits of synthetic analogues of natural antitumor agents such as dystamycin, CC-1065, duocarmycin, and netropsin [139–141]. They also act as adenosine A2A receptor antagonists [142]. Based on the essential value of carboxamides, hit C8 was analyzed in detail for its binding to class I HDAC isoforms. In the catalytic pockets of HDACs, sulfamoylphenyl ring of C8 actively participated in the hydrogen bonding with HIS140, HIS148, HIS178, ASP264 and TYR303 residues of HDAC1; HIS145, HIS146, HIS183 and TYR308 of HDAC2; HIS134, HIS135, HIS172, ASP259 and TYR298 of HDAC3 and HIS142, HIS143, HIS180 and TYR306 of HDAC8 (Fig 4). These interactions were mainly mediated by the conformational readjustments of linker region, while benzofuran ring was uniquely involved in hydrophobic interactions. Consequently, the O-atoms of SO2 coordinated with metal ion in HDAC hydrophobic pockets. Indeed, study of hydrogen bond network between catalytic metal ion and sulfonamide group may be useful to synthesize more potent inhibitors for HDACs.
The proposed aromatic/heterocyclic compounds possessed isoform-specific interactions. For example, crystal structure of HDAC1 exhibited binding with C3, C5 and C6 hits, HDAC2 showed binding with C1-C10, HDAC3 with C5, C6 and C7, while HDAC8 was complexed to C1, C2, C3, C7 and C10. Interestingly, C1, C4, C6 and C9 compounds exhibited halogens as capping groups which indicated that these inhibitors may prefer class I HDACs for binding. The importance of halogenated capping in the selectivity of inhibitors has been well documented. In a series of aroyl pyrrolyl hydroxamide (APHA) compounds screened against maize HD1-B and HD1-A (homologues of mammalian class I and class II HDACs), non-halogenated derivatives showed no selectivity for class I or II [143]. Modification of the capping group region led to 176-fold selectivity for class I HDAC over II. These data underscore that halogens impart class selectivity. However, further analysis is warranted to understand the significance of capping region in parallel to variations in the choice of linker region and metal binding groups in HDACi selectivity.
Moreover, compounds with conserved scaffolds exhibited the comparable binding pattern and energetics. Here, C1, C2 and C7 analogues (group I) possessed similar linker and Zn+2 ion binding regions with distinguished capping region substituents i.e. difluoromethoxy, carbamoylmethoxy and dimethoxyphenyl, respectively (Fig 3A). Likewise, compounds C5 and C6 (group II) were distinct only in the capping site where the benzodioxin in C5 was replaced by chlorophenyl in C6. (Fig 3A). Energy profiles of group I analogues (C1, C2 and C7) against HDAC isoforms depicted more selectivity for HDAC2 and HDAC8 (Fig 3B). These acetamides derivatives showed similar binding patterns at Zn+2 containing pockets of HDAC2 and 8 (Figures G and H in S1 File), while at the outward surface of binding tunnel, only C1 and C2 showed binding to the PHE (PHE210 of HDAC2 and PHE208 of HDAC8) and TYR (TYR308 of HDAC2 and TYR306 of HDAC8) residues (Figures G and H in S1 File). Similarly, group II (C5 and C6) hydrazides showed binding selectivity against HDAC1, 2 and 3 (Fig 3B). Metal binding regions of C5 and C6 hits formed hydrogen bonds with active site residues of HDAC enzymes, whereas unlike C6, C5 exhibited binding of benzodioxin with HIS178, HIS183, HIS172 of HDAC1, 2 and 3, respectively (Figures I—K in S1 File). Furthermore, predicted bioactivity analysis of these compounds also showed variability where C7 (group I) having dimethoxyphenyl group and C6 (group II) with chlorophenyl substituent exhibited the highest values (12.5 and 8), respectively. (Fig 5).
MD simulations of C8 with HDAC2 and HDAC8 impressively illustrated the conformational readjustments in the corresponding catalytic sites. Generally, certain conformational changes take place in the vicinity of HDAC catalytic sites to accommodate the inhibitor [144,145]. Binding of C8 was mediated by coordination with metal ion and hydrogen bond acceptors such as HIS145, HIS146, HIS183 and TYR308 in HDAC2 and HIS142, HIS143, HIS180 and TYR306 in HDAC8. These residues showed no structural dynamics during the inhibitor binding. Notably, hydrophobic residues namely ILE40-THR43, PHE210 and LEU333 exhibited more fluctuations in HDAC2 bound to C8 hit. Particularly, PHE210 exhibited the highest peak (0.8-2Å) in RMSF plot. This residue is conserved across the class I HDACs. Previously, an active contribution of PHE210 was detected in the binding of NSC746457 [146] and YF479 inhibitor [147] to HDAC2. Moreover, its involvement is also visible in TSA (Trichostatin A)-HDAC2 crystal structure [148]. The consistent trend in residual conformations suggested an active contribution of PHE210 in the induction of inhibitor binding to HDAC2. In HDAC8, more pronounced fluctuations were observed in ASP87, ASP88, SER193-SER199 and GLY320 residues. However, the residues holding the Zn+2 ion in the catalytic sites of HDAC2 (ASP181, HIS183 and ASP269) and HDAC8 (HIS142, HIS143, ASP178, HIS180, ASP267 and TYR306) were quite stable.
With these structural insights, our nominated drug-like compounds may prove to be operational in combinatorial cancer therapy (Fig 13). Further studies are needed to delineate the inhibitory effects of proposed inhibitors through in-vitro and in-vivo assays.
Fig 13. Disease association and inhibition mechanism of class I HDACs.

Over expression of HDACs has been perceived in numerous cancers. Binding of drug-like molecules to the catalytic cavity of HDACs eradicates these enzymes from transcription initiation site and stimulates the inclusion of HATs. Acetylation of histones by means of HATs reactivates the transcription of tumor suppressor genes. Expression of these genes controls the abnormal cell growth by cell cycle arrest and mitotic cell death or escalation of apoptosis and autophagy. Tumor suppressor genes enhance the sensitivity of chemotherapy and restrict the process of angiogenesis by decreasing VEGF.
Conclusions
We employed QSAR approach to generate models of 10 chemical compounds (C1-C10) which were derivatives of benzamides, acetamides, carboxamides and hydrazides. Pharmacophore models demonstrated the improved values which stated the higher predictability and reliability of screened hits. The predicted hits were tested for their inhibitory effects through docking and MD simulation assays. C8 and C9 targeted all four members of class I HDACs, whereas other compounds showed member specific interactions. The associated conformational changes in the close proximity of binding regions assisted in the interaction of C8 to HDAC2 and HDAC8. RMSD trends for HDAC2 were much similar in both bound and unbound forms, while HDAC8 bound to C8 attained more stability than apo HDAC8. On the basis of in-silico binding analysis, these hits may prove as more potent drugs against class I HDACs than previously known analogs to reverse the disequilibrium of acetylation and deacetylation events.
Supporting Information
(PDF)
Acknowledgments
We acknowledge members of Functional Informatics Lab, National Center for Bioinformatics for their indispensable support and encouragement.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors have no support or funding to report. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Legube G, Trouche D. Regulating histone acetyltransferases and deacetylases. EMBO Rep. 2003;4(10): 944–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hayes JJ, Tullius TD, Wolffe AP. The structure of DNA in a nucleosome. Proc Natl Acad Sci. 1990;87(19): 7405–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arents G, Moudrianakis EN. Topography of the histone octamer surface: repeating structural motifs utilized in the docking of nucleosomal DNA. Proc Natl Acad Sci. 1993;90(22): 10489–10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gregory PD, Wagner K, Hörz W. Histone acetylation and chromatin remodeling. Exp Cell Res. 2001;265(2): 195–202. [DOI] [PubMed] [Google Scholar]
- 5. Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72(1): 73–84. [DOI] [PubMed] [Google Scholar]
- 6. Norton VG, Imai BS, Yau P, Bradbury EM. Histone acetylation reduces nucleosome core particle linking number change. Cell. 1989;57(3): 449–457. [DOI] [PubMed] [Google Scholar]
- 7. Vettese-Dadey M, Grant P, Hebbes T, Crane-Robinson C, Allis C, Workman J. Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. The EMBO J. 1996;15(10): 2508 [PMC free article] [PubMed] [Google Scholar]
- 8. Sidoli S, Cheng L, Jensen ON. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 2012;75(12): 3419–3433. 10.1016/j.jprot.2011.12.029 [DOI] [PubMed] [Google Scholar]
- 9. Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143): 407–412. [DOI] [PubMed] [Google Scholar]
- 10. Baradari V, Huether A, Höpfner M, Schuppan D, Scherübl H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr-Relat Cancer. 2006;13(4): 1237–1250. [DOI] [PubMed] [Google Scholar]
- 11. Hahn CK, Ross KN, Warrington IM, Mazitschek R, Kanegai CM, Wright RD, et al. Expression-based screening identifies the combination of histone deacetylase inhibitors and retinoids for neuroblastoma differentiation. Proc Natl Acad Sci. 2008;105(28): 9751–9756. 10.1073/pnas.0710413105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol. 2012;52: 303–319. [DOI] [PubMed] [Google Scholar]
- 13. Thomas EA. Focal nature of neurological disorders necessitates isotype-selective histone deacetylase (HDAC) inhibitors. Mol Neurobiol. 2009;40(1): 33–45. 10.1007/s12035-009-8067-y [DOI] [PubMed] [Google Scholar]
- 14. De Ruijter A, Van Gennip A, Caron H, Kemp S, van Kuilenburg A. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gregoretti I, Lee Y-M, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338(1): 17–31. [DOI] [PubMed] [Google Scholar]
- 16. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1): 38–51. [DOI] [PubMed] [Google Scholar]
- 17. Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30(1): 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17(99): 1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, et al. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51(1): 57–67. 10.1016/j.molcel.2013.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res. 2010;62(1): 18–34. 10.1016/j.phrs.2010.02.010 [DOI] [PubMed] [Google Scholar]
- 21. Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1(1): 19–25. 10.1016/j.molonc.2007.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. The Prostate. 2004;59(2): 177–189. [DOI] [PubMed] [Google Scholar]
- 23. Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ, et al. Expression profile of histone deacetylase 1 in gastric cancer tissues. Cancer Sci. 2001;92(12): 1300–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res Tr. 2005;94(1): 11–16. [DOI] [PubMed] [Google Scholar]
- 25. Wilson AJ, Byun D-S, Popova N, Murray LB, L'Italien K, Sowa Y, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281(19): 13548–13558. [DOI] [PubMed] [Google Scholar]
- 26. Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. Apmis. 2005;113(4): 264–268. [DOI] [PubMed] [Google Scholar]
- 27. Huang B, Laban M, Leung CH, Lee L, Lee C, Salto-Tellez M, et al. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12(4): 395–404. [DOI] [PubMed] [Google Scholar]
- 28. Zhu P, Martin E, Mengwasser J, Schlag P, Janssen K-P, Göttlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer cell. 2004;5(5): 455–463. [DOI] [PubMed] [Google Scholar]
- 29. Hayashi A, Horiuchi A, Kikuchi N, Hayashi T, Fuseya C, Suzuki A, et al. Type‐specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E‐cadherin. Int J Cancer. 2010;127(6): 1332–1346. 10.1002/ijc.25151 [DOI] [PubMed] [Google Scholar]
- 30. Fenrick R, Hiebert SW. Role of histone deacetylases in acute leukemia. J Cell Biochem. 1998;72(S30-31): 194–202. [DOI] [PubMed] [Google Scholar]
- 31. Mai A, Massa S, Rotili D, Cerbara I, Valente S, Pezzi R, et al. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med Res Rev. 2005;25(3): 261–309. [DOI] [PubMed] [Google Scholar]
- 32. Di Gennaro E, Bruzzese F, Caraglia M, Abruzzese A, Budillon A. Acetylation of proteins as novel target for antitumor therapy: review article. Amino acids. 2004;26(4): 435–441. [DOI] [PubMed] [Google Scholar]
- 33. Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91: 137–168. [DOI] [PubMed] [Google Scholar]
- 34. Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96(2): 293–304. [DOI] [PubMed] [Google Scholar]
- 35. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9): 769–784. [DOI] [PubMed] [Google Scholar]
- 36. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27(32): 5459–5468. 10.1200/JCO.2009.22.1291 [DOI] [PubMed] [Google Scholar]
- 37. Monneret C. Histone deacetylase inhibitors. Eur J Med Chem. 2005;40(1): 1–13. [DOI] [PubMed] [Google Scholar]
- 38. Pan L, Lu J, Huang B. HDAC inhibitors: a potential new category of anti-tumor agents. Cell Mol Immunol. 2007;4(5): 337–343. [PubMed] [Google Scholar]
- 39. Schaefer EW, Loaiza-Bonilla A, Juckett M, DiPersio JF, Roy V, Slack J, et al. A phase 2 study of vorinostat in acute myeloid leukemia. haematologica. 2009;94(10): 1375–1382. 10.3324/haematol.2009.009217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Molife L, Attard G, Fong P, Karavasilis V, Reid A, Patterson S, et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). ANN ONCOL. 2010;21(1): 109–113. 10.1093/annonc/mdp270 [DOI] [PubMed] [Google Scholar]
- 41. Mackay HJ, Hirte H, Colgan T, Covens A, MacAlpine K, Grenci P, et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur J Cancer. 2010;46(9): 1573–1579. 10.1016/j.ejca.2010.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1(3–4): 117–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim H-J, Bae S-C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3(2): 166 [PMC free article] [PubMed] [Google Scholar]
- 44. Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong T-C, Prince HM, et al. Panobinostat in patients with relapsed/refractory Hodgkin's lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol. 2012;30(18): 2197–2203. 10.1200/JCO.2011.38.1350 [DOI] [PubMed] [Google Scholar]
- 45. KIM YB, KI SW, YOSNIDA M, HORINOUCHI S. Mechanism of cell cycle arrest caused by histone deacetylase inhibitors in human carcinoma cells. J Antibiot. 2000;53(10): 1191–1200. [DOI] [PubMed] [Google Scholar]
- 46. Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone Deacetylase (HDAC) Inhibitor LBH589 Increases Duration of γ-H2AX Foci and Confines HDAC4 to the Cytoplasm in Irradiated Non–Small Cell Lung Cancer. Cancer Res. 2006;66(23): 11298–11304. [DOI] [PubMed] [Google Scholar]
- 47. Wang D-F, Helquist P, Wiech NL, Wiest O. Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J Med Chem. 2005;48(22): 6936–6947. [DOI] [PubMed] [Google Scholar]
- 48. Vadivelan S, Sinha B, Rambabu G, Boppana K, Jagarlapudi SA. Pharmacophore modeling and virtual screening studies to design some potential histone deacetylase inhibitors as new leads. J Mol Graphics Modell. 2008;26(6): 935–946. [DOI] [PubMed] [Google Scholar]
- 49. Tang H, Wang XS, Huang X-P, Roth BL, Butler KV, Kozikowski AP, et al. Novel inhibitors of human histone deacetylase (HDAC) identified by QSAR modeling of known inhibitors, virtual screening, and experimental validation. J Chem Inf Model. 2009;49(2): 461–476. 10.1021/ci800366f [DOI] [PubMed] [Google Scholar]
- 50. Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Kollias G, Igglessi-Markopoulou O. Predictive QSAR workflow for the in silico identification and screening of novel HDAC inhibitors. Mol Divers. 2009;13(3): 301–311. 10.1007/s11030-009-9115-2 [DOI] [PubMed] [Google Scholar]
- 51. Nair SB, Teli MK, Pradeep H, Rajanikant G. Computational identification of novel histone deacetylase inhibitors by docking based QSAR. Comput Biol Med. 2012;42(6): 697–705. 10.1016/j.compbiomed.2012.04.001 [DOI] [PubMed] [Google Scholar]
- 52. Park H, Kim S, Kim YE, Lim SJ. A Structure‐Based Virtual Screening Approach toward the Discovery of Histone Deacetylase Inhibitors: Identification of Promising Zinc‐Chelating Groups. ChemMedChem. 2010;5(4): 591–597. 10.1002/cmdc.200900500 [DOI] [PubMed] [Google Scholar]
- 53. Kalyaanamoorthy S, Chen Y-PP. Energy based pharmacophore mapping of HDAC inhibitors against class I HDAC enzymes. BBA-Proteins Proteom. 2013;1834(1): 317–328. [DOI] [PubMed] [Google Scholar]
- 54. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28(1): 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13): 1605–1612. [DOI] [PubMed] [Google Scholar]
- 56. Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 2001;29(1): 221–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Remiszewski SW, Sambucetti LC, Atadja P, Bair KW, Cornell WD, Green MA, et al. Inhibitors of human histone deacetylase: synthesis and enzyme and cellular activity of straight chain hydroxamates. J Med Chem. 2002;45(4): 753–757. [DOI] [PubMed] [Google Scholar]
- 58. Kim D-K, Lee JY, Kim J-S, Ryu J-H, Choi J-Y, Lee JW, et al. Synthesis and biological evaluation of 3-(4-substituted-phenyl)-N-hydroxy-2-propenamides, a new class of histone deacetylase inhibitors. J Med Chem. 2003;46(26): 5745–5751. [DOI] [PubMed] [Google Scholar]
- 59. Bouchain G, Leit S, Frechette S, Khalil EA, Lavoie R, Moradei O, et al. Development of potential antitumor agents. Synthesis and biological evaluation of a new set of sulfonamide derivatives as histone deacetylase inhibitors. J Med Chem. 2003;46(5): 820–830. [DOI] [PubMed] [Google Scholar]
- 60. Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004;64(3): 1079–1086. [DOI] [PubMed] [Google Scholar]
- 61. Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem. 2007;50(23): 5543–5546. [DOI] [PubMed] [Google Scholar]
- 62. Witter DJ, Harrington P, Wilson KJ, Chenard M, Fleming JC, Haines B, et al. Optimization of biaryl selective HDAC1&2 inhibitors (SHI-1: 2). Bioorg Med Chem Lett. 2008;18(2): 726–731. [DOI] [PubMed] [Google Scholar]
- 63. Zhou N, Moradei O, Raeppel S, Leit S, Frechette S, Gaudette F, et al. Discovery of N-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino) methyl] benzamide (MGCD0103), an orally active histone deacetylase inhibitor. J Med Chem. 2008;51(14): 4072–4075. 10.1021/jm800251w [DOI] [PubMed] [Google Scholar]
- 64. Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, et al. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett. 2008;18(6): 1814–1819. 10.1016/j.bmcl.2008.02.025 [DOI] [PubMed] [Google Scholar]
- 65. Kinzel O, Llauger-Bufi L, Pescatore G, Rowley M, Schultz-Fademrecht C, Monteagudo E, et al. Discovery of a potent class I selective ketone histone deacetylase inhibitor with antitumor activity in vivo and optimized pharmacokinetic properties. J Med Chem. 2009;52(11): 3453–3456. 10.1021/jm9004303 [DOI] [PubMed] [Google Scholar]
- 66. Olsen CA, Ghadiri MR. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic α3β-tetrapeptides. J Med Chem. 2009;52(23): 7836–7846. 10.1021/jm900850t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6(1): 25–33. 10.1038/nchembio.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Auzzas L, Larsson A, Matera R, Baraldi A, Deschenes-Simard Bt, Giannini G, et al. Non-natural macrocyclic inhibitors of histone deacetylases: design, synthesis, and activity. J Med Chem. 2010;53(23): 8387–8399. 10.1021/jm101092u [DOI] [PubMed] [Google Scholar]
- 69. Vaidya AS, Karumudi B, Mendonca E, Madriaga A, Abdelkarim H, van Breemen RB, et al. Design, synthesis, modeling, biological evaluation and photoaffinity labeling studies of novel series of photoreactive benzamide probes for histone deacetylase 2. Bioorg Med Chem Lett. 2012;22(15): 5025–5030. 10.1016/j.bmcl.2012.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hirata Y, Hirata M, Kawaratani Y, Shibano M, Taniguchi M, Yasuda M, et al. Anti-tumor activity of new orally bioavailable 2-amino-5-(thiophen-2-yl) benzamide-series histone deacetylase inhibitors, possessing an aqueous soluble functional group as a surface recognition domain. Bioorg Med Chem Lett. 2012;22(5): 1926–1930. 10.1016/j.bmcl.2012.01.053 [DOI] [PubMed] [Google Scholar]
- 71. Olson DE, Wagner FF, Kaya T, Gale JP, Aidoud N, Davoine EL, et al. Discovery of the first histone deacetylase 6/8 dual inhibitors. J Med Chem. 2013;56(11): 4816–4820. 10.1021/jm400390r [DOI] [PubMed] [Google Scholar]
- 72. Feng T, Wang H, Su H, Lu H, Yu L, Zhang X, et al. Novel N-hydroxyfurylacrylamide-based histone deacetylase (HDAC) inhibitors with branched CAP group (Part 2). Bioorgan Med Chem. 2013;21(17): 5339–5354. [DOI] [PubMed] [Google Scholar]
- 73. Kalin JH, Bergman JA. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J Med Chem. 2013;56(16): 6297–6313. 10.1021/jm4001659 [DOI] [PubMed] [Google Scholar]
- 74. Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(D1): D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Swain M. Chemicalize. org. J Chem Inf Model. 2012;52(2): 613–615. [Google Scholar]
- 76.Cheminformatics M. Molinspiration. 2013.
- 77. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2): 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Summers JB, Kim KH, Mazdiyasni H, Holms JH, Ratajczyk JD, Stewart AO, et al. Hydroxamic acid inhibitors of 5-lipoxygenase: quantitative structure-activity relationships. J Med Chem. 1990;33(3): 992–998. [DOI] [PubMed] [Google Scholar]
- 79. Wu TY, Hassig C, Wu Y, Ding S, Schultz PG. Design, synthesis, and activity of HDAC inhibitors with a N-formyl hydroxylamine head group. Bioorg Med Chem Lett. 2004;14(2): 449–453. [DOI] [PubMed] [Google Scholar]
- 80. Jones P, Altamura S, Chakravarty PK, Cecchetti O, De Francesco R, Gallinari P, et al. A series of novel, potent, and selective histone deacetylase inhibitors. Bioorg Med Chem Lett. 2006;16(23): 5948–5952. [DOI] [PubMed] [Google Scholar]
- 81. Mahboobi S, Sellmer A, Höcher H, Garhammer C, Pongratz H, Maier T, et al. 2-Aroylindoles and 2-aroylbenzofurans with N-hydroxyacrylamide substructures as a novel series of rationally designed histone deacetylase inhibitors. J Med Chem. 2007;50(18): 4405–4418. [DOI] [PubMed] [Google Scholar]
- 82. Kozikowski AP, Chen Y, Gaysin A, Chen B, D'Annibale MA, Suto CM, et al. Functional differences in epigenetic modulators superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J Med Chem. 2007;50(13): 3054–3061. [DOI] [PubMed] [Google Scholar]
- 83. Methot JL, Chakravarty PK, Chenard M, Close J, Cruz JC, Dahlberg WK, et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1: 2). Bioorg Med Chem Lett. 2008;18(3): 973–978. 10.1016/j.bmcl.2007.12.031 [DOI] [PubMed] [Google Scholar]
- 84. Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J Med Chem. 2008;51(15): 4370–4373. 10.1021/jm8002894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen Y, Lopez-Sanchez M, Savoy DN, Billadeau DD, Dow GS, Kozikowski AP. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J Med Chem. 2008;51(12): 3437–3448. 10.1021/jm701606b [DOI] [PubMed] [Google Scholar]
- 86. Tapadar S, He R, Luchini DN, Billadeau DD, Kozikowski AP. Isoxazole moiety in the linker region of HDAC inhibitors adjacent to the Zn-chelating group: effects on HDAC biology and antiproliferative activity. Bioorg Med Chem Lett. 2009;19(11): 3023–3026. 10.1016/j.bmcl.2009.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dallavalle S, Cincinelli R, Nannei R, Merlini L, Morini G, Penco S, et al. Design, synthesis, and evaluation of biphenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Eur J Med Chem. 2009;44(5): 1900–1912. 10.1016/j.ejmech.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 88. Gupta PK, Reid RC, Liu L, Lucke AJ, Broomfield SA, Andrews MR, et al. Inhibitors selective for HDAC6 in enzymes and cells. Bioorg Med Chem Lett. 2010;20(23): 7067–7070. 10.1016/j.bmcl.2010.09.100 [DOI] [PubMed] [Google Scholar]
- 89. He R, Chen Y, Chen Y, Ougolkov AV, Zhang J-S, Savoy DN, et al. Synthesis and biological evaluation of triazol-4-ylphenyl-bearing histone deacetylase inhibitors as anticancer agents. J Med Chem. 2010;53(3): 1347–1356. 10.1021/jm901667k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cho YS, Whitehead L, Li J, Chen CH-T, Jiang L, Vögtle M, et al. Conformational refinement of hydroxamate-based histone deacetylase inhibitors and exploration of 3-piperidin-3-ylindole analogues of dacinostat (LAQ824). J Med Chem. 2010;53(7): 2952–2963. 10.1021/jm100007m [DOI] [PubMed] [Google Scholar]
- 91. Oger F, Lecorgne A, Sala E, Nardese V, Demay F, Chevance S, et al. Biological and biophysical properties of the histone deacetylase inhibitor suberoylanilide hydroxamic acid are affected by the presence of short alkyl groups on the phenyl ring. J Med Chem. 2010;53(5): 1937–1950. 10.1021/jm901561u [DOI] [PubMed] [Google Scholar]
- 92. Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, et al. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl) benzamides. Bioorg Med Chem Lett. 2010;20(10): 3142–3145. 10.1016/j.bmcl.2010.03.091 [DOI] [PubMed] [Google Scholar]
- 93. Terracciano S, Di Micco S, Bifulco G, Gallinari P, Riccio R, Bruno I. Synthesis and biological activity of cyclotetrapeptide analogues of the natural HDAC inhibitor FR235222. Bioorgan Med Chem. 2010;18(9): 3252–3260. [DOI] [PubMed] [Google Scholar]
- 94. Mahboobi S, Sellmer A, Winkler M, Eichhorn E, Pongratz H, Ciossek T, et al. Novel chimeric histone deacetylase inhibitors: a series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity. J Med Chem. 2010;53(24): 8546–8555. 10.1021/jm100665z [DOI] [PubMed] [Google Scholar]
- 95. Kemp MM, Wang Q, Fuller JH, West N, Martinez NM, Morse EM, et al. A novel HDAC inhibitor with a hydroxy-pyrimidine scaffold. Bioorg Med Chem Lett. 2011;21(14): 4164–4169. 10.1016/j.bmcl.2011.05.098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Botta CB, Cabri W, Cini E, De Cesare L, Fattorusso C, Giannini G, et al. Oxime amides as a novel zinc binding group in histone deacetylase inhibitors: synthesis, biological activity, and computational evaluation. J Med Chem. 2011;54(7): 2165–2182. 10.1021/jm101373a [DOI] [PubMed] [Google Scholar]
- 97. Neelarapu R, Holzle DL, Velaparthi S, Bai H, Brunsteiner M, Blond SY, et al. Design, synthesis, docking, and biological evaluation of novel diazide-containing isoxazole-and pyrazole-based histone deacetylase probes. J Med Chem. 2011;54(13): 4350–4364. 10.1021/jm2001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Huang D, Li X, Wei Y, Xiu Z. A novel series of l-2-benzyloxycarbonylamino-8-(2-pyridyl)-disulfidyloctanoic acid derivatives as histone deacetylase inhibitors: design, synthesis and molecular modeling study. Eur J Med Chem. 2012;52: 111–122. 10.1016/j.ejmech.2012.03.009 [DOI] [PubMed] [Google Scholar]
- 99. Lai M-J, Huang H-L, Pan S-L, Liu Y-M, Peng C-Y, Lee H-Y, et al. Synthesis and biological evaluation of 1-arylsulfonyl-5-(N-hydroxyacrylamide) indoles as potent histone deacetylase inhibitors with antitumor activity in vivo. J Med Chem. 2012;55(8): 3777–3791. 10.1021/jm300197a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wagner FF, Olson DE, Gale JP, Kaya T, Weïwer M, Aidoud N, et al. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J Med Chem. 2013;56(4): 1772–1776. 10.1021/jm301355j [DOI] [PubMed] [Google Scholar]
- 101. Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem. 2003;46(24): 5097–5116. [DOI] [PubMed] [Google Scholar]
- 102. Chen J-B, Chern T-R, Wei T-T, Chen C-C, Lin J-H, Fang J-M. Design and synthesis of dual-action inhibitors targeting histone deacetylases and 3-hydroxy-3-methylglutaryl coenzyme A reductase for cancer treatment. J Med Chem. 2013;56(9): 3645–3655. 10.1021/jm400179b [DOI] [PubMed] [Google Scholar]
- 103. Marson CM, Matthews CJ, Yiannaki E, Atkinson SJ, Soden PE, Shukla L, et al. Discovery of potent, isoform-selective inhibitors of histone deacetylase containing chiral heterocyclic capping groups and a N-(2-aminophenyl) benzamide binding unit. J Med Chem. 2013;56(15): 6156–6174. 10.1021/jm400634n [DOI] [PubMed] [Google Scholar]
- 104. Wang T, Sepulveda M, Gonzales P, Gately S. Identification of novel HDAC inhibitors through cell based screening and their evaluation as potential anticancer agents. Bioorg Med Chem Lett. 2013;23(17): 4790–4793. 10.1016/j.bmcl.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 105. Yu C-W, Chang P-T, Hsin L-W, Chern J-W. Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J Med Chem. 2013;56(17): 6775–6791. 10.1021/jm400564j [DOI] [PubMed] [Google Scholar]
- 106. Blackburn C, Barrett C, Chin J, Garcia K, Gigstad K, Gould A, et al. Potent histone deacetylase inhibitors derived from 4-(aminomethyl)-N-hydroxybenzamide with high selectivity for the HDAC6 isoform. J Med Chem. 2013;56(18): 7201–7211. 10.1021/jm400385r [DOI] [PubMed] [Google Scholar]
- 107. Guerra-Bubb JM, Bowers AA, Smith WB, Paranal R, Estiu G, Wiest O, et al. Synthesis and HDAC inhibitory activity of isosteric thiazoline-oxazole largazole analogs. Bioorg Med Chem Lett. 2013;23(21): 6025–6028. 10.1016/j.bmcl.2013.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhang X, Bao B, Yu X, Tong L, Luo Y, Huang Q, et al. The discovery and optimization of novel dual inhibitors of topoisomerase II and histone deacetylase. Bioorgan Med Chem. 2013;21(22): 6981–6995. [DOI] [PubMed] [Google Scholar]
- 109. Taddei M, Cini E, Giannotti L, Giannini G, Battistuzzi G, Vignola D, et al. Lactam based 7-amino suberoylamide hydroxamic acids as potent HDAC inhibitors. Bioorg Med Chem Lett. 2014;24(1): 61–64. 10.1016/j.bmcl.2013.11.072 [DOI] [PubMed] [Google Scholar]
- 110.ACD/ChemSketch Freeware, Advanced Chemistry Development, Inc., Toronto, ON, Canada, www.acdlabs.com, 2014.
- 111. Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45(1): 160–169. [DOI] [PubMed] [Google Scholar]
- 112. Shahlaei M. Descriptor selection methods in quantitative structure–activity relationship studies: a review study. Chem Rev. 2013;113(10): 8093–8103. 10.1021/cr3004339 [DOI] [PubMed] [Google Scholar]
- 113. Verma J, Khedkar VM, Coutinho EC. 3D-QSAR in drug design-a review. Curr Top Med Chem. 2010;10(1): 95–115. [DOI] [PubMed] [Google Scholar]
- 114.IBM SPSS statistics for windows (2012). Corporation, I. B. M, 22 edn.,
- 115.Aurora Fine Chemicals Ltd–Europe. http://www.aurorafinechemicals.com.
- 116.Princeton Biomolecular Research, Inc. http://www.princetonbio.com.
- 117.Loeffler HH, Winn M. Large biomolecular simulation on hpc platforms III. AMBER, CHARMM, GROMACS, LAMMPS and NAMD. Technical report, STFC Daresbury Laboratory, Warrington WA4 4AD, UK, 2012.
- 118. Bibi N, Parveen Z, Rashid S. Identification of potential Plk1 targets in a cell-cycle specific proteome through structural dynamics of kinase and Polo box-mediated interactions. PloS one. 2013;8(8): e70843 10.1371/journal.pone.0070843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kausar S, Asif M, Bibi N, Rashid S. Correction: Comparative Molecular Docking Analysis of Cytoplasmic Dynein Light Chain DYNLL1 with Pilin to Explore the Molecular Mechanism of Pathogenesis Caused by Pseudomonas aeruginosa PAO. PloS one. 2013;8(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Discovery Studio Modeling Environment (2013). Accelrys Software Inc, 4.0 edn., San Diego: [Google Scholar]
- 121. Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11): 3099–3105. 10.1021/ci300367a [DOI] [PubMed] [Google Scholar]
- 122. Sander T. OSIRIS property explorer Allschwil: Actelion Pharmaceuticals Ltd; 2001. [Google Scholar]
- 123. Boda K, Seidel T, Gasteiger J. Structure and reaction based evaluation of synthetic accessibility. J Comput Aid Mol Des. 2007;21(6): 311–325. [DOI] [PubMed] [Google Scholar]
- 124. Seidel T, Ibis G, Bendix F, Wolber G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov Today: Technol. 2011;7(4): e221–e228. [DOI] [PubMed] [Google Scholar]
- 125. Tropsha A. Best practices for QSAR model development, validation, and exploitation. Mol Inform. 2010;29(6‐7): 476–488. [DOI] [PubMed] [Google Scholar]
- 126. Andrews P, Craik D, Martin J. Functional group contributions to drug-receptor interactions. J Med Chem. 1984;27(12): 1648–1657. [DOI] [PubMed] [Google Scholar]
- 127. Reynolds CH, Bembenek SD, Tounge BA. The role of molecular size in ligand efficiency. Bioorg Med Chem Lett. 2007;17(15): 4258–4261. [DOI] [PubMed] [Google Scholar]
- 128. Reynolds CH, Tounge BA, Bembenek SD. Ligand binding efficiency: trends, physical basis, and implications. J Med Chem. 2008;51(8): 2432–2438. 10.1021/jm701255b [DOI] [PubMed] [Google Scholar]
- 129. Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6(11): 881–890. [DOI] [PubMed] [Google Scholar]
- 130. Ryckmans T, Edwards MP, Horne VA, Correia AM, Owen DR, Thompson LR, et al. Rapid assessment of a novel series of selective CB 2 agonists using parallel synthesis protocols: a lipophilic efficiency (LipE) analysis. Bioorg Med Chem Lett. 2009;19(15): 4406–4409. 10.1016/j.bmcl.2009.05.062 [DOI] [PubMed] [Google Scholar]
- 131. Keserü GM, Makara GM. The influence of lead discovery strategies on the properties of drug candidates. Nat Rev Drug Discov. 2009;8(3): 203–212. 10.1038/nrd2796 [DOI] [PubMed] [Google Scholar]
- 132. Mortenson PN, Murray CW. Assessing the lipophilicity of fragments and early hits. J Comput Aid Mol Des. 2011;25(7): 663–667. [DOI] [PubMed] [Google Scholar]
- 133. Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2): 105–121. 10.1038/nrd4163 [DOI] [PubMed] [Google Scholar]
- 134. Bio-Loom for Windows version 1.5. BioByte Corporation, CA, USA: [Google Scholar]
- 135. Kuntz I, Chen K, Sharp K, Kollman P. The maximal affinity of ligands. Proc Natl Acad Sci. 1999;96(18): 9997–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Giannini G, Vesci L, Battistuzzi G, Vignola D, Milazzo FM, Guglielmi MB, et al. ST7612AA1, a Thioacetate-ω (γ-lactam carboxamide) Derivative Selected from a Novel Generation of Oral HDAC Inhibitors. J Med Chem. 2014;57(20): 8358–8377. 10.1021/jm5008209 [DOI] [PubMed] [Google Scholar]
- 137. Attenni B, Ontoria JM, Cruz JC, Rowley M, Schultz-Fademrecht C, Steinkühler C, et al. Histone deacetylase inhibitors with a primary amide zinc binding group display antitumor activity in xenograft model. Bioorg Med Chem Lett. 2009;19(11): 3081–3084. 10.1016/j.bmcl.2009.04.011 [DOI] [PubMed] [Google Scholar]
- 138. Hranjec M, Sović I, Ratkaj I, Pavlović G, Ilić N, Valjalo L, et al. Antiproliferative potency of novel benzofuran-2-carboxamides on tumour cell lines: cell death mechanisms and determination of crystal structure. Eur J Med Chem. 2013;59: 111–119. 10.1016/j.ejmech.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 139. Baraldi PG, Romagnoli R, Beria I, Cozzi P, Geroni C, Mongelli N, et al. Synthesis and antitumor activity of new benzoheterocyclic derivatives of distamycin A. J Med Chem. 2000;43(14): 2675–2684. [DOI] [PubMed] [Google Scholar]
- 140. Baraldi PG, Romagnoli R, Bianchi N, Gambari R. Benzoyl nitrogen mustard derivatives of benzoheterocyclic analogues of netropsin: synthesis and biological activity. Bioorgan Med Chem. 2003;11(11): 2381–2388. [DOI] [PubMed] [Google Scholar]
- 141. Wang Y, Li L, Ye W, Tian Z, Jiang W, Wang H, et al. CC-1065 analogues bearing different DNA-binding subunits: synthesis, antitumor activity, and preliminary toxicity study. J Med Chem. 2003;46(4): 634–637. [DOI] [PubMed] [Google Scholar]
- 142. Saku O, Saki M, Kurokawa M, Ikeda K, Takizawa T, Uesaka N. Synthetic studies on selective adenosine A 2A receptor antagonists: Synthesis and structure–activity relationships of novel benzofuran derivatives. Bioorg Med Chem Lett. 2010;20(3): 1090–1093. 10.1016/j.bmcl.2009.12.028 [DOI] [PubMed] [Google Scholar]
- 143. Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, et al. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl) pyrrolyl hydroxyamides. J Med Chem. 2005;48(9): 3344–3353. [DOI] [PubMed] [Google Scholar]
- 144. Park H, Lee S. Homology modeling, force field design, and free energy simulation studies to optimize the activities of histone deacetylase inhibitors. J Comput Aid Mol Des. 2004;18(6): 375–388. [DOI] [PubMed] [Google Scholar]
- 145. Yan C, Xiu Z, Li X, Li S, Hao C, Teng H. Comparative molecular dynamics simulations of histone deacetylase‐like protein: Binding modes and free energy analysis to hydroxamic acid inhibitors. Proteins: Struct, Funct, Bioinf. 2008;73(1): 134–149. [DOI] [PubMed] [Google Scholar]
- 146. Hou J, Feng C, Li Z, Fang Q, Wang H, Gu G, et al. Structure-based optimization of click-based histone deacetylase inhibitors. Eur J Med Chem. 2011;46(8): 3190–3200. 10.1016/j.ejmech.2011.04.027 [DOI] [PubMed] [Google Scholar]
- 147. Zhang T, Chen Y, Li J, Yang F, Wu H, Dai F, et al. Antitumor action of a novel histone deacetylase inhibitor, YF479, in breast cancer. Neoplasia. 2014;16(8): 665–677. 10.1016/j.neo.2014.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cronin CN, Hilgers MT, Knuth MW, Navre ME, Sang BC, Skene RJ, et al. Crystallization of histone deacetylase 2. Google Patents; 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.