

Abstract
Streptococcus agalactiae infection causes high mortality in cardiovascular disease (CVD) patients, especially in case of setting prosthetic valve during cardiac surgery. However, the pathogenesis mechanism of S. agalactiae associate with CVD has not been well studied. Here, we have demonstrated the pathogenicity of S. agalactiae in rat cardiomyocytes (H9C2). Interestingly, both live and dead cells of S. agalactiae were uptaken by H9C2 cells. To further dissect the process of S. agalactiae internalization, we chemically inhibited discrete parts of cellular uptake system in H9C2 cells using genistein, chlorpromazine, nocodazole and cytochalasin B. Chemical inhibition of microtubule and actin formation by nocodazole and cytochalasin B impaired S. agalactiae internalization into H9C2 cells. Consistently, reverse‒ transcription PCR (RT‒PCR) and quantitative real time‒PCR (RT-qPCR) analyses also detected higher levels of transcripts for cytoskeleton forming genes, Acta1 and Tubb5 in S. agalactiae‒infected H9C2 cells, suggesting the requirement of functional cytoskeleton in pathogenesis. Host survival assay demonstrated that S. agalactiae internalization induced cytotoxicity in H9C2 cells. S. agalactiae cells grown with benzyl penicillin reduced its ability to internalize and induce cytotoxicity in H9C2 cells, which could be attributed with the removal of surface lipoteichoic acid (LTA) from S. agalactiae. Further, the LTA extracted from S. agalactiae also exhibited dose‒dependent cytotoxicity in H9C2 cells. Taken together, our data suggest that S. agalactiae cells internalized H9C2 cells through energy‒dependent endocytic processes and the LTA of S. agalactiae play major role in host cell internalization and cytotoxicity induction.
Introduction
Group B Streptococci (GBS), or Streptococcus agalactiae is considered as a leading cause of life‒threatening invasive bacterial infections in pregnant women, infants, adults and immuno-compromised individuals [1]. These bacteria are Gram‒positive, β‒hemolytic, chain‒forming cocci that are normal residents of the vaginal microflora of 25% of healthy women. GBS causes cellulitis, arthritis and urinary tract infections and is also considered as the most common causative agent of sepsis and meningitis [2‒4]. The S. agalactiae colonize genital tracts of pregnant women and approximately 50% of newborn babies to these infected mothers are highly susceptible to GBS infection [5, 6].
The molecular events that dictate transition of commensal GBS to invasive pathogens is poorly understood. Successful progression of S. agalactiae infection requires initial adherence to extracellular matrix followed by entry into the host cells and appropriate expression of virulence gene products in response to the host/external environment during infection [7, 2]. S. agalactiae produces several virulence factors including pore‒forming toxins, sialic acid‒rich capsule polysaccharide (CPS), C5a peptidase, hyaluronidase and various surface proteins. A total of nine different capsular polysaccharide have been identified and characterized (Ia, Ib, and II-VIII) from S. agalactiae. Of these, type III serotype is most frequently associated with neonatal infections, whereas other serotypes were known to be involved in infections to adult [5]. S. agalactiae is also an aggressive infective endocarditis (IE) pathogen, which causes valve damage, heart failure, and thromboembolism [8, 9, 10]. S. agalactiae colonization in myocardium may lead to infective myocarditis atherosclerosis. Several case reports are available on S. agalactiae- induced endocarditis in adults, elderly persons and people with chronic immunosuppressive diseases [9, 11–15]. Only fewer reports were available on acute purulent myocarditis caused by S. agalactiae [16–18]. S. agalactiae infection causes high mortality (34–50%) in cardiovascular disease (CVD) patients especially in case of setting prosthetic valve during cardiac surgery [19]. However, there is high risk of S. agalactiae infection to CDV individuals, the mechanisms underlying the colonization and pathogenesis of S. agalactiae in cardiac cells has not been well studied.
In this study, we have investigated the pathogenesis of a S. agalactiae strain isolated from the blood of a valvular heart disease patient [20] in rat cardiomyocytes (H9C2). We found that S. agalactiae was internalized into H9C2 cells through cytoskeleton‒mediated energy-dependent endocytic processes. Further, Lipoteichoic acid, the cell wall component S. agalactiae, play major role in internalization and cytotoxicity induction in H9C2 cells.
Materials and Methods
Bacterial Strains and Growth Conditions
The S. agalactiae CVD001A strain was isolated from the blood of a CVD patient and has been previously identified by 16S rDNA sequencing. The 16S rDNA nucleotide sequence of S. agalactiae CVD001A was available in GenBank under accession number JQ001868.1 [20]. S. gordonii ATCC 12403 was obtained from American Type Culture Collection (ATCC) and was exclusively used as a non‒invasive negative control to show significant differences in the invasion of S. agalactiae into the host H9C2 cells. The Streptococcus sp. and Escherichia coli DH5α strains were grown in Brain Heart Infusion (BHI) and Luria–Bertani (LB) broth, respectively at 37°C overnight with agitation at 200 rpm. Bacterial cells grown to exponential phase were used for infection assays.
Cell Culture
H9C2 cell line, which was originally derived from the embryonic rat ventricle, was obtained from National Centre for Cell Science, Pune, India. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) F12 Ham supplemented with 10% Fetal Bovine Serum (FBS), 100 U ml-1 of penicillin, 100 μg ml-1 streptomycin and 2.5 μg ml-1 of amphotericin B in a humified incubator with 5% CO2 and 37°C. Cardiomyoblast differentiation was induced using the method described by Menard et al. [21]. Briefly, H9C2 cells were seeded in 75 cm2 tissue culture flasks at a density of 3.5 х 104 cells in growth medium. At 50‒60% of confluency, high serum media was replaced by low serum media (1% FBS) and incubated for 4 days in dark with daily supplement of 1 μM All‒Trans‒retinoic acid (ATRA) (Sigma, USA). On 5th day of treatment, differentiated cells were trypsinized with 0.025% trypsin‒EDTA solution and seeded for further experiments.
Bacterial Infection Assay
We evaluated the internalization of fluorescently‒ labeled live and heat‒killed S. agalactiae into H9C2 cells by live cell imaging using High Content Screening (HCS) system (Operetta, Perkin Elmer, USA). The live S. agalactiae cells were stained with 5 μg ml-1 of acridine orange (AO) in phosphate-buffered saline (PBS) for 5 min at room temperature and washed repeatedly with 1 X PBS to remove the residual stain. Heat‒killed cell suspension of S. agalactiae was prepared by incubating the cells at 100°C for 15 min and then the viability of heat‒killed cells were assessed by plating on to BHI agar plate. Heat‒killed S. agalactiae cells were then stained with propidium iodide (PI) at a final concentration of 48 μM for 5 min at room temperature in dark and washed repeatedly with 1 X PBS to remove residual stain. For infection assay, approximately 1 х 104 differentiated H9C2 cells were seeded in a 96‒well view plate black (Perkin Elmer, USA) in growth medium. The H9C2 cells were infected with fluorescently‒labeled live and heat‒killed S. agalactiae at different multiplicity of infection (MOI) viz., 1:10, 1:100 and 1:1000 in antibiotics and serum free media for 2 h. After infection, the cells were washed five times with 1 X PBS and incubated in their respective media containing 10 μg ml-1 of penicillin and 200 μg ml-1 of gentamycin to inhibit the growth of extracellular bacteria. The antibiotic concentration used to inhibit growth of extracellular bacterium was based on the determined minimum inhibitory concentration (MIC) for S. agalactiae and sub‒minimum cytotoxic concentration (Sub-MCC) for H9C2 cells. After incubation, the infected H9C2 cells were washed with 1 X PBS and stained with Hoechst 33342 (5μM) for 30 min at room temperature in dark. Subsequently, the plate was kept in the live cell chamber provided in HCS at 37°C with continuous supply of 5% CO2 and the images were captured immediately. In addition, the internalization of live and heat‒killed S. agalactiae cells into the H9C2 cells were quantified using microtiter plate reader (SpectraMax® M2, Molecular Devices, USA) at excitation wavelength of 502 nm & 535 nm and emission wavelength of 525 nm & 617 nm for AO and PI, respectively. The AO‒stained S. gordonii ATCC 12403 and non‒pathogenic E coli DH5α strain were used as experimental controls for infection assay. All the experiments were performed in triplicates and the mean values were represented with the standard deviation.
Viable Cell Count Analysis
The intracellular colony forming units (CFUs) from S. agalactiae‒infected H9C2 cells was enumerated by viable cell count analysis. H9C2 cells were infected with different inoculum of S. agalactiae (104, 105 and 106 CFU) for 2 h at 37°C in antibiotic free media followed by five times wash with 1 X PBS. The infected H9C2 cells were washed with respective antibiotics in growth medium to remove the extracellular bacteria as described previously. The infected cells were recovered and lysed with 0.1% Triton X-100 (Sigma, USA). The lysate was then transferred to sterile 1.5‒ml microfuge tubes and vortexed vigorously for 10 s to disrupt streptococcal chains and further confirmed by Gram‒staining. Aliquots of the lysates were plated on to BHI agar plates for CFU enumeration. These experiments were performed in triplicates and the mean values were represented with standard deviation.
Endocytic Inhibition Assay
The uptake of fluorescently‒labeled live S. agalactiae by H9C2 cells was studied in the presence/ absence of endocytic inhibitor, sodium azide (NaN3). H9C2 cells were pre‒treated with NaN3 at a final concentration of 10 mM for 1 h prior to the infection. After treatment with NaN3, H9C2 cells were washed repeatedly with 1X PBS for 10 min and suspended in antibiotic free growth medium. The NaN3‒treated and untreated H9C2 cells were infected with S. agalactiae at MOI of 1:100 for 2 h. After infection, the extracellular bacteria were removed and the population of H9C2 cells internalized with S. agalactiae in the presence and absence of endocytic inhibitor, NaN3 were quantified using flow cytometry. Approximately, 50,000 cells were gated on live cells by forward scatter (FSC)/ side scatter (SSC) using fluorescence-activated cell sorter (FACSAria III, Beckton Dickinson, San Jose, CA, USA). The population of H9C2 cells exhibiting green fluorescence for live cell uptake was quantified using FACS at 488 nm employing a blue laser with emission in the 530⁄30 band pass filter. The results obtained were analyzed using FACSDiva version 6.1.3 software package (BD, San Jose, CA, USA).
Characterization of Endocytic Mechanisms
To further dissect the process of S. agalactiae internalization into H9C2 cells, we chemically inhibited discrete part of cellular uptake system in H9C2 cells. The different endocytic chemical inhibitors used in this study were: genistein (50‒200 μM), a inhibitor for tyrosine kinases involved in caveolae‒mediated endocytosis [22]; chlorpromazine (20‒50 μM), an inhibitor of clathrin‒mediated endocytosis [23]; nocodazole (5‒30 μM), a microtubule‒disrupting agent [24, 25] and cytochalasin B (50‒100 μM), an actin‒disrupting agent [26]. For endocytic inhibition assay, H9C2 cell were pre‒treated with each endocytic inhibitors for 30 min at 37°C. For microtubule distruption, the H9C2 cells were pre‒treated with nocodazole for 1 h on ice and then warmed to 37°C for 30 min prior to the assay [27]. All infection assays were performed in the presence of each inhibitor at 37°C for 2 h. H9C2 cells treated with each inhibitor without streptococci were used as experimental control. The effect of each inhibitor on the viability of H9C2 cells were studied by trypan blue exclusion assay to ensure that the inhibitor concentration used in this study did not affect the viability of H9C2 cells. The endocytic inhibition of S. agalactiae in H9C2 cells in presence of different endocytic inhibitors were determined by CFU enumeration. All assays were performed in triplicate and the mean values were represented with standard deviation.
S. agalactiae‒Induced Cytotoxicity in H9C2 Cells
S. agalactiae-induced cytotoxicity in H9C2 cells was determined by confocal microscopy, MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] assay and flow cytometry analyses. H9C2 cells were infected with S. agalactiae for 6 h and morphological changes induced in H9C2 cells by S. agalactiae infection within 6 h was quantified using Ready-made image analysis tool in Operetta compact high content imaging system (PerkinElmer, Massachusetts, USA). For MTT assay, approximately 50,000 H9C2 cells were seeded per well in 12‒well tissue culture plate and infected with S. agalactiae at different MOI (1:10; 1:100 and 1:1000) for 2 h at 37°C. After infection, 20 μL of MTT solution (1 mg ml-1) was added to each well containing the infected population of cells in 200 μL of growth medium (10% final solution) and incubated at room temperature for 4 h. After incubation, the medium was removed and formazan crystals were dissolved in 200 μL of DMSO. Immediately, absorbance was determined spectrophotometrically at 550 and 690 nm in microtiter plate reader (SpectraMax® M2, Molecular Devices, USA). A subtraction analysis of the dual wavelength (D550 to D690) was performed to increase accuracy of cytotoxicity measurement.
Annexin V-FITC detection kit (Calbiochem, USA) was used to detect S. agalactiae‒induced apoptosis in H9C2 cells. Approximately 5 х 104 H9C2 cells were seeded per well in 12‒well tissue culture plate and infected with S. agalactiae at MOI of 1: 100. After infection, H9C2 cells were washed in 1 X PBS and re‒suspended in binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Annexin V-FITC was added to the mixture at a final concentration of 100 ng ml-1 and incubated in dark at 37°C for 10 min. After incubation, the cells were washed with 1 X PBS and re‒suspended in 300 μl of binding buffer. To this, 10 μl of propidium iodide (PI) was added and subjected to flow cytometry analysis according to the manufacturer’s instruction. The results obtained were analyzed using FACSDiva version 6.1.3 software package (BD, San Jose, CA, USA).
RNA Extraction and cDNA Synthesis
H9C2 cells were infected with S. agalactiae at MOI of 1: 100 for 2 h. The total RNA was extracted from the S. agalactiae-infected and uninfected H9C2 cells using the RNeasy minikit (Qiagen, Germany). The isolated RNA was treated with DNase I (5U/μl) at 37°C for 30 min to remove DNA contamination. The cDNA was synthesized from the purified RNA using Revert Aid First‒strand cDNA synthesis kit (Thermo Scientific, USA) according to the manufacturer’s instructions. The synthesized cDNA was used for further experiment.
Semi-Quantitative Reverse Transcription (RT)-PCR
The cytoskeleton forming genes, Acta1 (Actin, alpha 1, skeletal muscle gene) and Tubb5 (tubulin, beta 5 class I) were PCR amplified individually in a 25 μl reaction volume mixture containing 1 X PCR buffer, 2 mM MgCl2, 0.8 mM of each dNTPs, 0.1 μM of respective primers and 0.625 U of Taq DNA polymerase using cDNA as template. Gapdh (glyceraldehyde‒3‒phosphate dehydrogenase) gene was used as the internal control for all experiments. The list of primers used in RT-PCR experiments are shown in Table 1.The PCR reactions were carried out in thermal cycler (Eppendorf, USA), which consisted one step cycle of 94°C, 30 cycles of 94°C for 30 s, 55°C for 1 min and 72°C for 30 s followed by an final extension at 72°C for 5 min. The amplified products were analyzed on 2% agarose gel.
Table 1. List of primers used in the study.
| Gene name & Symbol | Primer Sequences (5’-3’) | |
|---|---|---|
| Semi-quantitative RT-PCR | RT-qPCR | |
| Tubulin, beta 5 class I (Tubb5) | F: CACAGGTGGCAAGTATGTCC R: GGTCTCATCCGTGTTCTAACC | F: CTGGGACTATGGACTCCGTT R: ACCAACTCAGCTCCCTCTGT |
| Actin, alpha 1, skeletal muscle (Acta1) | F: GACCACAGCTGAACGTGAGA R: GACCACAGCTGAACGTGAGA | F: CCAGAGTCAGAGCAGCAGAC R: CGTGTGGCTCAGTAGGAGAG |
| Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) | F: CGTCTTCACCACCATGGAGA R: CGGCCATCACGCCACAGTTT | F: CACCATCCGGGTTCCTATAA R: AGGGAGGAGCAGAGAGCAC |
Quantitative Real Time‒PCR (RT‒qPCR)
Primer pairs producing ~100‒bp products of Tubb5, Acta 1 and Gapdh were designed using Primer Express (version 3.0) software. The lists of primers used in RT‒qPCR are shown in Table 1. The expression levels of Tubb5, Acta 1 and Gapdh in S. agalactiae infected and uninfected H9C2 cells were quantitated by CFX96 real-time PCR machine (BioRad, CA, USA). The assays were performed individually for each genes providing 100 ng of synthesized cDNA as template using Quanti Fast SYBR green PCR kit (Qiagen, Germany). Two independent experiments were performed in triplicate and the gene expression was quantified. The gene expression analysis was performed using relative quantification (ΔΔCT) method. Transcript levels were first normalized to the housekeeping gene, Gapdh and expressed as relative level to that of normalized surface. The expression of Tubb5, Acta 1 and Gapdh in uninfected H9C2 cells was used as a calibrator to determine the relative gene expression change in S. agalactiae‒ infected H9C2 cells. Nuclease free water was used as no template control (NTC).
Surface Removal of Teichoic Acid from S. agalactiae Cells
S. agalactiae cells were grown in BHI broth at 37°C for 16 h. The grown cells were sub‒ cultured into fresh BHI broth supplemented with different concentrations of benzyl penicillin (0.5, 1, 2, 3 and 4 μg ml-1) to remove the surface lipoteichoic acid (LTA). Cells were harvested after 3 h and washed with 1X PBS at 37°C for 5 min. The LTA‒reduced S. agalactiae cells were used for internalization and cytotoxicity studies. The S. agalactiae cells grown in the absence of benzyl penicillin was used as experimental control.
Extraction and Purification of Lipoteichoic Acid
Lipoteichoic acid (LTA) was extracted from S. agalactiae using hot aqueous phenol method [28, 29]. S. agalactiae cells were grown in BHI broth for overnight at 37°C. The overnight grown bacterial cells were harvested by centrifugation at 10, 000 rpm for 15 min at 25°C. The harvested cells were washed in phosphate buffer (10 mM NaH2PO4, 1 mM MgCl2, pH 7.5) and concentrated to a final concentration of approximately 0.4 g cells ml-1. The cell suspension was stirred with an equal volume of pre‒warmed 85% phenol at 65°C for 90 min followed by centrifugation at 10,000 rpm for 20 min at 25°C. The aqueous layer was removed and extracted with an equal volume of chloroform/isoamyl alcohol (24: 1, v/v) followed by centrifugation at 10,000 rpm for 20 min at 25°C and the aqueous layer was retained. The crude extract obtained was treated with RNase (20 μg ml-1) and DNase (20 μg ml-1) at 30°C for 8 h followed by repeated extraction with cold phenol: chloroform: ethanol (25: 24: 1, by vol.). Finally, LTA was purified on Sephadex gel using fast performance liquid chromatography (FPLC) system (BioRad, CA, USA). The purified LTA was dialyzed against 10,000 volumes of Tris buffer (10 mM Tris/HCl, 1 mM MgCl2, pH 7.0 ±5.0, 4°C) for 24 h to remove solvents and nucleic acid fragments. The yield of purified LTA was estimated from chromatographic elution profiles by determining the amount of phosphorus present excluding that ascribed to RNA, and the results were expressed as milligrams of LTA phosphorus per gram (dry weight) of bacterial cells. The dry weight of LTA was calculated from the known phosphorus content of the sample multiplied by a conversion factor (i.e., 1 mg of phosphorus = 5.81 mg of TA) obtained from the experimentally determined chain length and molar ratio of highly purified TA [30].
Determination of LTA‒Induced Cytotoxicity in H9C2 Cells
H9C2 cells were treated with different concentrations of purified LTA (i.e., 50, 25, 12.5, 6.25, 3.13 and 1.56 μg) in growth medium supplemented with 1% FBS and antibiotics for 2 h at 37°C with 5% CO2. LTA‒induced cytotoxicity in H9C2 cells were determined by confocal microscopy and MTT assay.
Statistical Analysis
All experiments were repeated at least three times and the data were expressed as Mean ± SD. Data were analyzed for statistical significance using Student’s unpaired t-test and p-value of <0.05 was used as threshold for significance.
Results
S. agalactiae Internalization into H9C2 Cells
In vitro bacterial infection assay and fluorescence analysis revealed that H9C2 cells could efficiently internalize both live and heat‒killed cells of S. agalactiae within 2 h of infection. No significant uptake was observed in H9C2 cells infected with S. gordonii ATCC 12403. However, H9C2 cells could not internalize fluorescently‒labeled live E. coli DH5α cells even at MOI of 1:1000, incubated for more than 24 h. The endocytic uptake of S. agalactiae by H9C2 cells was found to be increased with increase in MOI (Fig 1A & 1B). Consistently, the intracellular viable CFUs recovered from S. agalactiae‒infected H9C2 cells were also found to be increased with increase in MOI. The total intracellular CFUs recovered from S. agalactiae‒infected H9C2 cells at MOI of 1:10, 1: 100, 1: 1000 were found to be 3.3 x 103, 1.5 x104, and 3.3 x 105 ml-1 respectively. Whereas, no significant increase in intracellular viable CFUs were observed in H9C2 cells infected with S. gordonii ATCC 12403 and E. coli DH5α (Fig 1C).
Fig 1. Endocytic uptake of S. agalactiae by H9C2 cells.
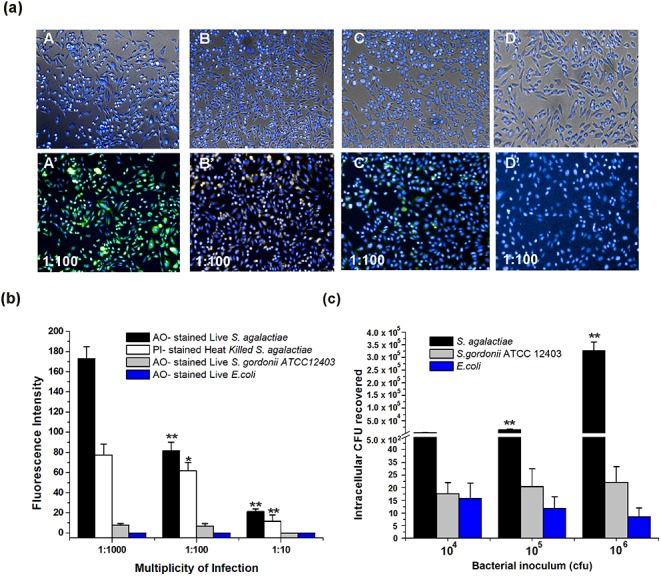
(a) Infection assay: A, B, C, D- merged images of bright field and Hoechst 33342 ‒stained H9C2 cells infected with live & heat‒killed S. agalactiae, S. gordonii ATCC 12403 and E. coli, respectively. A’, B’ C’, D’- merged images of H9C2 cells (counter-stained with Hoechst 33342 after infection) and endocytosed fluorescently labeled live‒ & heat‒killed S. agalactiae (AO & PI‒stained), S. gordonii ATCC 12403 (AO‒stained) and E. coli (AO‒stained), respectively. (b) Quantification of endocytic uptake of fluorescently‒labelled live & heat‒killed S. agalactiae, S. gordonii ATCC 12403 and E. coli by H9C2 cells. (c) Viable cell count of bacteria from infected H9C2 cells. H9C2 cells were infected with different inoculum of S. agalactiae, S. gordonii ATCC 12403 and E. coli (i.e., 104, 105 and 106) for 2 h. The recovered intracellular bacteria from H9C2 cells are represented as CFU/well (Mean ± SD) obtained from three independent experiments. Statistically significant differences are indicated by an asterisk (* p<0.05 or **p<0.01).
Endocytosis of S. agalactiae by H9C2 Cells
Endocytic inhibition studies with sodium azide (NaN3) by flow cytometry revealed that after 2 h of infection, 72% of population of H9C2 cells were internalized with S. agalactiae, whereas only 11.8% the population of NaN3 ‒treated H9C2 cells were internalized with S. agalactiae (Fig 2A). These results suggest that S. agalactiae could enter the H9C2 cells through an energy‒dependent endocytic mechanism.
Fig 2. Energy dependent endocytic uptake of S. agalactiae by H9C2 cells.

(a) Quantification of endocytic uptake of fluorescently‒labeled live S. agalactiae (AO-stained) by H9C2 cells in the presence (green) or absence (brown) of sodium azide by flow cytometry. P1-unstained population of H9C2 cells, P2- population of H9C2 cells internalized with fluorescently‒labelled S. agalactiae. (b) Determination of intracellular relative viable CFU of S. agalactiae recovered from H9C2 cells in the presence of different endocytic inhibitors such as genistein, chlorpromazine, cytochalasin B and nocodazole. The results were expressed in relative percentage compared with the experimental control. Statistically significant differences are indicated by an asterisk (* p<0.05 or **p<0.01).
Further characterization of endocytic processes of S. agalactiae in presence of different chemical endocytic inhibitors revealed that the disruption of microtubules and actin formation by nocodazole and cytochalasin B severely impaired S. agalactiae internalization into H9C2 cells, whereas the S. agalactiae internalization was not significantly inhibited by chlorpromazine and genistein (S1A Fig). Similar results were observed in CFU enumeration. The intracellular bacterial CFUs recovered from S. agalactiae‒infected H9C2 cells were drastically reduced with increase in concentration of cytochalasin B and nocodazole. The relative CFU recovered from H9C2 cells for cytochalasin B and nocadazole treatment at their higher concentration were 16% and 33%, respectively. Chlorpromazine and genistein treatment showed no significant decrease in intracellular viable CFUs compared to experimental control (without inhibitor) (Fig 2B). Flow cytometry analysis revealed that 68% population of control H9C2 cells (without inhibitor) was internalized with S. agalactiae, whereas, only 2.2% and 1.4% population of H9C2 cells were internalized with S. agalactiae for cytochalasin B and nocodazole treatment, respectively. Consistent with confocal microcopy and CFU enumeration studies, flow cytometry data also showed that S. agalactiae internalization into H9C2 cells was not inhibited by chlorpromazine and genistein (S1B Fig). All these data suggest that S. agalactiae internalization into H9C2 cells was mediated by cytoskeleton‒ mediated energy dependent endocytic processes.
S. agalactiae–Induced Cytotoxicity in H9C2 Cells
Confocal microscopic analysis revealed that H9C2 cells infected with both live and heat‒killed S. agalactiae showed changes in morphology within 6 h of infection. The morphological changes were prominent within 6 h in nearly 80% of population of H9C2 cells infected with S. agalactiae. The representative images for changes in H9C2 cells morphology within 6 h after S. agalactiae infection are shown in Fig 3A. MTT assay clearly demonstrated that the internalized S. agalactiae induced dose‒dependent cytotoxicity in H9C2 cells, i,e., increase in bacterial numbers had led to increased cytotoxicity in H9C2 cells. The maximum cytotoxicity was observed in H9C2 cells infected with live and heat‒killed S. agalactiae at MOI of 1: 1000 at 2 h (Fig 3B). Flow cytometry analysis revealed that both live and heat‒killed S. agalactiae induced cytotoxicity in 99.5% (39.5% early apoptotic & 59.8% late apoptotic) and 69.1% (56.7% early apoptotic, 8.2% late apoptotic cells and 4.2% dead cells) of population of H9C2 cells after 2 h post‒infection period (Fig 3C). These results suggested that both live and heat‒killed S. agalactiae internalization induce cytotoxicity in H9C2 cells.
Fig 3. S. agalactiae‒induced cytotoxicity in H9C2 cells.

(a) Morphological changes induced by S. agalactiae in H9C2 cells after 6 h of infection, Top panel: S. agalactiae-infected and uninfected H9C2 cells after 6 h of infection. Middle and bottom panel: (i), (ii), (iii) & (iv) are representative confocal micrographs showing changes in H9C2 cell morphology within 6 h of infection with live (ASG) and heat‒killed (PSG) S. agalactiae. Nuclei (N) of H9C2 cells were shown by Hoechst 33342 staining. (b) Determination of live and heat‒killed S. agalactiae‒induced cytotoxicity in H9C2 cells by MTT assay. Statistically significant differences are indicated by an asterisk (* p<0.05 or **p<0.01). (c) Quantification of live and heat‒killed S. agalactiae‒induced cytotoxicity in H9C2 cells by flow cytometry. Q3‒live population of H9C2 cells (blue dots); Q4‒early apoptotic H9C2 cells (green); Q2‒late apoptotic H9C2 cells (yellow).
S. agalactiae-Infection Elevates Gene Expression of Acta1 and Tubb5 in H9C2 Cells
Semi quantitative RT-PCR analysis for cytoskeleton forming genes detected relatively higher levels of transcripts for Tubb5 and Acta1 genes in S. agalactiae‒infected H9C2 cells compared to uninfected control (Fig 4A). RT Q-PCR analysis revealed that the gene copy number of Tubb5 and Acta 1 in S. agalactiae‒infected H9C2 cells were nearly 5 folds and 22 folds greater than the uninfected control, respectively (Fig 4B). These experimental data provided greater evidence that Acta1 is the major factor contributing for S. agalactiae pathogenesis followed by Tubb5.
Fig 4. S. agalactiae‒infection induced gene expression of Tubb5 and Acta1 in H9C2 cells.

(a) Semi quantitative RT-PCR analysis for expression of cytoskeleton forming genes, Tubb5 and Acta1 in S. agalactiae‒infected (B) and uninfected (A) H9C2 cells (b) RT Q-PCR analysis for expression of Tubb5 and Acta1 in S. agalactiae‒infected and uninfected H9C2 cells. Gene expression values of the uninfected H9C2 cells (control) were set equal to 1 and the relative change in gene expression in S. agalactiae‒infected H9C2 cells were calculated. RT Q-PCR was performed twice independently and the triplicate measurements of gene expression per gene were included, which yielded similar results. Data shown are the mean relative change in expression ± standard deviation from one such experiment. Statistically significant differences are indicated by an asterisk (* p<0.05 or **p<0.01).
Role of Lipoteichoic Acid in S. agalactiae Internalization and Cytotoxicity
Growth studies revealed that S. agalactiae cells can grow normally up to 2 μg concentration of benzyl penicillin with 52% reduction in LTA (S2A Fig). Infection assay by flow cytometry revealed that untreated S. agalactiae internalized 91.1% populations of H9C2 cells at MOI of 1:1000, whereas only 23.3% populations of H9C2 cells were internalized with LTA‒reduced S. agalactiae. Thus, removal of LTA from S. agalactiae impaired with its ability to internalize into H9C2 cells (Fig 5A). MTT assay also clearly demonstrated that LTA‒reduced S. agalactiae induced lesser cytotoxicity to H9C2 cells compared to control S. agalactiae (Fig 5B).
Fig 5. S. agalactiae LTA‒induced cytotoxicity in H9C2 cells.

(a) Quantification of benzyl penicillin (BP)‒treated and untreated S. agalactiae internalization into H9C2 cells. Q3‒uninfected population of H9C2 cells (blue dots); Q4‒population of H9C2 cells internalized with AO‒stained S. agalactiae (green dots) (b) Cytotoxicity analysis of BP-treated and untreated S. agalactiae on H9C2 cells. S. agalactiae grown in the presence of benzyl penicillin showed reduced cytotoxicity in H9C2 cells compared to untreated control (c) Determination of LTA-induced cytotoxicity in H9C2 cells after 6 h by MTT assay. Statistically significant differences are indicated by an asterisk (* p<0.05 or **p<0.01).
Further, we tested the effect of purified LTA from S. agalactiae on the viability of H9C2 cells. LTA‒treated H9C2 cells showed morphological changes similar to that of S. agalactiae-infected H9C2 cells i,e., detachment of H9C2 cells from the substratum and rounded up within 6 h (S2B Fig). MTT assay also revealed that LTA induced dose‒dependent cytotoxicity in H9C2 cells (Fig 5C). All these data clearly demonstrated that the cell wall component of S. agalactiae, LTA could play major role in internalization and cytotoxicity induction in H9C2 cells.
Discussion
Though, S. agalactiae infections in cardiovascular diseases such as infective endocarditis, myocarditis and pericarditis have been widely reported. However, the association of this bacterium with cardiac cells remains unknown. In the present study, we have used H9C2 cell line as a model system to study the S. agalactiae pathogenesis, as it is being a surrogate for cardio myoblast and it has the properties of both skeletal and cardiac muscle cells. H9C2 cells can be easily differentiated into cardiac muscle cells by reducing the serum level with the addition of All-Trans Retinoic acid (ATRA) [21, 31]. Differentiated H9C2 cell line also has an advantage of being animal-free alternative and accurately mimics the cellular responses of human cardiomyoctes cell lines. It is also proven to be an excellent in vitro model system for prospective molecular studies in heart development and disease [32]
Ability of S. agalactiae to adhere and invade normal myocardium serves as the primary mechanism in bacterial myocarditis. Here, we have presented the evidence for the internalization and cytotoxicity induction by S. agalactiae in H9C2 cells. We have shown that both live and heat‒killed S. agalactiae internalized into H9C2 cells through energy‒dependent endocytic processes. The chemical endocytic inhibitors specific for discrete eukaryotic cell functions were used to ascertain the specific host endocytic mechanism involved in S. agalactiae internalization. The experiments performed with cytochalasin B and nocodazole have demonstrated the requirement of functional cytoskeletal elements (microfilaments and microtubules) for S. agalactiae internalization into H9C2 cells. Transcript profiling of genes responsible for formation of cytoskeletal elements also showed elevated gene expression of Acta 1 and Tubb5 in S. agalactiae‒infected H9C2 cells, which further substantiated the role of actin and tubulin microfilament in S. agalactiae internalization into H9C2 cells. Consistent with our findings, earlier study has also shown that viridans group streptococci can enter the human cardiomicrovascular endothelial cells (HCMEC) through the polymerization of actin filaments [33].
The streptococci internalization has also been reported to induce apoptosis in human epithelial cells through caspase dependent pathways [34]. Ulett et al. [35] have reported that S. agalactiae induced apoptosis in murine and human macrophages. It is interesting to note that heat‒killed S. agalactiae also induced cytotoxicity in H9C2 cells, which suggest that the not only live replicating cells, deads cells can also internalize and induce cytotoxicity in cardiac cells. Earlier report has shown that the administration of heat‒killed S. agalactiae caused rapid death of adult rats [36]. Heat‒killed Streptococcus suis type 2 strain also shown to induce the production of pro-inflammatory cytokines, tumor necrosis factor alpha (TNF-a) and interleukin-6 (IL-6) in murine macrophages [37].
Further, we attempted to investigate the molecular determinant responsible for pathogen internalization and cytotoxicity induction. We found that the cell surface component of S. agalactiae, LTA play a major role in S. agalactiae internalization and cytotoxicity induction. Teichoic acid (TA) is a ubiquitous antigenic component and virulence factors of various Gram‒positive bacteria including groups A to G streptococci [38, 39]. These are phosphate-rich molecules found in a wide range of Gram‒positive bacteria. There are two types of TAs: the lipo-TAs (LTAs), which are anchored to the plasma membrane and extend from the cell surface into the peptidoglycan layer; and the wall TAs (WTAs), which are covalently attached to peptidoglycan and extend through and beyond the cell wall. LTA is heat‒stable component of cell membrane and wall of most Gram‒positive bacteria [40, 41]. In our study, the removal of LTA from S. agalactiae impaired with its ability to internalize into H9C2 cells. Therefore, we believe that heat‒stable LTA is the target molecule involved in internalization of heat‒killed S. agalactiae into H9C2 cells as well. Previous studies have shown that LTA is well-known ligand for pattern recognition receptors such as, Toll‒like receptors (TLRs) [42]. LTA is a central inducer of inflammatory responses caused by Gram‒positive bacteria, which occurs mainly through the activation of TLR2. LTA can be efficiently internalized into human monocytes and peripheral dendritic cells through receptor‒mediated endocytosis [42, 43]. Earlier reports have also shown that LTA of GBS mediates invasion and induces cytotoxicity in adult & neonatal epithelial cells, HeLa, Giardi heart cells, human embryonic brain and amnion [44, 45]. LTA is involved in signal transduction pathways and induced ERK1/2, JNK, p38, AKT phosphorylation and IL-1 beta gene expression in H9C2 cells [46]. The ability of LTA to modulate host cellular responses might be responsible for induction of cytotoxicity in H9C2 cells. Kristian et al., [47] have reported that deletion of dltA gene involved in TA D-alanylation in an invasive GAS diminished its ability to adhere and invade cultured human pharyngeal epithelial cells. Similarly, we also found that LTA‒reduced S. agalactiae showed lesser ability to internalize and induce cytotoxicity to H9C2 cells. LTA purified from S. agalactiae also exhibited cytotoxicity in H9C2 cells, which further evidenced the role of LTA in pathogenesis. To conclude, S. agalactiae could enter the cardiac cells through energy dependent endocytic processes; wherein, the surface determinant of S. agalactiae, LTA plays major role in S. agalactiae internalization and cytotoxicity induction in rat cardiomyocytes.
Supporting Information
The specific endocytic entry mechanism of S. agalactiae into H9C2 cells was studied in the presence of different endocytic inhibitors (a) confocal micrographs of H9C2 cells infected with fluorescently‒labeled S. agalactiae in the presence and absence of different endocytic inhibitors. (b) Quantification of endocytic uptake of live S. agalactiae (AO‒stained) by H9C2 cells in presence of different endocytic inhibitors such as genistein (violet), chlorpromazine (sky blue), cytochalasin B (brown), nocodazole (yellow) and no inhibitors (dark green) by flow cytometry. P1‒unstained population of H9C2 cells and P2‒ population of H9C2 cells internalized with fluorescently‒labeled S. agalactiae (AO-stained).
(TIF)
(a) Effect of benzyl penicillin on LTA reduction and growth of S. agalactiae (b) Merged confocal micrographs of bright field and Hoechst 33342‒stained control and LTA‒treated H9C2 cells after 6 h. X and Y represent magnified portion of confocal micrographs of control and LTA treated H9C2 cells, respectively.
(TIF)
Acknowledgments
Authors acknowledge the DST-PURSE, UGC-CAS, UGC-CEGS, UGC-NRCBS, DBT-IPLS, and DST-FIST programs at SBS, MKU. The Authors also acknowledge Dr. A. Mahesh for technical support in flow cytometry and Ms. M. Niraimathi for her technical support in confocal imaging.
Data Availability
All relevant data are within the paper.
Funding Statement
The authors have no support or funding to report.
References
- 1. High KP, Edwards MS, Baker C J. Group B streptococcal infections in elderly adults. Clin Infect Dis 2005;41: 839–847. [DOI] [PubMed] [Google Scholar]
- 2. Sunkara B, Bheemreddy S, Lorber B, Lephart PR, Hayakawa K, Sobel JD, et al. Group B Streptococcus infections in non-pregnant adults: the role of immunosuppression. Int J Infect Dis 2012;16: e182–e186. 10.1016/j.ijid.2011.11.008 [DOI] [PubMed] [Google Scholar]
- 3. Farley MM, Strasbaugh LJ. Group B streptococcal disease in non-pregnant adults. Clin Infect Dis 2001;33: 556–561. [DOI] [PubMed] [Google Scholar]
- 4. Rajagopal L. Understanding the regulation of Group B Streptococcal virulence factors. Future Microbiol 2009;4: 201–221. 10.2217/17460913.4.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schuchat A. Epidemiology of group B streptococcal disease in the United States: shifting paradigms. Clin Microbiol Rev 1998;11: 497–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spellerberg B. Pathogenesis of neonatal Streptococcus agalactiae infections. Microbes Infect 2000;2: 1733–1742. [DOI] [PubMed] [Google Scholar]
- 7. Beckmann C, Waggoner JD, Harris TO, Tamura GS, Rubens CE. Identification of novel adhesins from group B streptococci by use of phage display reveals that C5a peptidase mediates fibronectin binding. Infect Immun 2002;70: 2869–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friman G, Wesslen L, Fohlman J, Karjalainen J, Rolf C. The epidemiology of infectious myocarditis, lymphocytic myocarditis and dilated cardiomyopathy. Eur Heart J 1995;16: 36–41. [DOI] [PubMed] [Google Scholar]
- 9. Sambola A, Miro JM, Tornos MP, Almirante B, Moreno-Torrico A, Gurgui M, et al. Streptococcus agalactiae infective endocarditis: analysis of 30 cases and review of the literature, 1962–1998. Clin Infect Dis 2002;34: 1576–1584. [DOI] [PubMed] [Google Scholar]
- 10. Rollan MJ, San Roman JA, Vilacosta I, Sarria C, Lopez J, Acuna M, Bratos JL. Clinical profile of Streptococcus agalactiae native valve endocarditis. Am Heart J 2003;146: 1095–1098. [DOI] [PubMed] [Google Scholar]
- 11. Scully BE, Neu HC, Spriggs D. Streptococcus agalactiae (group B) endocarditis—a description of twelve cases and review of the literature. Infect 1987; 15: 169–176. [DOI] [PubMed] [Google Scholar]
- 12. Wenger NK. Women in cardiology: the US experience. Heart 2005;91: 277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Narkwiboonwong T, Luveera V, Phiboonbanakit D, Trakulhun K. Acute Endocarditis Caused by Streptococcus agalactiae . J Infect Dis Antimicrob Agents 2011;28: 179–82. [Google Scholar]
- 14. Machado C, Almeida C, Santos E, Pelicano N, Dourado R, Martins D. Streptococcus agalactiae endocarditis. Rev Port Cardiol 2012; 31:619–621. [DOI] [PubMed] [Google Scholar]
- 15. Cheng Q, Stafslien D, Purushothaman SS, Cleary P. The group B streptococcal C5a peptidase is both a specific protease and an invasin. Infect Immun 2002;70: 2408–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brockman MD, Rhoades ER. Myocardial abscess and fatal sepsis caused by group B Streptococcus (S. agalactiae) in an adult. South Med J 1979;72: 1629 [DOI] [PubMed] [Google Scholar]
- 17. Abad C. Purulent abscess myocarditis with fatal outcome, caused by Streptococcus agalactiae . Med Clin1996;106: 119. [PubMed] [Google Scholar]
- 18. Bateman AC, Richards M, Pallett AP. Fatal myocarditis associated with a Lancefield group B streptococcus. J Infect 1998;36: 354–355. [DOI] [PubMed] [Google Scholar]
- 19. Siciliano RF, Cais D P, Navarro RC, Strabelli TM. Acute Streptococcus agalactiae endocarditis: outcomes of early surgical treatment. Heart Lung 2010;39: 331–334. 10.1016/j.hrtlng.2009.06.018 [DOI] [PubMed] [Google Scholar]
- 20. Dinakaran V, John L, Rathinavel A, Gunasekaran P, Rajendhran J. Prevalence of bacteria in the circulation of cardiovascular disease patients, Madurai, India. Heart Lung Circ, 2012;21: 281–283. 10.1016/j.hlc.2012.02.007 [DOI] [PubMed] [Google Scholar]
- 21. Menard C, Pupier S, Mornet D, Kitzmann M, Nargeot J, Lory P. Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J Biol Chem. 1999;274: 29063–29070. [DOI] [PubMed] [Google Scholar]
- 22. Sit KH, Bay BH, Wong KP. Effect of genistein, a tyrosine-specific protein kinase inhibitor, on cell rounding by pH upshifting. In Vitro Cell Dev-An.1993;29:395–402. [DOI] [PubMed] [Google Scholar]
- 23. Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev 2002;54: 561–587. [DOI] [PubMed] [Google Scholar]
- 24. Liu SM, Magnusson KE, Sundqvist T. Microtubules are involved in transport of macromolecules by vesicles in cultured bovine aortic endothelial cells. J Cell Physiol. 1993;156: 311–316. [DOI] [PubMed] [Google Scholar]
- 25. Goltz JS, Wolkoff AW, Novikoff PM, Stockert RJ, Satir P. A role for microtubules in sorting endocytic vesicles in rat hepatocytes. PNAS 1992;89: 7026–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacLean-Fletcher S, Pollard TD. Mechanism of action of cytochalasin B on actin. Cell 1980;20:329–341. [DOI] [PubMed] [Google Scholar]
- 27. Rosenshine I, Ruschkowski S, Finlay BB. Inhibitors of cytoskeletal function and signal transduction to study bacterial invasion. Method Enzymol 1994;236: 467–476. [DOI] [PubMed] [Google Scholar]
- 28. Sijtsma L, Wouters JT, Hellingwerf KJ. Isolation and characterization of lipoteichoic acid, a cell envelope component involved in preventing phage adsorption, from Lactococcus lactis subsp. cremoris SK110. J Bacteriol 1990;172: 7126–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hogg SD, Whiley RA, De Soet JJ. Occurrence of lipoteichoic acid in oral streptococci. Int J Sys Bacteriol 1997; 47: 62–66. [DOI] [PubMed] [Google Scholar]
- 30. Slabyj BM, Panos C. Membrane lipoteichoic acid of Streptococcus pyogenes and its stabilized L-form and the effect of two antibiotics upon its cellular content. J Bacteriol 1976;127: 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Branco AF, Pereira SL, Moreira AC, Holy J, Sardão VA, Oliveira PJ. Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9C2 cell line. Cardiovasc Toxicol 2011;11: 191–203. 10.1007/s12012-011-9111-5 [DOI] [PubMed] [Google Scholar]
- 32. Watkins SJ, Borthwick GM, Arthur HM. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Dev-An 2011;47:125–131. [DOI] [PubMed] [Google Scholar]
- 33. Stinson MW, Alder S, Kumar S. Invasion and killing of human endothelial cells by viridans group streptococci. Infect Immun 2003;71: 2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsai PJ, Lin YS, Kuo CF, Lei HY, Wu JJ. Group A Streptococcus induces apoptosis in human epithelial cells. Infect Immun.1999;67: 4334–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ulett GC, Maclean KH, Nekkalapu S, Cleveland JL, Adderson EE. Mechanisms of group B streptococcal-induced apoptosis of murine macrophages. J Immunol 2005;175: 2555–2562. [DOI] [PubMed] [Google Scholar]
- 36. Warejcka DJ, Goodrum KJ, Spitznagel JK. Toxicity of group B Streptococcus agalactiae in adult rats. Infect Immun 1985;48: 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Segura M, Stankova J, Gottschalk M. Heat-killed Streptococcus suis capsular type 2 strains stimulate tumor necrosis factor alpha and interleukin-6 production by murine macrophages. Infect Immun 1999; 67: 4646–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCarty M. The occurrence of polyglycerolphosphate as an antigenic component of various Gram‒positive bacterial species. J Exp Med 1959;109: 361–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weidenmaier C, Kokai-Kun JF, Kristian SA, Chanturiya T, Kalbacher H, Gross M, et al. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat med 2004;10: 24–245. [DOI] [PubMed] [Google Scholar]
- 40. Wicken AJ, Knox KW. Lipoteichoic acids: a new class of bacterial antigen. Science 1975;187: 1161–1167. [DOI] [PubMed] [Google Scholar]
- 41. Wicken AJ, Knox KW. Bacterial cell surface amphiphiles. BBA-Biomembranes 1980:604: 1–26. [DOI] [PubMed] [Google Scholar]
- 42. Andersson LI, Hellman P, Eriksson H. Receptor-mediated endocytosis of particles by peripheral dendritic cells. Hum Immunol, 2008;69: 625–633. 10.1016/j.humimm.2008.07.010 [DOI] [PubMed] [Google Scholar]
- 43. Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling; role of CD14 and CD36. J Leukocyte Biol 2008;84: 280–291. 10.1189/jlb.0907656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Teti G, Tomasello F, Chiofalo MS, Orefici G, Mastroeni P. Adherence of group B streptococci to adult and neonatal epithelial cells mediated by lipoteichoic acid. Infect Immun 1987;55: 3057–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goldschmidt JC, Panos C. Teichoic acids of Streptococcus agalactiae: chemistry, cytotoxicity, and effect on bacterial adherence to human cells in tissue culture. Infect Immun 1984;43: 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gutierrez‐Venegas G, Luna OA. Ventura‐Arroyo J A, Hernandez‐Bermudez C. Myricetin suppresses lipoteichoic acid‐induced interleukin‐1β and cyclooxygenase‐2 expression in human gingival fibroblasts. Microbiol Immunol 2010;57: 849–856. [DOI] [PubMed] [Google Scholar]
- 47. Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, et al. D-alanylation of teichoic acids promotes group a streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol 2005;187: 6719–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The specific endocytic entry mechanism of S. agalactiae into H9C2 cells was studied in the presence of different endocytic inhibitors (a) confocal micrographs of H9C2 cells infected with fluorescently‒labeled S. agalactiae in the presence and absence of different endocytic inhibitors. (b) Quantification of endocytic uptake of live S. agalactiae (AO‒stained) by H9C2 cells in presence of different endocytic inhibitors such as genistein (violet), chlorpromazine (sky blue), cytochalasin B (brown), nocodazole (yellow) and no inhibitors (dark green) by flow cytometry. P1‒unstained population of H9C2 cells and P2‒ population of H9C2 cells internalized with fluorescently‒labeled S. agalactiae (AO-stained).
(TIF)
(a) Effect of benzyl penicillin on LTA reduction and growth of S. agalactiae (b) Merged confocal micrographs of bright field and Hoechst 33342‒stained control and LTA‒treated H9C2 cells after 6 h. X and Y represent magnified portion of confocal micrographs of control and LTA treated H9C2 cells, respectively.
(TIF)
Data Availability Statement
All relevant data are within the paper.