

Abstract
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by memory loss and personality changes, leading to dementia. Histophatological hallmarks are represented by aggregates of beta-amyloid peptide (Aβ) in senile plaques and deposition of hyperphosphorylated tau protein in neurofibrillary tangles in the brain. Rare forms of early onset familial Alzheimer's disease are due to gene mutations. This has prompted researchers to develop genetically modified animals that could recapitulate the main features of the disease. The use of these models is complemented by non-genetically modified animals.
Area covered
This review summarizes the characteristics of the most used transgenic (Tg) and non-Tg models of AD. The authors have focused on models mainly used in their laboratories including: APP Tg2576, APP/PS1, 3xAD, single h-Tau, non-Tg mice treated with acute injections of Aβ or tau, and models of physiological aging.
Expert opinion
Animal models of disease might be very useful for studying the pathophysiology of the disease and for testing new therapeutics in preclinical studies but they do not reproduce the entire clinical features of human AD. When selecting a model, researchers should consider the various factors that might influence the phenotype. They should also consider the timing of testing/treating animals since the age at which each model develops certain aspects of the AD pathology varies.
Keywords: Transgenic models, Alzheimer’s disease, Aging, Synaptic plasticity, Memory, Behavior
1. Introduction
In 1906, the German psychiatrist Alois Alzheimer described a peculiar form of mental illness characterized by “progressive cognitive impairment, focal symptoms, hallucinations, delusions, and psychosocial incompetence” [1] in the patient Augustine Deter [2]. The histopathological examination of her brain revealed the presence of neurofibrillary tangles with “characteristic thickness and peculiar impregnability” and “numerous small miliary foci… determined by the storage of a peculiar material in the cortex” recognizable with the typical senile plaques. In 1909 the Italian physician Gaetano Perusini examined four cases of patients affected by dementia onset at the age of 50–60 years and confirmed the clinical and histopathological hallmarks of the new disease named “Alzheimer’s disease” (AD) by Emil Kraepelin [3]. In the following years, several case reports of dementia with the characteristic histopathological signs were diagnosed and epidemiological studies recognized AD as the main form of dementia in the elderly [4] expected to grow exponentially in the decades to follow.
In the ‘80s, researchers identified beta-amyloid protein (Aβ) as the main component of brain plaques and tau protein as the main component of neurofibrillary tangles. At the same time, the discovery of rare forms of early onset Familial Alzheimer's disease (FAD) inherited in an autosomal dominant fashion [5] highlighted the relevance of genetic factors in the pathogenesis of the disease [6]. Mutations in the gene for Amyloid Precursor Protein (APP) on chromosome 21 were identified in several families (London, Dutch, Swedish and Flemish mutations) affected by FAD and, afterwards, other mutations in the gene for presenilin 1 (PS1) in chromosome 14 and presenilin 2 (PS2) in chromosome 1 were observed in AD families, even if they could also be present in healthy subjects. However, all these mutations induced an increase of Aβ production from APP, even if it has been recently demonstrated that PS mutations may cause neurodegeneration and dementia through not only an increase of Aβ42/40 ratio but also a loss of the physiological PS function [7].
APP is a type-1 transmembrane glycoprotein formed by 365 to 770 aminoacids, with the isoform APP695 predominant in human neuronal tissue, and the isoforms APP751 and APP770 widely expressed in non-neuronal cells. APP undergoes a complex proteolitic cleavage catalyzed by secretases. APP can be initially cleaved by α- or β-secretases. The α-secretase cleavage generates a soluble extracellular domain, sAPPα, and a carboxy-terminal fragments (CTF), containing 83 amino acids (AAs) (CTF83); whereas, the β-secretase cleavage generates sAPPβ and 99 AAs (CTF99). In turn, subsequent cleavage by -secretase generates a p3 fragment and a 57–59 AA CTF from C83 and a 40–42 AA fragment (Aβ40 or Aβ42) together with APP intracellular domain (AICD) fragment from CTF99. Thus, β-secretase and -secretase, of which PS1 and PS2 are a subcomponent, catalyze the production of Aβ.
The knowledge of this pathway, together with the genetic mutations and the post-mortem Aβ-plaques in the AD brains, laid the basis for the so called “Amyloid hypothesis” that has dominated the scientific scene in the last 30 years. Thus, a number of therapeutic strategies aimed at reducing Aβ production in the AD brain have been developed.
This also prompted the neuroscience community to find a “model” of disease that, even if it does not reproduce the complete human disease, exhibits the characteristic histological lesions (amyloid plaques, neurofibrillary tangles and neuronal loss) and the main symptom of AD: memory loss associated to a deficit in synaptic plasticity mechanisms. In particular, rodent models of AD have been used in the last 20 years to study the pathogenetic mechanisms, the progression of the disease, and the efficacy of new drugs in preclinical studies. In this review we will mainly discuss only those models we have used in our laboratories: single transgenic (Tg) APP Tg2576, double Tg APP/PS1, triple Tg 3xAD, single h-Tau, non-Tg models obtained with acute injections of Aβ or tau, models of physiological aging.
2. Transgenic models for the study of AD
To date, animal models used in preclinical studies can be distinguished in: i) Tg models of AD, consisting in single or multi Tg animals overexpressing APP, PS and/or Tau mutations; ii) non-Tg models obtained by toxins injection in the brain, including direct injection of Aβ or tau, and models of aging.
Most of the Tg models are mice, whereas non-Tg models could also be rats, dogs and monkeys. Moreover, it could be useful to note that C57Bl/6 represents the most diffuse wild type background of the mouse Tg models.
The first attempt to create a Tg animal was based on the amyloid hypothesis, thus reproducing the deposits of Aβ in the brain by overexpressing the isoform β-APP751 containing the Kunitz protease inhibitor domain [8, 9], the human APP C-100 fragment [10], or the entire human APP sequence [11, 12]. Notwithstanding these mice could be considered a good model of Aβ hyperproduction, they did not resemble other features of an AD human brain. Thus, these are excellent models to better understand the pathophysiologic role of Aβ in AD or to test drugs aimed to modulate or reduce Aβ levels, but they might not be appropriate for the study of other aspects of AD since they lack other relevant yet critical factors.
In 1995, Games et al. [13] created the PDAPP mouse expressing high levels of human APP cDNA with a FAD-associated mutation (substitution of valine at position 717 with phenylalanine). This mouse expressed high levels of APP and developed several features of human AD such as extracellular amyloid fibrils organized in plaques, dystrophic neuritis, apoptosis, subcellular degenerative changes, synaptic loss and gliosis that spread progressively from hippocampus to cortex [13, 14]. More importantly, PDAPP mice presented the main feature of a patient with AD, memory loss. In the water maze and in the radial maze, PDAPP mice were impaired before and after amyloid plaque deposition [15, 16]. Object-recognition performance decreased with age and was associated with amyloid deposition [15]. Alterations in emotionality or fear and exploratory activity were found at 11 months of age [17]. Overall, these mice presented an age-related impairment of memory with a peak at 12–15 months of age [16, 18, 19].
In 1996, Karen Hsiao and colleagues created another Tg mouse model of AD, the Tg2576 line, carrying the double Swedish mutation (K670N and M671L) [20]. These mice displayed an increase of APP production (> 5-fold) with consequent overproduction of Aβ40 and Aβ42 and plaques formation in the frontal, temporal, and entorhinal cortices, hippocampus, presubiculum, and cerebellum at about 11–13 months of age. Other than the increase in Aβ production, they can also display hyperphosphorylated tau at old age. A battery of behavioral studies (i.e., Y-maze, visible platform, Morris water maze (MWM), circular platform, passive avoidance, and active avoidance) performed at different ages (3, 9, 14, and 19 months) demonstrated that Tg2576 mice were impaired in Y-maze spontaneous alternation and visible platform at 9 months of age, while deficit in sensorimotor tasks started at 14 months. In the fear conditioning test (FC), they presented normal levels of conditional freezing to an auditory conditional stimulus, confirming that amygdala function was normal, whereas they were impaired in the hippocampal-dependent conditioning for the context [21]. However, they did not present a profound cognitive impairment, even at old ages [22]. The deficit in memory was associated with a severe impairment of in-vitro and in-vivo long-term potentiation (LTP) in both the CA1 and dentate gyrus regions of the hippocampus [23], without structural alterations of the synapse, but with reduced ability of neurons to integrate and propagate information [24]. Some studies have found anxiety-like disturbances in Tg2576 mice, but results are contradictory. For instance, Elevated-Plus-Maze has revealed a reduction [25, 26] or an increase [27] of anxiety-like behavior.
In our experience, Tg2576 mice present an impairment of LTP and short-term memory at about 9–10 months of age, whereas contextual FC is impaired at 4–6 months of age. MWM (both learning curve and reference memory), and novel object recognition (NOR), are impaired at about 10–12 months. The advantages with using these mice consist in: i) their well-known characterization (they have been used in several laboratories as a model of AD for almost 20 years); ii) the relatively simple management of the colony (good fertility when using Tg2576 males and C57Bl/6 females, easier genotyping of a single transgene). The disadvantage is that the AD phenotype occurs late. Indeed, we usually wait the age of 12 months to perform experiments to be sure that animals present both synaptic and memory dysfunction.
The onset of the AD phenotype occurs early in double Tg mice in which Tg2576 are crossed with PS1 (M146L) (line 6.2) [28, 29]. Indeed, because FADs are also associated with PS1 and PS2 mutations [30], mouse models of overexpression of either M146L or M146V FAD-associated presenilin mutations have been created. However, when expressing only PS1 and PS2, mice failed to reproduce the AD phenotype in vivo [31, 32]. The PS1 variant (A246E) induced an increase of Aβ42/Aβ40 ratio in cell cultures but not amyloid pathology in mice [7, 33, 34]. However, crossing PS1 M146L with Tg2576 mice (or other APP mutants) caused an increase of amyloid production and deposition [28]. In particular, mice overexpressing APP (K670N:M671L) together with PS1 (M146L) have been extensively used to better understand the pathogenic mechanisms underlying synaptic dysfunction and memory loss in AD, and to validate new therapeutic approaches [35–46]. These mice presented a robust age-dependent Aβ deposition in plaques preceded by an increase of soluble Aβ40 and Aβ42. In several papers we have reported that APP/PS1 have abnormal LTP as early as 3 months of age, paralleling short-term memory and contextual FC impairment and plaque onset.
Conversely, long-term memory and basal synaptic transmission (BST) were impaired at 6 months, as amyloid burden increases. As for single APP, there is conflicting literature on the emotional changes in APP/PS1 mice. Some studies, including ours, have demonstrated normal fear and anxiety levels [38, 47, 48], whereas others decreased anxiety in APP/PS1 mice [49].
These mice have the advantage of presenting the AD-related phenotype at early age. However, they do not show some aspects of the disease such as neuronal loss and tau deposition.
Recently, mice containing 3 different mutations – 3XTg – such as APPSwe, PS1 M146V, and hyperphosphorylated tau (tauP301L) have been generated [50]. These mice presented Aβ pathology at 6 months of age (increased Aβ40 and Aβ42 levels, intracellular accumulation of Aβ, and amyloid plaques) that preceded tau pathology with neurofibrillary tangles formation at about 12 months of age. LTP and spatial memory impairment [50–53] were also evident. In our recent studies [54], 3XTg at 8–9 months of age showed an increase of Aβ42 levels and an increase of inflammatory mediators in the hippocampus, and an impairment of cognitive functions assessed by the MWM test and the NOR test. Increased age-related anxiety and fearfulness have been reported in some studies [55–57]. 3XTg did not present a decrease of synapse number and density in CA1 pyramidal layer but a decrease of perforated junctional areas [58]. Neuronal loss, potentially due to intraneuronal Aβ accumulation [59], has been found in a 5XTg mouse model containing APP (Swedish K670N/M671L, Florida I716V, and London V717I) and PSEN1 (M146L and L286V) mutations. However, reduction in neurons has been found in cortical layer 5 but not CA1 layer of the hippocampus [60].
More recently, we have started using Tg mice in which the mouse tau gene is replaced by the human tau gene (a.k.a. hTau mice) [61]. These animals display tau oligomers at 10–11 months, whereas neurofibrillary tangles are present at later ages [62, 63]. Additionally, they present memory loss associated with defects in LTP [64].
Histopathological changes, synaptic dysfunction, memory loss and other behavioral are not the only features of the disease that can be mimicked using both Tg and non-Tg models of the disease. For instance there is extensive evidence from these animal models suggesting a key role of proinflammatory cytokine overproduction as a possible driving force for progression of pathology in AD [65]. Such a role would occur both early in the disease and at later stages to accelerate its progression. Indeed, manipulations that lead to overproduction of cytokines worsen the disease outcomes, whereas selective suppression of proinflammatory cytokine overproduction leads to a reduction in disease relevant end points.
3. Non-transgenic models for the study of AD
Non-Tg models for the study of AD are mainly obtained by injecting Aβ or tau directly into the brain via intracerebroventricular (i.c.v.) or intrahippocampal injections [66, 67]. This allows studying the role of acute Aβ or tau increase and could be very useful when researchers want to use animals different than mice for experimental reasons (i.e. studies on non-humans primates) or do not have the resources to breed a Tg colony. However, acute models do not reproduce the gradual rise in Aβ occurring in many years in humans. To this end, we should also point at the fact that it is not known whether a chronic exposure to Aβ is relevant to the impairment of memory mechanisms. Indeed, all the studies performed so far point towards an acute effect of Aβ onto memory, regardless of time exposure to the peptide. This does not exclude though that other aspects of the disease (i.e. spreading of the pathology throughout the brain) are dependent upon a more chronic exposure to the peptide. Moreover both acute Aβ infusion models and transgenic APP models have limitations and, unfortunately, resemble some but not all the features of the human disease. For instance, while Tg mice mostly reflect genetic forms of the disease because they overexpress mutated forms of APP, AD is primarily a sporadic disorder. This can be partially mimicked in vivo by icv or intrahippocampal injections of Aβ, even if they do not reflect neither the concentration nor the time course of changes seen in humans. Additionally, Tg models overexpressing APP do not only show elevation of Aβ, but also elevation of full length APP and other fragments of APP processing that might interfere with the phenotype observed and provide misleading results.
For these reasons, we believe it is better to combine both Tg and non-Tg models to overcome limitations of the different models. The use of acute injections, for instance, gives the possibility to better understand how Aβ impairs specific signaling pathways leading to synaptic and memory dysfunctions, and this is crucial when designing new therapeutic strategies. Additionally, acute injection could be used to identify the targets of specific soluble Aβ species (from monomers to oligomers of different molecular weights) since they might exert a different role in synaptic plasticity and memory impairment. In this case, Tg mice do not represent a good tool because they overproduce different Aβ forms (monomers, dimers, trimers, oligomers, fibrils up to plaques) making very difficult the evaluation of the specific pathogenic role of these aggregates. Intrahippocampal or icv injections of a specific Aβ species, in turn, are a more appropriate model than transgenic models.
In summary, these non-Tg models allow a) to investigate the effects of Aβ and tau in animals for which Tg models are not available, b) to exclude the confounding effects of overexpression of APP and its fragments, c) to investigate the different role of Aβ and tau species (monomers vs. oligomers vs. insoluble) at different concentrations, d) to investigate the difference between an acute or a chronic administration (in this last case one could also implant mini-pumps for a chronic delivery of the peptide), e) to clarify aspects of the molecular mechanisms underlying Aβ and tau pathology that cannot be investigated using Tg models. In our laboratories we have often used intrahippocampal injections of Aβ and tau [68–71] with satisfactory results especially when we wanted to study the physiological role of low concentrations of the peptide [72, 73], but also with injections of high (nM) concentrations of Aβ and tau to study the effect of a drug on memory loss [74–76].
Recently, we have also used animal models of aging [77–79]. In this case, notwithstanding genetically- or drug-induced animal models of aging were available [80–83], we preferred to use a physiological model of aging to provide more realistic information on the natural development of the aging process. However, it is important to notice that a model of aging is not a model of AD as it could be misinterpreted. In our aged C57Bl/6 wild type animals we found an impairment of LTP at about 22 months, whereas BST was unaffected up to 26 months. In addition these mice were demonstrated to have severely impaired spatial learning and reference memory as tested by the MWM [84, 85] and recognition memory as tested by NOR [86, 87]. We also found an increase of apoptosis and, more interestingly, a modification of APP processing and Aβ levels (intensification of the amyloidogenic pathway of APP cleavage with increase of full-length APP and sAPPβ toward the formation of Aβ42 and an increase of the Aβ42:Aβ40 ratio) consistently with other studies [88]. As expected, we did not find senile plaques in normal old mice.
4. Expert opinion
It is noteworthy that both Tg and non-Tg models do not reproduce the entire clinical features of human AD. At present we have a number of very interesting tools to study AD but, we should not forget that they are just models with their intrinsic limitations. Tg animals have allowed several advances in this field but, as discussed, they do not reproduce the real pathophysiology since they are genetically “forced” to imitate the disease thus resulting in a “adulterated” phenotype with exacerbated (i.e. plaque deposition) or completely missing (i.s. neuronal loss) aspects, a different timeframe of the pathogenetic events (i.e. Aβ pathology always preceding tau pathology) and several compensatory mechanisms that might mask the real effect of the gene mutation. Moreover, it is not uncommon that they develop side behavioral attitudes (aggressiveness, stereotypies, inability to take care of the offspring, etc.).
In summary, there are several aspects to consider before beginning a study using Tg models of disease:
First, you must have a very good knowledge of the selected model. In addition to carefully read the available literature, you have always to remember that several factors might influence the phenotype especially when you work in vivo, from the genetic background, to the breeding conditions (light/dark cycle, housing, diet, renewal of the colony). Moreover, some strains present hearing loss or retinopathies, making their use not possible for behavioral studies such as FC (mice should hear the sound and recognize the space around them) or spatial memory test (mice should see the cues). One of the most frequent problems faced by researchers is the timing of testing/treating animals. As pointed out in the previous paragraph, each model develops the pathology at different ages, thus the aim of the study should be very clear. Obviously, if you want to understand whether a treatment might counteract a certain aspect the disease you should wait for the appropriate age.
In general, our advice is to perform a prior screening of your models before starting the experiments. Moreover you should keep in mind what is impaired when. For example, in our conditions: i) LTP is impaired at about 9–10 months in Tg2576, at 3 months in APP/PS1, at 22 months in physiological models of aging; ii) BST is impaired at about 12 months in Tg2576, at 6 months in APP/PS1, after 26 months in physiological models of aging; iii) short-term memory is impaired at 12 months in Tg2576, at 3 months in APP/PS1, at 22 months in physiological models of aging; iv) long-term memory is impaired at 12 months in Tg2576, at 6 months in APP/PS1, at 24 months in physiological models of aging. See Figure 1 for a summary. The increase of Aβ levels starts approximately with the impairment of synaptic plasticity and continues to rise with age leading to plaque deposition. As already pointed out, in aged mice there was an increase of Aβ load but plaques were not present even at 28 months of age.
Figure 1.
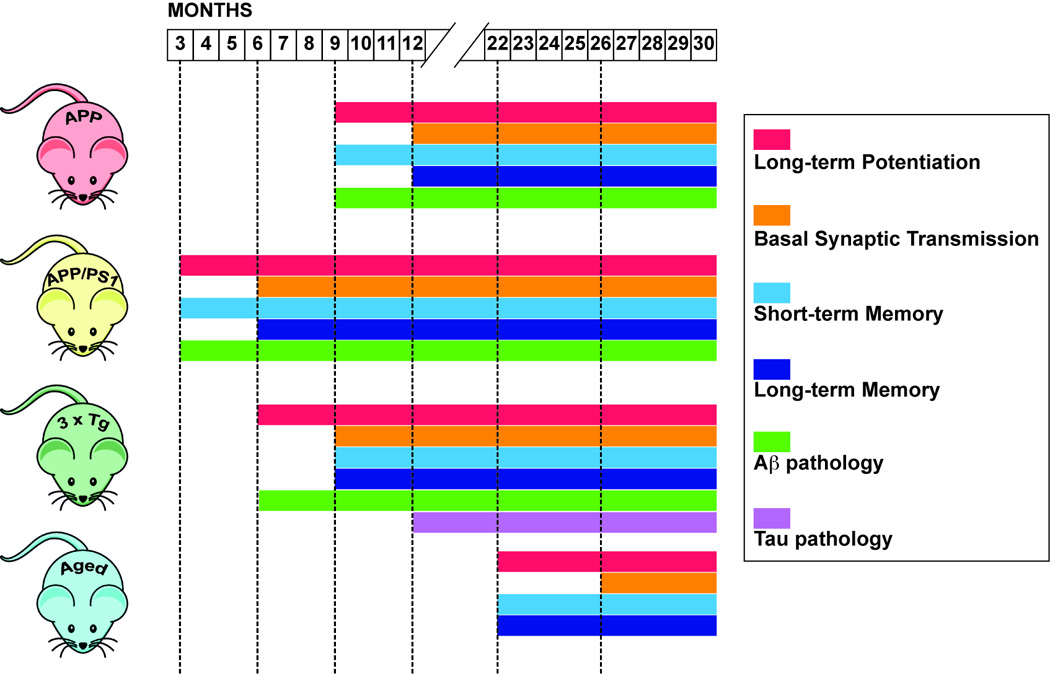
Gantt chart showing the onset and progression of synaptic impairment, memory loss and Aβ and tau pathology in 3 mouse Tg models of AD (single APP Tg2576, double APP/PS1, triple 3xTg) and in a physiological model of aging.
When the aim is to validate a therapy, another important point is to keep in mind the aim of the treatment: To delay the onset of the disease? To slow down the progression of the disease? To act on functional parameters (synaptic plasticity and/or memory), on plaques and neurofibrillary tangles formation or both? To investigate the acute, chronic or long-lasting effect of a drug? In general, we suggest to test the initial functional deficit (before structural damage as evidenced by BST impairment and plaques formation) based on the concept that AD is thought to begin as a synaptic disorder produced, at least in part, by soluble Aβ [89].
On the other hand, if you use a non-Tg model with Aβ injections, it is very important to consider that an acute injection could be useful to study the mechanisms underlying Aβ toxicity, but it will hardly reproduce the chronic AD phenotype.
When approaching a pre-clinical study we need to keep in mind that animal models reproduce only some aspects of the disease and, at the moment, it is not possible to recapitulate the entire human clinical picture. Furthermore, even if behavioral tests used in rodents have been designed to parallel the neuropsychological evaluation used in humans, some cognitive domains (such as language) cannot be investigated in animals. Several aspects should be considered when designing a pre-clinical study: a very good knowledge of the selected model, the various factors that might influence the phenotype, the timing of testing/treating animals because each model develops some aspects of the pathology at different ages. Although with their intrinsic limitations, both Tg and non-Tg AD models allow investigating synaptic plasticity, memory, histopathological modifications and molecular mechanisms underlying the disease. They represent therefore an invaluable tool to improve our knowledge of the disease, to better understand its pathophysiology and to establish new therapeutic strategies.
Acknowledgments
The authors are supported boy National Institutes of Health grants AG034248 and NS49442. They are also supported by a grant from the Alzheimer's Association (IIRG-09-134220).
Footnotes
Financial and Competing Interests Disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
- 1.Alzheimer A. Über einen eigenartigen schweren Erkrankungsprozeß der Hirnrinde. Neurologisches Centralblatt. 1906;23:1129–1136. [Google Scholar]
- 2.Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer's disease. Lancet. 1997;349:1546–1549. doi: 10.1016/S0140-6736(96)10203-8. [DOI] [PubMed] [Google Scholar]
- 3.Kraepelin E. Psychiatrie: Ein Lehrbuch für Studierende und Ärzte. Leipzig: Barth. 1910:593–632. [Google Scholar]
- 4.Rocca WA, Amaducci LA, Schoenberg BS. Epidemiology of clinically diagnosed Alzheimer's disease. Ann Neurol. 1986;19:415–424. doi: 10.1002/ana.410190502. [DOI] [PubMed] [Google Scholar]
- 5.St George-Hyslop PH. Molecular genetics of Alzheimer's disease. Biol Psychiatry. 2000;47:183–199. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- 6.Tang YP, Gershon ES. Genetic studies in Alzheimer's disease. Dialogues Clin Neurosci. 2003;5:17–26. doi: 10.31887/DCNS.2003.5.1/yptang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia D, Watanabe H, Wu B, et al. Presenilin-1 Knockin Mice Reveal Loss-of-Function Mechanism for Familial Alzheimer's Disease. Neuron. 2015;85:967–981. doi: 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quon D, Wang Y, Catalano R, et al. Formation of beta-amyloid protein deposits in brains of transgenic mice. Nature. 1991;352:239–241. doi: 10.1038/352239a0. [DOI] [PubMed] [Google Scholar]
- 9.Higgins LS, Holtzman DM, Rabin J, et al. Transgenic mouse brain histopathology resembles early Alzheimer's disease. Ann Neurol. 1994;35:598–607. doi: 10.1002/ana.410350514. [DOI] [PubMed] [Google Scholar]
- 10.Sandhu FA, Salim M, Zain SB. Expression of the human beta-amyloid protein of Alzheimer's disease specifically in the brains of transgenic mice. J Biol Chem. 1991;266:21331–21334. [PubMed] [Google Scholar]
- 11.Lamb BT, Sisodia SS, Lawler AM, et al. Introduction and expression of the 400 kilobase amyloid precursor protein gene in transgenic mice [corrected] Nat Genet. 1993;5:22–30. doi: 10.1038/ng0993-22. [DOI] [PubMed] [Google Scholar]
- 12.Pearson BE, Choi TK. Expression of the human beta-amyloid precursor protein gene from a yeast artificial chromosome in transgenic mice. Proc Natl Acad Sci U S A. 1993;90:10578–10582. doi: 10.1073/pnas.90.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Games D, Adams D, Alessandrini R, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. * First description of a Tg AD mouse model, the PDAPP mouse.
- 14.Masliah E, Sisk A, Mallory M, et al. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dodart JC, Meziane H, Mathis C, et al. Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behav Neurosci. 1999;113:982–990. doi: 10.1037//0735-7044.113.5.982. [DOI] [PubMed] [Google Scholar]
- 16.Chen G, Chen KS, Knox J, et al. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- 17.Gerlai R, Fitch T, Bales KR, et al. Behavioral impairment of APP(V717F) mice in fear conditioning: is it only cognition? Behav Brain Res. 2002;136:503–509. doi: 10.1016/s0166-4328(02)00198-5. [DOI] [PubMed] [Google Scholar]
- 18.Dodart JC, Mathis C, Saura J, et al. Neuroanatomical abnormalities in behaviorally characterized APP(V717F) transgenic mice. Neurobiol Dis. 2000;7:71–85. doi: 10.1006/nbdi.1999.0278. [DOI] [PubMed] [Google Scholar]
- 19.Reilly JF, Games D, Rydel RE, et al. Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc Natl Acad Sci U S A. 2003;100:4837–4842. doi: 10.1073/pnas.0330745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 21.Corcoran KA, Lu Y, Turner RS, et al. Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer's disease. Learn Mem. 2002;9:243–252. doi: 10.1101/lm.51002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. King DL, Arendash GW. Behavioral characterization of the Tg2576 transgenic model of Alzheimer's disease through 19 months. Physiol Behav. 2002;75:627–642. doi: 10.1016/s0031-9384(02)00639-x. * Article describing the behavioral characterization of sensorimotor and cognitive performance of the Tg2576 mouse model of AD at different ages.
- 23.Chapman PF, White GL, Jones MW, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 24.Stern EA, Bacskai BJ, Hickey GA, et al. Cortical synaptic integration in vivo is disrupted by amyloid-beta plaques. J Neurosci. 2004;24:4535–4540. doi: 10.1523/JNEUROSCI.0462-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lalonde R, Lewis TL, Strazielle C, et al. Transgenic mice expressing the betaAPP695SWE mutation: effects on exploratory activity, anxiety, and motor coordination. Brain Res. 2003;977:38–45. doi: 10.1016/s0006-8993(03)02694-5. [DOI] [PubMed] [Google Scholar]
- 26.Gil-Bea FJ, Aisa B, Schliebs R, et al. Increase of locomotor activity underlying the behavioral disinhibition in tg2576 mice. Behav Neurosci. 2007;121:340–344. doi: 10.1037/0735-7044.121.2.340. [DOI] [PubMed] [Google Scholar]
- 27.Bedrosian TA, Herring KL, Weil ZM, et al. Altered temporal patterns of anxiety in aged and amyloid precursor protein (APP) transgenic mice. Proc Natl Acad Sci U S A. 2011;108:11686–11691. doi: 10.1073/pnas.1103098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holcomb L, Gordon MN, McGowan E, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. * First description of the APP/PS1 double Tg mouse model of AD.
- 29. Trinchese F, Liu S, Battaglia F, et al. Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann Neurol. 2004;55:801–814. doi: 10.1002/ana.20101. ** Article describing the evolution of AD-related pathology including histopathology, synaptic function and memory over time in APP/PS1 mice.
- 30.Ertekin-Taner N. Genetics of Alzheimer's disease: a centennial review. Neurol Clin. 2007;25:611–667. doi: 10.1016/j.ncl.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duff K, Eckman C, Zehr C, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 32.Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci U S A. 1999;96:11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 34.Borchelt DR, Ratovitski T, van Lare J, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 35.Gong B, Vitolo OV, Trinchese F, et al. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gong B, Cao Z, Zheng P, et al. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- 37.Trinchese F, Fa' M, Liu S, et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–2807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puzzo D, Staniszewski A, Deng SX, et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer's disease mouse model. J Neurosci. 2009;29:8075–8086. doi: 10.1523/JNEUROSCI.0864-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez-Gonzalez R, Pascual C, Antequera D, Bolos M, Redondo M, Perez DI, Pérez-Grijalba V, Krzyzanowska A, Sarasa M, Gil C, Ferrer I, Martinez A, Carro E. Phosphodiesterase 7 inhibitor reduced cognitive impairment and pathological hallmarks in a mouse model of Alzheimer's disease. Neurobiol Aging. 2013;34:2133–2145. doi: 10.1016/j.neurobiolaging.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Guo J, Zhao X, Chen Z, Wang G, Liu A, Wang Q, Zhou W, Xu Y, Wang C. Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav Brain Res. 2013;250:230–237. doi: 10.1016/j.bbr.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 41.Liu MY, Wang S, Yao WF, Zhang ZJ, Zhong X, Sha L, He M, Zheng ZH, Wei MJ. Memantine improves spatial learning and memory impairments by regulating NGF signaling in APP/PS1 transgenic mice. Neuroscience. 2014;273:141–151. doi: 10.1016/j.neuroscience.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 42.McClean PL, Hölscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer's disease. Neuropharmacology. 2014;76:57–67. doi: 10.1016/j.neuropharm.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 43.Sierksma AS, van den Hove DL, Pfau F, Philippens M, Bruno O, Fedele E, Ricciarelli R, Steinbusch HW, Vanmierlo T, Prickaerts J. Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology. 2014;77:120–130. doi: 10.1016/j.neuropharm.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 44.Zhang MY, Zheng CY, Zou MM, Zhu JW, Zhang Y, Wang J, Liu CF, Li QF, Xiao ZC, Li S, Ma QH, Xu RX. Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol Aging. 2014;35:2713–2725. doi: 10.1016/j.neurobiolaging.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Li H, Ruberu K, Muñoz SS, Jenner AM, Spiro A, Zhao H, Rassart E, Sanchez D, Ganfornina MD, Karl T, Garner B. Apolipoprotein D modulates amyloid pathology in APP/PS1 Alzheimer's disease mice. Neurobiol Aging. 2015 doi: 10.1016/j.neurobiolaging.2015.02.010. pii: S0197-4580(15)00110-4. [DOI] [PubMed] [Google Scholar]
- 46.Schrott LM, Jackson K, Yi P, Dietz F, Johnson GS, Basting TF, Purdum G, Tyler T, Rios JD, Castor TP, Alexander JS1. Acute oral Bryostatin-1 administration improves learning deficits in the APP/PS1 transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. 2015;12:22–31. doi: 10.2174/1567205012666141218141904. [DOI] [PubMed] [Google Scholar]
- 47.Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001;891:42–53. doi: 10.1016/s0006-8993(00)03186-3. [DOI] [PubMed] [Google Scholar]
- 48.Lok K, Zhao H, Zhang C, He N, Shen H, Wang Z, Zhao W, Yin M. Effects of accelerated senescence on learning and memory, locomotion and anxiety-like behavior in APP/PS1 mouse model of Alzheimer's disease. J Neurol Sci. 2013;335:145–154. doi: 10.1016/j.jns.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 49.Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW. Lifelong immunization with human beta-amyloid (1–42) protects Alzheimer's transgenic mice against cognitive impairment throughout aging. Neuroscience. 2005;130:667–684. doi: 10.1016/j.neuroscience.2004.09.055. [DOI] [PubMed] [Google Scholar]
- 50. Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. * Article describing the generation and features of the triple-transgenic model (3xTg-AD) harboring PS1(M146V), APP(Swe), and tau(P301L) transgenes.
- 51.Billings LM, Oddo S, Green KN, et al. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 52.Kazim SF, Blanchard J, Dai CL, Tung YC, LaFerla FM, Iqbal IG, Iqbal K. Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2014;71:110–130. doi: 10.1016/j.nbd.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Stevens LM, Brown RE. Reference and working memory deficits in the 3xTg-AD mouse between 2 and 15-months of age: A cross-sectional study. Behav Brain Res. 2015;278:496–505. doi: 10.1016/j.bbr.2014.10.033. [DOI] [PubMed] [Google Scholar]
- 54.Cantarella G, Di Benedetto G, Puzzo D, et al. Neutralization of TNFSF10 ameliorates functional outcome in a murine model of Alzheimer's disease. Brain. 2015;138:203–216. doi: 10.1093/brain/awu318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giménez-Llort L, Blázquez G, Cañete T, Johansson B, Oddo S, Tobeña A, LaFerla FM, Fernández-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007;31:125–147. doi: 10.1016/j.neubiorev.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Pietropaolo S, Feldon J, Yee BK. Age-dependent phenotypic characteristics of a triple transgenic mouse model of Alzheimer disease. Behav Neurosci. 2008;122:733–747. doi: 10.1037/a0012520. [DOI] [PubMed] [Google Scholar]
- 57.Sterniczuk R, Antle MC, Laferla FM, Dyck RH. Characterization of the 3xTg-AD mouse model of Alzheimer's disease: part 2. Behavioral and cognitive changes. Brain Res. 2010;1348:149–155. doi: 10.1016/j.brainres.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 58.Bertoni-Freddari C, Sensi SL, Giorgetti B, et al. Decreased presence of perforated synapses in a triple-transgenic mouse model of Alzheimer's disease. Rejuvenation Res. 2008;11:309–313. doi: 10.1089/rej.2008.0660. [DOI] [PubMed] [Google Scholar]
- 59.Oakley H, Cole SL, Logan S, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jawhar S, Trawicka A, Jenneckens C, et al. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2012;33:196, e29–e40. doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 61.Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 62.Ma QL, Zuo X, Yang F, et al. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J Biol Chem. 2013;288:4056–4065. doi: 10.1074/jbc.M112.393751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castillo-Carranza DL, Gerson JE, Sengupta U, et al. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014;40:S97–S111. doi: 10.3233/JAD-132477. [DOI] [PubMed] [Google Scholar]
- 64. Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29:10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. * Article demonstrating the presence of neurofibrillary tangles associated with synaptic function and memory loss in a mouse expressing human tau.
- 65. Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Martin Watterson D. Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol. 2007;82:277–296. doi: 10.1016/S0074-7742(07)82015-0. * Review summarizing how neuroinflammation is involved in progression of the disease mechanisms that can be targeted for drug discovery in AD and other neurodegenerative diseases.
- 66.Balducci C, Forloni G. In vivo application of beta amyloid oligomers: a simple tool to evaluate mechanisms of action and new therapeutic approaches. Curr Pharm Des. 2014;20:2491–2505. doi: 10.2174/13816128113199990497. [DOI] [PubMed] [Google Scholar]
- 67. Puzzo D, Lee L, Palmeri A, Calabrese G, Arancio O. Behavioral assays with mouse models of Alzheimer's disease: practical considerations and guidelines. Biochem Pharmacol. 2014;88:450–467. doi: 10.1016/j.bcp.2014.01.011. ** A very thorough explanation of the behavioral assessment and methodology that can be used in Tg and non-Tg mouse models of AD.
- 68.Moe JG, Puzzo D, Chatterjee I, et al. Evaluation of the effect of extracellular tau oligomers on synaptic function. Washington DC (USA) Society for Neurosciences Abstract. 2008 543.17/R12. [Google Scholar]
- 69.Moe JG, Chatterjee I, Davidowitz EJ, et al. Modulation of synaptic function by extracellular tau enriched in oligomers. Alzheimers Dement. 2009;5:P499. [Google Scholar]
- 70.Moe JG, Chatterjee I, Puzzo D, et al. Validation of extracellular tau oligomer target for drug discovery in a novel animal model. San Diego (USA) Society for Neurosciences Abstract. 2010;(527):8. [Google Scholar]
- 71.Moe JG, Chatterjee I, Puzzo D, et al. Extracellular oligomeric tau inhibits memory formation in mice. Alzheimers Dement. 2010;6:S277. [Google Scholar]
- 72. Puzzo D, Privitera L, Leznik E, et al. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. ** First evidence of the positive role of physiological concentrations of amyloid-beta in hippocampal synaptic plasticity and memory.
- 73.Puzzo D, Privitera L, Fa' M, et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann Neurol. 2011;69:819–830. doi: 10.1002/ana.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Puzzo D, Privitera L, Palmeri A. Hormetic effect of amyloid-β peptide in synaptic plasticity and memory. Neurobiol Aging. 2012;33:1484, e15–e24. doi: 10.1016/j.neurobiolaging.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 75.Fiorito J, Saeed F, Zhang H, et al. Synthesis of quinoline derivatives: discovery of a potent and selective phosphodiesterase 5 inhibitor for thetreatment of Alzheimer's disease. Eur J Med Chem. 2013;60:285–294. doi: 10.1016/j.ejmech.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watterson DM, Grum-Tokars VL, Roy SM, et al. Development of Novel In Vivo Chemical Probes to Address CNS Protein Kinase Involvement in Synaptic Dysfunction. PLoS One. 2013;8:e66226. doi: 10.1371/journal.pone.0066226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Palmeri A, Privitera L, Giunta S, et al. Inhibition of phosphodiesterase-5 rescues age-related impairment of synaptic plasticity and memory. Behav Brain Res. 2013;240:11–20. doi: 10.1016/j.bbr.2012.10.060. [DOI] [PubMed] [Google Scholar]
- 78.Puzzo D, Loreto C, Giunta S, et al. Effect of phosphodiesterase-5 inhibition on apoptosis and beta amyloid load in aged mice. Neurobiol Aging. 2014;35:520–531. doi: 10.1016/j.neurobiolaging.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Puzzo D, Bizzoca A, Loreto C, et al. Role of F3/contactin expression profile in synaptic plasticity and memory in aged mice. Neurobiol Aging. 2015;36:1702–1715. doi: 10.1016/j.neurobiolaging.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 80.Takeda T, Hosokawa M, Takeshita S, et al. A new murine model of accelerated senescence. Mech Ageing Dev. 1981;17:183–194. doi: 10.1016/0047-6374(81)90084-1. [DOI] [PubMed] [Google Scholar]
- 81.Treuting PM, Hopkins HC, Ware CA, et al. Generation of genetically altered mouse models for aging studies. Exp Mol Pathol. 2002;72:49–55. doi: 10.1006/exmp.2001.2405. [DOI] [PubMed] [Google Scholar]
- 82.Sprott RL. Introduction: animal models of aging: something old, something new. ILAR J. 2011;52:1–3. doi: 10.1093/ilar.52.1.1. [DOI] [PubMed] [Google Scholar]
- 83.Yeoman M, Scutt G, Faragher R. Insights into CNS ageing from animal models of senescence. Nat Rev Neurosci. 2012;13:435–445. doi: 10.1038/nrn3230. [DOI] [PubMed] [Google Scholar]
- 84.Li Q, Zhao HF, Zhang ZF, et al. Long-term administration of green tea catechins prevents age-related spatial learning and memory decline in C57BL/6 J mice by regulating hippocampal cyclic amp-response element binding protein signaling cascade. Neuroscience. 2009;159:1208–1215. doi: 10.1016/j.neuroscience.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 85.Bergado JA, Almaguer W, Rojas Y, et al. Spatial and emotional memory in aged rats: a behavioral-statistical analysis. Neuroscience. 2011;172:256–269. doi: 10.1016/j.neuroscience.2010.10.064. [DOI] [PubMed] [Google Scholar]
- 86.Wang W, Li S, Dong HP, et al. Differential impairment of spatial and nonspatial cognition in a mouse model of brain aging. Life Sci. 2009;85:127–135. doi: 10.1016/j.lfs.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 87.Soontornniyomkij V, Risbrough VB, Young JW, et al. Hippocampal calbindin-1 immunoreactivity correlate of recognition memory performance in aged mice. Neurosci Lett. 2012;516:161–165. doi: 10.1016/j.neulet.2012.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Placanica L, Zhu L, Li YM. Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS One. 2009;4:e5088. doi: 10.1371/journal.pone.0005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. * Interesting review article arguing that AD begins with alterations of hippocampal synaptic efficacy caused by diffusible oligomeric assemblies of Aβ.