

Abstract
In species with a heterogametic sex, population genetics theory predicts that DNA sequences on the X chromosome can evolve faster than comparable sequences on autosomes. Both neutral and nonneutral evolutionary processes can generate this pattern. Complex traits like gene expression are not predicted to have accelerated evolution by these theories, yet a “faster-X” pattern of gene expression divergence has recently been reported for both Drosophila and mammals. Here, we test the hypothesis that accelerated adaptive evolution of cis-regulatory sequences on the X chromosome is responsible for this pattern by comparing the relative contributions of cis- and trans-regulatory changes to patterns of faster-X expression divergence observed between strains and species of Drosophila with a range of divergence times. We find support for this hypothesis, especially among male-biased genes, when comparing different species. However, we also find evidence that trans-regulatory differences contribute to a faster-X pattern of expression divergence both within and between species. This contribution is surprising because trans-acting regulators of X-linked genes are generally assumed to be randomly distributed throughout the genome. We found, however, that X-linked transcription factors appear to preferentially regulate expression of X-linked genes, providing a potential mechanistic explanation for this result. The contribution of trans-regulatory variation to faster-X expression divergence was larger within than between species, suggesting that it is more likely to result from neutral processes than positive selection. These data show how accelerated evolution of both coding and noncoding sequences on the X chromosome can lead to accelerated expression divergence on the X chromosome relative to autosomes.
Keywords: cis-regulation, trans-regulation, selection, gene expression, faster-X
Introduction
Empirical studies have shown that X-linked sequences can evolve more rapidly than autosomal sequences in species with heterogametic XY sex chromosomes such as Drosophila and mammals (Torgerson and Singh 2003; Khaitovich et al. 2005; Baines and Harr 2007; Begun et al. 2007; Connallon 2007; Langley et al. 2012; Mackay et al. 2012; Hu et al. 2013; Garrigan et al. 2014). A similar pattern has been observed for Z-linked sequences in species with ZW sex chromosomes (Mank, Nam, et al. 2010; Sackton et al. 2014). This accelerated sequence divergence, commonly called “faster-X” in XY species and “faster-Z” in ZW species, is often attributed to beneficial alleles having a greater probability of fixation on the X chromosome than on autosomes because recessive alleles are not masked in the heterogametic sex (Charlesworth et al. 1987); but, nonadaptive processes can also explain this pattern. For example, differences in effective population size between the X chromosome and autosomes can cause a faster-X pattern of sequence evolution because the presence of fewer X chromosomes than autosomes in a given population can cause purifying selection to be less effective on the X chromosome (Mank, Vicoso, et al. 2010). Sexual selection can also cause a greater probability of fixation for mutations on the X chromosome than autosomes by altering the numbers of males and/or females available for breeding (Caballero 1995; Charlesworth 2001; Laporte and Charlesworth 2002; Vicoso and Charlesworth 2009a). The presence (Charlesworth et al. 1987) and method (Mank, Vicoso, et al. 2010) of dosage compensation, chromosome and sex-based differences in recombination rates (Vicoso and Charlesworth 2009b; Campos et al. 2014), male-biased mutational processes (Crow 2000; Kirkpatrick and Hall 2004; Ellegren and Parsch 2007; Xu et al. 2012), and the nonrandom distribution of functional categories of genes on chromosomes (e.g., sex-biased, gene ontology groups) (Baines and Harr 2007; Baines et al. 2008; Grath and Parsch 2012) have also all been implicated in faster substitution rates for the X chromosome than autosomes.
The faster-X (or faster-Z) pattern of sequence evolution appears to be stronger in birds and mammals than in Drosophila (Mank, Vicoso, et al. 2010), and it has been attributed primarily to differences in effective population size between the X (or Z) chromosome and autosomes as well as sexual selection in these species. In Drosophila, some studies have found evidence of faster-X sequence evolution, whereas others have not (reviewed in Presgraves [2008]). Most recently, evidence for more nonsynonymous changes on the X chromosome than autosomes was observed among species in the melanogaster clade, but not for species in the pseudoobscura clade (Avila et al. 2014). Comparisons between intraspecific polymorphism and interspecific divergence suggest that adaptive evolution contributes to the faster-X effect pattern of sequence evolution in Drosophila when it does occur (Mank, Nam, et al. 2010; Mank, Vicoso, et al. 2010; Campos et al. 2014).
Although the faster-X theory describes the evolution of DNA sequences, a faster-X pattern has also recently been reported for gene expression divergence in both mammals (Brawand et al. 2011) and Drosophila (Kayserili et al. 2012; Llopart 2012; Meisel et al. 2012). In Drosophila, this pattern appears to be strongest for genes with male-biased expression (Grath and Parsch 2012; Llopart 2012; Meisel et al. 2012), consistent with models involving selection acting on alleles revealed only in the hemizygous sex. The molecular mechanisms responsible for greater expression divergence on the X chromosome compared with the autosomes remain unknown, but the prevailing hypothesis is that accelerated sequence divergence on the X chromosome resulted in accelerated cis-regulatory divergence, and this cis-regulatory divergence has caused the observed faster-X expression divergence (Kayserili et al. 2012; Meisel et al. 2012; Meisel and Connallon 2013). This idea is based on the fact that cis- regulatory sequences controlling a gene’s expression are located on the same chromosome as the affected gene, whereas trans-regulatory factors that interact with these cis-regulatory sequences can be located anywhere in the genome (Brem et al. 2002; reviewed in Wray et al. 2003). Here, we directly test the hypothesis that cis-regulatory divergence is responsible for the faster evolution of gene expression on the X chromosome and investigate the role of neutral and nonneutral processes in this evolution.
Results and Discussion
To investigate the molecular mechanisms underlying faster-X evolution of gene expression, we compared total mRNA abundance as well as cis- and trans-regulation between 1) two divergent strains of D. melanogaster, the North American Zhr line and an African isofemale line z30 (referred to as mel-mel); 2) D. simulans (Tsimbazaza) and D. sechellia (droSec1) (referred to as sim-sech); and 3) D. melanogaster (Zhr) and D. simulans (Tsimbazaza) (referred to as mel-sim). These measurements were based on RNA-seq data collected from whole adult females from each strain as well as from F1 hybrid females produced by crossing each pair of strains. Although using females limited our power to study male-biased genes, the presence of both maternally and paternally inherited X chromosomes in females allowed us to measure relative cis-regulatory activity for X-linked genes. Genomic DNA was also sequenced from each strain and used to construct strain-specific genomes. These genomes were used to quantify sequence divergence between pairs of strains as well as to align RNA-seq reads for quantifying total and allele-specific gene expression. After controlling for differences in sequencing depth across samples, equalizing power for statistical tests on a gene-by-gene basis among the mel-mel, sim-sech, and mel-sim data sets, and excluding genes with low sequencing coverage or low proportions of allele-specific reads, 4,851 genes were deemed suitable for comparing cis- and trans-regulatory evolution in all three comparisons (Coolon et al. 2014). Of these genes, 998 (21%) are X-linked.
Faster-X Sequence Divergence
To determine whether the specific set of genes we examined showed faster-X sequence evolution in the mel-mel, sim-sech, and/or mel-sim comparisons, we contrasted percent sequence divergence for coding (all transcribed sites) and noncoding regions of individual genes between genes on the X chromosome and genes on the autosomes (see Materials and Methods). We found that sequence divergence was significantly greater on the X chromosome than on autosomes for both coding (fig. 1A) and noncoding sequences (fig. 1B) in both interspecific comparisons (Mann–Whitney U test, Psim-sech coding = 9.0 × 10−8, Pmel-sim coding = 2.2 × 10−11, Psim-sech noncoding = 7.1 × 10−12, Pmel-sim noncoding = 0.0017), consistent with previous studies (Begun et al. 2007; Langley et al. 2012; Mackay et al. 2012; Hu et al. 2013). Greater differentiation of X-linked sequences between two strains of the same species (mel-mel) is not predicted by the faster-X theory, yet we also observed greater differentiation for the X chromosome than autosomes for the mel-mel comparison (Mann–Whitney U test, Pmel-mel coding < 1.0 × 10−15, Pmel-mel noncoding < 1.0 × 10−15). Prior studies of sequence differentiation between North American and African strains of D. melanogaster have also observed a faster-X pattern of sequence differentiation (Yukilevich et al. 2010; Langley et al. 2012), and North American and African strains of D. melanogaster have been suggested to be in the early stages of speciation (Hollocher, Ting, Pollack, et al. 1997; Hollocher, Ting, Wu, et al. 1997). Our mel-mel comparison therefore most likely captures a level of differentiation somewhere between intraspecific polymorphism and interspecific divergence.
Fig. 1.
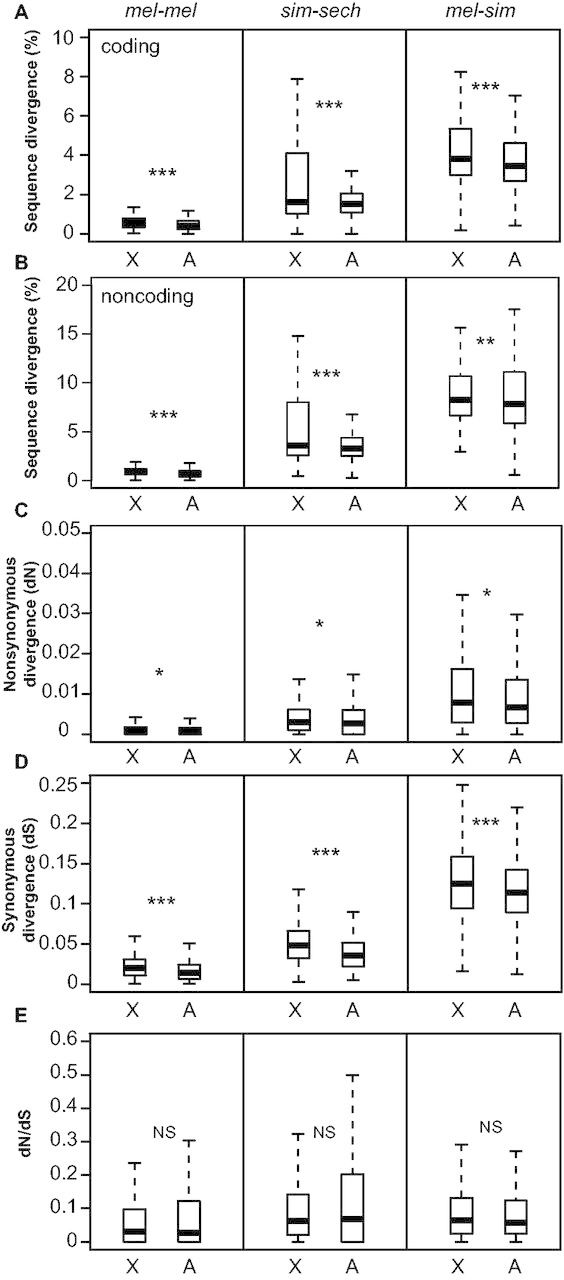
Faster-X divergence of DNA sequence. Percent sequence divergence is shown for coding (A) and noncoding (B) regions of each of the 4,851 genes analyzed in all three comparisons (mel-mel, sim-sech, mel-sim). Maximum-likelihood estimates of (C) nonsynonymous (dN) and (D) synonymous changes (dS) as well as (E) the ratio of nonsynonymous to synonymous changes (dN/dS) are shown. Boxplots show the distribution of these values separately for genes on the X chromosome and autosomes with the median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. Statistical significance of the difference in median values between X-linked and autosomal genes was determined using Mann–Whitney U tests, where NS indicates not significant, *P ≤ 0.05, **P ≤ 0.001, and ***P ≤ 1 × 10−4.
To investigate the evolutionary forces shaping the patterns of sequence variation we observed, we used PAML (Yang 2007) to generate maximum-likelihood estimates of coding sequence divergence at synonymous sites (dS), nonsynonymous sites (dN), and nonsynonymous relative to synonymous sites (dN/dS) for each comparison. We found that both dN (fig. 1C) and dS (fig. 1D) were significantly greater for genes on the X chromosome than genes located on autosomes in all comparisons (Mann–Whitney U test, Pmel-mel dN = 0.005, Psim-sech dN = 0.05, Pmel-sim dN = 0.003, Pmel-mel dS = 1.0 × 10−15, Psim-sech dS = 1.0 × 10−15, Pmel-sim dS = 1.9 × 10−7). However, we found no significant difference between the X chromosome and autosomes for the dN/dS ratio in any comparison (fig. 1E; Mann–Whitney U test, Pmel-mel dN/dS = 0.71, Psim-sech dN/dS = 0.98, Pmel-sim dN/dS = 0.07). The absence of accelerated dN/dS on the X chromosome for this set of genes is surprising given a recent study showing evidence of this pattern genome-wide between D. melanogaster and D. sechellia (Avila et al. 2014), but is consistent with the mixed-results of prior studies that performed similar analyses in the melanogaster species group (Thornton et al. 2006; Begun et al. 2007; Connallon 2007; Hu et al. 2013). A greater mutation rate on the X chromosome than on autosomes might explain the higher dN and dS values observed in all comparisons; however, the higher recombination rate on the X chromosome reducing the effects of background selection (Vicoso and Charlesworth 2009b) or less effective purifying selection on the X chromosome resulting from differences in effective population size between the X chromosome and autosomes could also produce this pattern. The extent to which each of these and/or other evolutionary processes (Vicoso and Charlesworth 2006) or demographic factors (Pool and Nielsen 2008) contribute to the observed patterns of sequence variation will require further investigation.
Faster-X Expression Divergence
To determine whether the 4,851 genes analyzed showed greater expression divergence on the X chromosome than on autosomes, we used the nonparametric Spearman’s correlation coefficient (ρ) to compare levels of mRNA expression between strains or species in each comparison and then compared the degree of divergence for genes on the X chromosome and genes on autosomes. Spearman’s ρ has previously been used to contrast expression divergence between the X chromosome and autosomes (Brawand et al. 2011; Kayserili et al. 2012; Meisel et al. 2012) and compares the rank order of gene expression levels for orthologous genes between samples. We found that expression divergence (1 − ρ) was greater for genes on the X chromosome than for genes on the autosomes in all three comparisons (fig. 2A), although this difference was only statistically significant in mel-mel and sim-sech (one-sided permutation test, Pmel-mel = 0.01, Psim-sech = 0.005, Pmel-sim = 0.1). The “faster-X” pattern of expression divergence in the sim-sech and mel-sim comparisons is consistent with previous comparisons between pairs of Drosophila species with greater evolutionary distances (Kayserili et al. 2012; Meisel et al. 2012), but the faster-X expression change observed in the mel-mel comparison conflicts with previous reports of intraspecific expression polymorphism in D. melanogaster that found no evidence for faster-X expression change (Hutter et al. 2008; Müller et al. 2011; Kayserili et al. 2012; Llopart 2012; Meisel et al. 2012). As described above for sequence variation, this difference likely results from the fact that the North American and African strains used for our mel-mel comparison are more divergent than those used in prior studies.
Fig. 2.

Faster-X divergence of gene expression. (A) Overall expression divergence (1 − ρ) is shown for X-linked genes and autosomal genes for each of the three comparisons. Error bars indicate the 2.5 and 97.5 percentiles of 1 − ρ from bootstrapping 10,000 times. Statistical significance for the difference between the X chromosome and autosomes was determined by permuting the chromosome assignment of individual genes relative to their expression levels 100,000 times. (B) The magnitude of expression divergence measured for each gene was determined and boxplots showing the distribution for the X chromosome and autosomes with the median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. Statistical significance of the difference between the X chromosome and autosomes was determined with Mann–Whitney U tests. (C) The proportion of genes with significant expression differences in each comparison is shown and error bars represent the 2.5 and 97.5 percentiles from bootstrapping 10,000 times. Statistical significance of the difference between the X chromosome and autosomes was determined with Fisher’s exact tests (NS indicates not significant, *P ≤ 0.05, **P ≤ 0.001, and ***P ≤ 1 × 10−4).
Spearman’s ρ captures differences in the magnitude of expression divergence for individual genes as well as differences in the number of genes that are differentially expressed. To examine these two factors separately, we quantified the magnitude of expression divergence on a gene-by-gene basis and examined the number of genes with evidence of a significant change in expression based on binomial exact tests. We found that the magnitude of expression differences for individual genes was higher for genes on the X chromosome than autosomes in all three cases, but only significantly so in sim-sech and mel-sim (fig. 2B, Mann–Whitney U test, Pmel-mel = 0.059, Psim-sech = 0.0015, Pmel-sim = 0.04). By contrast, the proportion of genes with a significant difference in expression was significantly higher on the X chromosome in all three comparisons (fig. 2C, Fisher’s exact test, Pmel-mel = 6.1 × 10−6, Psim-sech = 8.6 × 10−8, Pmel-sim = 0.0017), indicating that the faster-X pattern of gene expression we observed is primarily driven by a greater number of genes with expression differences on the X chromosome.
Faster-X Cis-Regulatory Divergence
To test the hypothesis that faster-X divergence in gene expression is driven by faster-X divergence in cis-regulation (Kayserili et al. 2012; Meisel et al. 2012; Meisel and Connallon 2013), we examined relative allele-specific expression in F1 hybrids, which provides a direct readout of relative cis-regulatory activity (Cowles et al. 2002; Wittkopp et al. 2004). We found that overall divergence (1 − ρ) in cis- regulatory activity was significantly greater for genes on the X chromosome than on autosomes in both sim-sech and mel-sim, but not in mel-mel (one-sided permutation test, Pmel-mel = 0.99, Psim-sech = 0.006, Pmel-sim = 0.046; fig. 3A). This result is consistent with prior studies showing that cis-regulatory changes contribute more to expression divergence between species than within species (Wittkopp et al. 2008; Coolon et al. 2014). It also underscores that differentiation in the mel-mel comparison is distinct from interspecific divergence despite showing a pattern of faster-X sequence and expression evolution (figs. 1 and 2). The magnitude of cis-regulatory divergence between genes on the X chromosome and autosomes showed no significant difference in any comparison (Mann–Whitney U test, Pmel-mel = 0.97, Psim-sec = 0.30, Pmel-sim = 0.48; fig. 3B), even after scaling by total regulatory divergence (% cis; Wittkopp et al. 2008), (Mann–Whitney U test, Pmel-mel = 0.97, Psim-sec = 0.88, Pmel-sim = 0.96; fig. 3C); however, the number of genes with evidence of a significant difference in cis-regulatory activity was significantly higher on the X chromosome than on autosomes in sim-sech and mel-sim (Fisher’s exact test, Pmel-mel = 0.41, Psim-sec = 0.002, Pmel-sim = 0.001; fig. 3D). Similar to our findings for total expression differences, these analyses indicate that the number of genes with cis-regulatory differences has a bigger impact on the overall cis-regulatory divergence we observed than the magnitude of cis-regulatory differences.
Fig. 3.
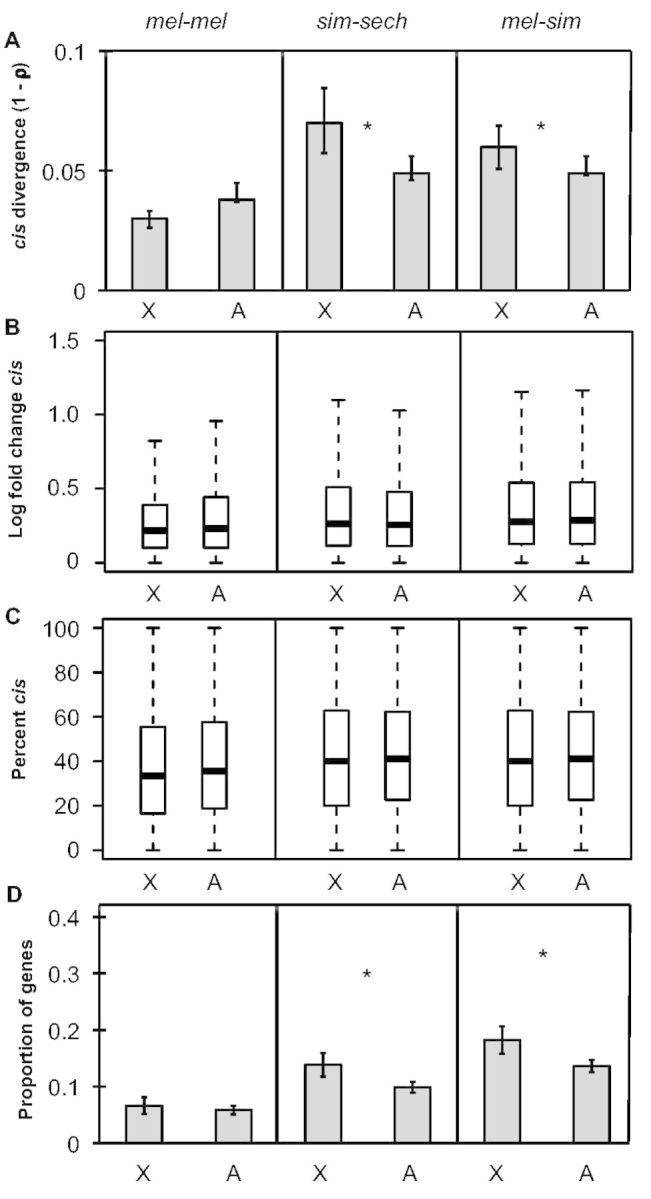
Faster-X divergence of cis-regulation. (A) Divergence in cis-regulatory activity (1 − ρ) is shown for genes on the X-chromosome and autosomes for all three comparisons and error bars represent the 2.5 and 97.5 percentiles from 10,000 bootstrap replicates. Statistical significance for the difference between the X chromosome and autosomes was determined with permutation tests (100,000 permutations). (B) Boxplots showing the magnitude of cis-regulatory divergence in F1 hybrids (abs(log2(allele 1/allele 2))) for the X chromosome and autosomes for each comparison are shown with median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. (C) The magnitude of cis-regulatory divergence scaled by total regulatory divergence ([|cis|/(|cis|+|trans|)] × 100, %cis; Wittkopp et al. 2008) was determined and boxplots with the median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. (B, C) Statistical significance of differences between the X chromosome and autosomes was determined using Mann–Whitney U tests. (D) The proportion of genes with significant cis-regulatory differences in F1 hybrids for each comparison is shown and error bars represent the 2.5 and 97.5 percentiles from bootstrapping 10,000 times. Statistical significance of the difference between the X chromosome and autosomes was determined with Fisher’s exact tests (NS indicates not significant, *P ≤ 0.05, **P ≤ 0.001, and ***P ≤ 1 × 10−4).
The data from the sim-sech and mel-sim comparisons are consistent with the hypothesis that cis-regulatory divergence contributes to the faster-X pattern of gene expression divergence between species. However, these cis-regulatory differences appear to be insufficient to fully explain the faster-X pattern of expression divergence: The magnitude of overall expression differences (compare fig. 2A with 3A) as well as the proportion of genes with significant expression differences (compare fig. 2C with 3D) are both smaller for cis-regulation than total expression differences.
Faster-X Trans-Regulatory Divergence
Changes in gene expression that are not explained by cis-regulatory differences can be attributed to trans-regulatory differences (Wittkopp et al. 2004), suggesting that trans-regulatory divergence might also contribute substantially to the faster-X pattern of expression divergence. Consistent with this hypothesis, overall trans-regulatory divergence (1 − ρ) was significantly higher on the X chromosome than on autosomes in mel-mel (permutation test, Pmel-mel = 8 × 10−5, Psim-sech = 0.43, Pmel-sim = 0.78; fig. 4A). The magnitude of trans-regulatory divergence was also greater for genes on the X chromosome than on autosomes for all three comparisons (Mann–Whitney U test, Pmel-mel = 0.02, Psim-sech = 0.0025, Pmel-sim = 0.0017; fig. 4B), but remained significant only for mel-mel and mel-sim after scaling for total regulatory divergence (% trans, Mann–Whitney U test, Pmel-mel = 0.024, Psim-sech = 0.12, Pmel-sim = 0.034; fig. 4C). The proportion of genes with evidence of significant trans-regulatory divergence was also significantly greater for X-linked than autosomal genes in all three comparisons (Fisher’s exact test, Pmel-mel = 1.2 × 10−6, Psim-sech = 1.9 × 10−5, Pmel-sim = 1.5 × 10−6; fig. 4D), with more genes showing evidence of trans-regulatory divergence than cis-regulatory divergence in each comparison (compare fig. 4D with 3D). These data indicate that faster-X trans-regulatory divergence is responsible for the faster-X pattern of expression divergence in mel-mel and contributes to this pattern in sim-sech and mel-sim. It remains to be seen whether trans-regulatory divergence also contributes significantly to the faster-X patterns of gene expression divergence observed among species with greater divergence times (Brawand et al. 2011; Kayserili et al. 2012; Meisel et al. 2012).
Fig. 4.
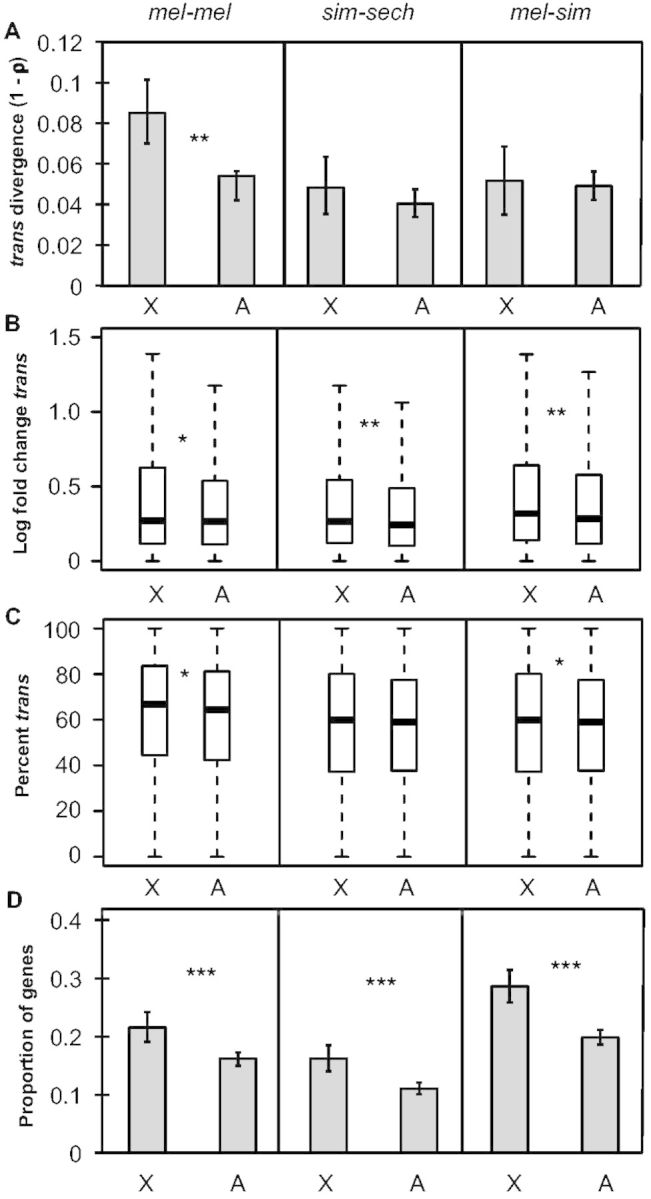
Faster-X divergence of trans-regulation. (A) Divergence in trans-regulatory activity (1 − ρ) is shown for genes on the X-chromosome and autosomes for all three comparisons and error bars represent the 2.5 and 97.5 percentiles from 10,000 bootstrap replicates. Statistical significance for the difference between the X chromosome and autosomes was determined with permutation tests (100,000 permutations). (B) Boxplots showing the magnitude of trans-regulatory divergence (abs(log2(species 1/species 2)) − abs(log2(allele 1/allele 2))) for the X chromosome and autosomes for each comparison are shown with the median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. (C) The magnitude of trans-regulatory divergence scaled by total regulatory divergence ([|trans|/(|cis|+|trans|)] × 100, %trans) was determined and boxplots with the median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. (B, C) Statistical significance of differences between X and autosome was determined using Mann–Whitney U tests. (D) The proportion of genes with significant trans-regulatory differences for each comparison is shown and error bars represent the 2.5 and 97.5 percentiles from bootstrapping 10,000 times. Statistical significance of the difference between the X chromosome and autosomes was determined with Fisher’s exact tests (NS indicates not significant, *P ≤ 0.05, **P ≤ 0.001, and ***P ≤ 1 × 10−4).
The large contribution of trans-regulatory divergence to differences seen between the North American and African strains of D. melanogaster is consistent with the excess of trans-regulatory variation previously observed segregating within species of Drosophila (Wittkopp et al. 2008; Coolon et al. 2014) and Saccharomyces (Emerson et al. 2010). However, the large contribution of trans-acting variation to the faster-X pattern of expression divergence is surprising because trans-regulatory factors controlling expression of X-linked genes are generally assumed to be distributed randomly across the genome. This is in stark contrast to cis-regulatory sequences, which are, by definition, always located on the X chromosome for X-linked genes. If this assumption about the genomic distribution of trans-acting factors is wrong, and trans-acting factors encoded by genes on the X chromosome preferentially regulate expression of X-linked genes, trans-regulatory divergence could indeed explain the faster-X pattern of expression divergence.
Preferential Regulation of X-Linked Genes by X-Linked Transcription Factors
Although many types of genes can have trans-acting effects on gene expression, genes encoding transcription factors (TFs) are commonly assumed to have the most direct effects (Yvert et al. 2003). Although X-linked TFs have previously been shown to disproportionately contribute to expression divergence genome-wide between species, we wanted to determine whether TFs encoded by genes on the X chromosome are likely to disproportionally affect expression of X-linked genes. To do this, we used a recently constructed gene regulatory network for D. melanogaster that contains 310,634 predicted regulatory interactions among 617 TFs and 12,286 target genes (Marbach et al. 2012). This network was constructed by integrating TF binding motif conservation, ChIP-chip and ChIP-seq for TFs and chromatin marks, and microarray and RNA-seq measures of gene expression (Marbach et al. 2012). We identified 4,379 genes common to our expression analyses and the published regulatory network and using the edges in this network; we compared the proportion of X-linked target genes for X-linked and autosomal TFs (fig. 5A). For the 213 TFs included in this set of 4,379 genes, 46 of which are X-linked (21.6%), we found that TFs encoded by genes on the X chromosome had a larger proportion of their target genes on the X chromosome than TFs encoded by genes on autosomes (Mann–Whitney U test, P4379 < 1.0 × 10−15). This was again true when we considered all 617 TFs in the network, 102 (16.5%) of which were X-linked (Mann–Whitney U test, Pall < 1.0 × 10−15), showing that this pattern is not limited to the set of genes that were analyzed. We also compared the proportion of TFs that are X-linked for target genes on the X chromosome and autosomes (fig. 5B). We found that target genes on the X chromosome were also regulated by a higher proportion of X-linked TFs than target genes on autosomes for both the 4,379 genes examined here (Mann–Whitney U test, P4379 = 1.4 × 10−11) and the full set of genes included in the network (Mann–Whitney U test, Pall < 1.0 × 10−15). Neither pattern was observed for any of the individual autosomes (supplementary figs. S1 and S2, Supplementary Material online), indicating that this finding does not result from a genome-wide pattern of preferential interactions between regulators and genes on the same chromosome. This finding that X-linked TFs seem to preferentially regulate X-linked genes suggests that accelerated sequence divergence of X-linked TFs (affecting their cis-regulatory sequences and/or coding sequences) might have a greater impact on expression of X-linked genes than autosomal genes.
Fig. 5.

X-linked TFs preferentially regulate X-linked target genes. (A) For TFs on either the X chromosome or autosomes, the proportion of downstream target genes on the X chromosome was determined using a Drosophila regulatory network (Marbach et al. 2012). Boxplots show the distribution of these values for the X-chromosome and the autosomes with the median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. Mann–Whitney U tests were used to compare the X chromosome and autosomes. (B) For target genes on the X chromosome and autosomes, the proportion of upstream TFs encoded by genes on the X chromosome. Boxplots show the distribution of these values for the X-chromosome and the autosomes with the median (center line) as well as 25th and 75th percentiles indicated (box upper and lower bounds) and whiskers extending 1.5× the interquartile range. Mann–Whitney U tests were performed to compare the X chromosome and autosomes (NS indicates not significant, *P ≤ 0.05, **P ≤ 0.001, and ***P ≤ 1 × 10−4).
To further explore this possibility, we tested whether the 46 TFs on the X chromosome showed greater sequence divergence than the 167 TFs on the autosomes. We found evidence of a faster-X pattern of sequence evolution for both coding (Mann–Whitney U test, Pmel-mel = 2.3 × 10−8, Psim-sech = 0.0007, Pmel-sim = 0.0016; supplementary fig. S3A, Supplementary Material online) and noncoding (Mann–Whitney U test, Pmel-mel = 8.04 × 10−7, Psim-sech = 0.00015, Pmel-sim = 1.4 × 10−5; supplementary fig. S3B, Supplementary Material online) sequences in all three comparisons, but a statistically significant faster-X pattern for dN and dS only in mel-mel and sim-sech (Mann–Whitney U test, Pmel-mel dN = 0.04, Psim-sech dN = 0.03, Pmel-sim dN = 0.12, Pmel-mel dS = 2.9 × 10−7, Psim-sech dS = 0.0003, Pmel-sim dS = 0.44; supplementary fig. S3C and D, Supplementary Material online). Although not statistically significant, median dN and dS were larger for X-linked than autosomal TFs even in the mel-sim comparison. The absence of a statistically significant faster-X pattern in mel-sim for dN or dS despite a significant faster-X pattern for coding sequence divergence presumably results from reduced power of the statistical tests when coding sequences were divided into these two categories. The dN/dS ratio was not significantly elevated for X-linked TFs compared with autosomal TFs in any comparison (Mann–Whitney U test, Pmel-mel = 0.99, Psim-sech = 0.27, Pmel-sim = 0.08; supplementary fig. S3E, Supplementary Material online), consistent with the absence of this pattern in the full set of genes. These data suggest greater amino acid sequence divergence of X-linked TFs compared with autosomal TFs, but provide no evidence that this excess is due to positive selection.
We also tested whether X-linked TFs showed greater divergence in total expression, cis-regulation, or trans-regulation than autosomal TFs. Comparing overall differences using 1 − Spearman’s ρ, magnitude of difference, and proportion of genes with differences, we found that none of these measures showed a significant difference between the X chromosome and autosomes in any comparison (P > 0.05 in all cases). That said, there is a finite number of TFs in the genome and it is possible that these statistical tests lack sufficient power to detect small differences. To test this possibility, we determined the magnitude of effect that could be detected with 80% power for each metric of expression or regulatory divergence. We found that, in all cases, the difference between the X chromosome and autosomes would need to have been 2–4 times greater than what we observed to achieve 80% detection power. This suggests that if there are differences between the X chromosome and autosomes for the regulation and/or expression of TFs, they are subtle. Given the proposed role of X-linked TFs in generating the faster-X expression pattern of expression divergence, elevated sequence divergence for X-linked TFs in the absence of elevated expression divergence could suggest that TFs on the X chromosome may have altered protein function.
Another prediction that follows from our hypothesis that greater divergence of X-linked TFs contributes to the faster-X pattern of expression divergence is that genes with a greater proportion of trans-acting regulators located on the X chromosome might tend to show greater differences in total expression and trans-regulatory divergence. Consistent with this prediction, we observed weak but statistically significant correlations between the proportion of trans-acting regulators of a gene that are X-linked and the magnitude of expression and trans-regulatory divergence in all but one comparison (trans-regulatory divergence in mel-sim), and even this case was marginally significant (supplementary fig. S4A and C, Supplementary Material online; permutation test, total expression: Pmel-mel < 1 × 10−4, Psim-sech = 2 × 10−4, Pmel-sim = 0.049; trans-regulation: Pmel-mel < 1 × 10−4, Psim-sech < 1 × 10−4, Pmel-sim = 0.07). The proportion of X-linked regulators for a gene is not expected to be related to its levels of cis-regulatory divergence, and we found no evidence of a significant correlation in any of the comparisons (supplementary fig. S4B, Supplementary Material online, permutation test, Pmel-mel = 0.73, Psim-sech = 0.23, Pmel-sim = 0.81).
Adaptive Cis-Regulatory Changes and Neutral Trans-Regulatory Changes Both Appear to Contribute to Faster-X Expression Divergence
Although we found no evidence of positive selection accelerating protein sequence divergence on the X chromosome, adaptive evolution might still contribute to faster-X expression divergence by increasing the probability of fixation for regulatory changes affecting expression of X-linked genes relative to genes on the autosomes. Prior work has argued that if adaptive processes play a role in faster-X evolution, they should have a larger impact on genes with male-biased or unbiased expression than those with female-biased expression because their phenotypic effects should be more pronounced in the hemizygous male sex (Baines et al. 2008). The sex-specific effects exhibited by many cis-regulatory variants (Massouras et al. 2012; Coolon et al. 2013; Meiklejohn et al. 2014; Stocks et al. 2015) might make them likely to contribute to this pattern of evolution. Faster-X sequence evolution was shown to be strongest for male-biased genes (Baines et al. 2008; Müller et al. 2011; Grath and Parsch 2012) and faster-X gene expression evolution has been shown to be either stronger (Meisel et al. 2012) or only present for genes with male-biased or unbiased expression (Llopart 2012), suggesting that selection does contribute to the faster-X pattern in Drosophila.
To test for differences among genes with male-biased, female-biased, and unbiased expression in our data set, we used published classifications of sex-bias for D. melanogaster genes (Zhang et al. 2007) and compared the proportion of genes with significant differences in total expression, cis-regulation, and trans-regulation between X-linked and autosomal genes for each of these gene sets when expressed in females (fig. 6). For the 39 X-linked and 183 autosomal female-biased genes, we found no evidence for faster-X expression (Fisher’s exact tests, Pmel-mel = 0.85, Psim-sech = 0.25, Pmel-sim = 0.63; fig. 6A), cis-regulatory (Fisher’s exact tests, Pmel-mel = 1, Psim-sech = 0.39, Pmel-sim = 1; fig. 6A), or trans-regulatory (Fisher’s exact tests, Pmel-mel = 0.26, Psim-sech = 0.06, Pmel-sim = 1; fig. 6A) divergence in any comparison. By contrast, for the 28 X-linked and 314 autosomal male-biased genes, we observed a significant faster-X pattern of total expression divergence (Fisher’s exact tests, Pmel-mel = 0.84, Psim-sech = 0.0036, Pmel-sim = 0.0024; fig. 6B) and cis-regulatory divergence (Fisher’s exact tests, Pmel-mel = 0.07, Psim-sech = 1.5 × 10−5, Pmel-sim = 0.0013; fig. 6B) in both interspecific comparisons (sim-sech and mel-sim) but not in the intraspecific comparison (mel-mel) (fig. 6B). There was no evidence of faster-X trans-regulatory divergence for male-biased genes in any comparison (Fisher’s exact tests, Pmel-mel = 0.49, Psim-sech = 0.25, Pmel-sim = 0.51; fig. 6B). For the 931 X-linked and 3,350 autosomal genes with unbiased expression, a faster-X pattern was observed for expression divergence (Fisher’s exact tests, Pmel-mel = 4.7 × 10−7, Psim-sech = 6.3 × 10−7, Pmel-sim = 0.0025; fig. 6C) and trans-regulatory divergence (Fisher’s exact tests, Pmel-mel = 6.5 × 10−7, Psim-sech = 1.3 × 10−5, Pmel-sim = 4.6 × 10−8; fig. 6C) in all comparisons. A faster-X pattern was also observed for cis-regulatory divergence in the two comparisons between species (Fisher’s exact tests, Pmel-mel = 0.32, Psim-sech = 0.025, Pmel-sim = 0.0023; fig. 6C). The absence of evidence for faster-X cis-regulatory differentiation among male-biased, female-biased, and unbiased genes in the mel-mel comparison is consistent with the analysis of the full data set described above (fig. 3D).
Fig. 6.

Genes with male-biased and unbiased expression have faster-X cis-regulatory divergence between species. The proportion of genes with significant differences in gene expression (E), cis-regulation (C), or trans-regulation (T) that were X-linked or on autosomes for mel-mel, sim-sech and mel-sim are shown for genes with (A) female biased expression, (B) male-biased expression, or (C) unbiased expression. Error bars represent the 2.5 and 97.5 percentiles from bootstrapping 10,000 times. Statistical significance of the difference between the X chromosome and autosomes was determined with Fisher’s exact tests (NS indicates not significant, *P ≤ 0.05, **P ≤ 0.001, and ***P ≤ 1 × 10−4).
The fact that a faster-X pattern of cis-regulatory divergence was seen for male-biased but not female-biased genes and between but not within species suggests that natural selection is responsible for generating this pattern. The faster-X pattern of trans-regulatory divergence, by contrast, was seen for unbiased genes both within and between species, suggesting that it might result from neutral processes. Taken together, these data suggest a model in which the faster-X pattern of expression divergence observed in all three comparisons results from a combination of two distinct molecular and evolutionary mechanisms: 1) Natural selection preferentially fixing cis-regulatory changes affecting male-biased genes on the X chromosome and 2) neutral processes, combined with the preferential regulation of X-linked genes by X-linked TFs, elevating trans-regulatory divergence throughout the X chromosome.
Materials and Methods
Data Analyzed
Sequence divergence, differences in total gene expression, and differences in cis- and trans-regulation for the mel-mel, sim-sech, and mel-sim comparisons were derived from genomic sequences and RNA-seq data provided in the supplementary material for Coolon et al. (2014). These data include 1) differences in expression between 7- and 10-day-old adult females from the two strains or species being compared (mel-mel, sim-sech, and mel-sim); 2) differences in allele-specific expression level in F1 hybrids produced by crossing D. melanogaster Zhr females with D. melanogaster z30 males, D. simulans females with D. sechellia males, and D. melanogaster (Zhr) females with D. simulans males; these data provide a readout of relative cis-regulatory activity; 3) measurements of % cis (cis-regulatory divergence scaled for total regulatory divergence; [|cis|/(|cis|+|trans|)] × 100; Wittkopp et al. 2008); 4) measurements of trans-regulatory activity, calculated as the difference between observed parental difference and cis-regulatory difference in F1 hybrids; 5) measurements of %trans (trans-regulatory divergence scaled for total regulatory divergence; [|trans|/(|cis|+|trans|)] × 100; Wittkopp et al. 2008); and 6) results of binomial and Fisher’s exact tests classifying each gene as having a significant expression difference between strains or species as well as significant cis- and/or trans-regulatory differences. The chromosomal location of each gene was determined using data from FlyBase (St Pierre et al. 2014). Gene sets with female-biased, male-biased, and unbiased expression were obtained from Zhang et al. (2007), with sex-biased genes identified based on statistically significant higher expression in one sex than the other.
Quantification of Percent Sequence Divergence
To determine percent sequence divergence in each comparison (mel-mel, sim-sec, and mel-sim), we created reverse chain files to LiftOver coordinates from D. melanogaster dm3 space to each of the other strain or species genomic space (zhr, z30, Tsimbazaza, droSec1; Coolon et al. 2014) using the chainSwap utility from the UCSC Genome Browser (Kent et al. 2002). We downloaded the D. melanogaster genomic annotations for coding (all transcribed sites) and noncoding (all intergenic sequences to the next gene and intronic sequences) regions for each gene from FlyBase (St Pierre et al. 2014). Using the chain files, we converted the dm3 genomic coordinates for each coding and noncoding region for each gene used for quantification in this study into their respective strain- or species-specific genomic coordinates. Using these coordinates, sequences for each region were extracted from each strain- or species-specific genome (provided in the supplementary material for Coolon et al. 2014). These sequences were aligned in pairs using Fast Statistical Alignment (FSA version 1.15.9) (Bradley et al. 2009) and the number of divergent sites per gene (coding and noncoding) was determined using a custom Perl script (pairwise_aln_FSA.pl).
Calculation of dN, dS, and dN/dS Sequence Metrics
We extracted the shortest transcript for all genes from the D. melanogaster dm3 genome build on FlyBase (St Pierre et al. 2014), and using the chain files described above converted the dm3 genomic coordinates for each transcript into their respective strain- or species-specific genomic coordinates using the liftOver tool from the UCSC Genome Browser (Kent et al. 2002). Genes with a valid start codon and an in-frame stop codon were retained for analyses. Coding sequences were aligned in pairs for each comparison (mel-mel, sim-sech, and mel-sim) with FSA (Bradley et al. 2009). We then used PAML version 4.7a (Yang 2007) to generate estimates of dN, dS, and dN/dS for each gene using the yn00 package and default settings. Ratios of dN/dS estimated using maximum-likelihood methods have great uncertainty when dS is very low so all genes with no observed synonymous differences were removed. After filtering, 4,351 genes were used in mel-mel (X: 926, A: 3,425), 3,328 in sim-sech (X: 529, A: 2,799), and 3,699 in the mel-sim (X: 622, A: 3,077) comparison.
Mining the D. melanogaster Regulatory Network
As stated in the main text, the supervised gene regulatory network was obtained from the online supplementary material from Marbach et al. (2012). This network consisted of 310,634 predicted regulatory interactions among 617 TFs (regulators) and their 12,286 target genes. The complete set of genes and their chromosomal locations was downloaded from FlyBase (St Pierre et al. 2014). We limited the predicted regulatory interactions to those whose TF and target gene were both present in the gene expression data (4,851 genes from Coolon et al. 2014). We then determined the proportion of X-linked targets for each regulator on each chromosome, as well as the proportion of X-linked regulators for each target on each chromosome, using custom Perl scripts (chromosomal_regulators_subset.pl and chromosomal_targets_subset.pl). This process was repeated with different focal chromosomes. To make sure that the features we observed were not due to the specific gene set used in expression quantification, we also determined these same proportions for the complete supervised gene regulatory network using custom Perl scripts (chromosomal_regulators.pl and chromosomal_targets.pl).
Statistical Analyses
As described more fully in Coolon et al. (2014), the total expression difference was calculated for each gene in each comparison as log2(genotype 1 read count/genotype 2 read count) from “mixed parental” samples containing equal numbers of total RNA-seq reads from the two strains or species being compared. The cis-regulatory difference was calculated for each gene as log2(allele 1 read count/allele 2 read count) using data from the F1 hybrids described above. The trans-regulatory difference for each gene in each comparison was calculated as the difference between the total expression and cis-regulatory differences: log2(genotype 1 read count/genotype 2 read count) − log2(allele 1 read count/allele 2 read count). % cis was then calculated as [|cis|/(|cis|+|trans|)] × 100, and % trans was calculated as [|trans|/(|cis|+|trans|)] × 100.
Spearman’s ρ was used to measure differences in total expression, cis-regulatory divergence, and trans-regulatory divergence on a genomic scale in the mel-mel, sim-sech, and mel-sim comparisons. To test for statistically significant differences in ρ between genes on the X chromosome and genes on the autosomes, we permuted the chromosomal location of genes relative to their expression level 100,000 times and repeated each analysis. A faster-X pattern was inferred when the observed data showed greater divergence on the X chromosome than at least 5,000 of the permuted data sets, corresponding to P = 0.05. This is a one-sided test of the hypothesis that genes on the X chromosome have diverged more between the strains or species examined than genes on the autosome. Error bars on measures of Spearman’s ρ for each sample were calculated by sampling 4,851 gene-specific read counts 10,000 times from the observed 4,851 genes with replacement using R, calculating ρ in each case, and identifying the 2.5% and 97.5% percentiles. Nonparametric Mann–Whitney U tests were used to compare percent sequence divergence between the X chromosome and autosomes for coding and noncoding sequences, dN, dS, and dN/dS ratios, the magnitude of differences in total expression, cis-regulation and trans-regulation between the X chromosome and autosomes for the mel-mel, sim-sech, and mel-sim comparisons before and after scaling for total regulatory divergence. Fisher’s exact tests were used to compare the number of significant tests for differences in total expression, cis-regulation and trans-regulation between the X chromosome and autosomes in all three comparisons and for female-biased, male-biased and unbiased gene sets. For X-linked TFs, the proportion of their targets on the X chromosome was compared with the proportion of their targets on autosomes, and for X-linked target genes, the proportion of their upstream regulators on the X chromosome was compared with the proportion of their upstream regulators on autosomes using nonparametric Mann–Whitney U tests. Correlations between the proportion of upstream regulators on the X chromosome and the magnitude of expression, cis- and trans-regulatory divergence were performed and the significance of these correlations was determined by permuting the magnitude of expression, cis- or trans-regulatory change relative to the proportion of regulators on the X chromosome 100,000 times and repeating each analysis. A significant result was inferred when the observed data showed greater correlation in at least 5,000 of the permuted data sets, corresponding to P = 0.05.
All statistical analyses were performed in R (version 3.1.0, CRAN) (R Development Core Team 2013) using a custom script that is available at http://sites.lsa.umich.edu/wittkopp-lab/publications/, last accessed June 16, 2015 and medians, P values and samples sizes for each test reported in supplementary table S1, Supplementary Material online.
Supplementary Material
Supplementary figures S1–S4 and table S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Brian Metzger, Richard Lusk, Fabien Duveau, and Alisha John, as well as the editor and three anonymous reviewers, for helpful comments on the manuscript. Funding for this work was provided by the National Institute of Health (5F32GM089009-02 to J.D.C. and 5R01GM095296 to B.R.G.), the National Science Foundation (NSF 0903629 to K.R.S. and MCB-1021398 to P.J.W.), and the Alfred P. Sloan Research Foundation (fellowship to P.J.W.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Institute of Health, National Science Foundation, or Sloan Foundation.
References
- Avila V, Marion de Proce S, Campos JL, Borthwick H, Charlesworth B, Betancourt AJ. 2014. Faster-X effects in two Drosophila lineages. Genome Biol Evol. 6:2968–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JF, Harr B. 2007. Reduced X-linked diversity in derived populations of house mice. Genetics 175:1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JF, Sawyer SA, Hartl DL, Parsch J. 2008. Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Mol Biol Evol. 25:1639–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al. 2007. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 5:2534–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L. 2009. Fast statistical alignment. PLoS Comput Biol. 5:e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478:343–348. [DOI] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L. 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296:752–755. [DOI] [PubMed] [Google Scholar]
- Caballero A. 1995. On the effective size of populations with separate sexes, with particular reference to sex-linked genes. Genetics 139:1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos JL, Halligan DL, Haddrill PR, Charlesworth B. 2014. The relation between recombination rate and patterns of molecular evolution and variation in Drosophila melanogaster. Mol Biol Evol. 31:1010–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 2001. The effect of life-history and mode of inheritance on neutral genetic variability. Genet Res. 77:153–166. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH. 1987. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 130:113–146 [Google Scholar]
- Connallon T. 2007. Adaptive protein evolution of X-linked and autosomal genes in Drosophila: implications for faster-X hypotheses. Mol Biol Evol. 24:2566–2572. [DOI] [PubMed] [Google Scholar]
- Coolon JD, McManus CJ, Stevenson KR, Graveley BR, Wittkopp PJ. 2014. Tempo and mode of regulatory evolution in Drosophila. Genome Res. 24:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, Webb W, Wittkopp PJ. 2013. Sex-specific effects of cis- regulatory variants in Drosophila melanogaster. Genetics 195:1419–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles CR, Hirschhorn JN, Altshuler D, Lander ES. 2002. Detection of regulatory variation in mouse genes. Nat Genet. 32:432–437. [DOI] [PubMed] [Google Scholar]
- Crow JF. 2000. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 1:40–47. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 8:689–698. [DOI] [PubMed] [Google Scholar]
- Emerson JJ, Hsieh L-C, Sung H-M, Wang T-Y, Huang C-J, Lu HH-S, Lu M-YJ, Wu S-H, Li W-H. 2010. Natural selection on cis and trans regulation in yeasts. Genome Res. 20:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan D, Kingan SB, Geneva AJ, Vedanayagam JP, Presgraves DC. 2014. Genome diversity and divergence in Drosophila mauritiana: multiple signatures of faster X evolution. Genome Biol Evol. 6:2444–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grath S, Parsch J. 2012. Rate of amino acid substitution is influenced by the degree and conservation of male-biased transcription over 50 Myr of Drosophila evolution. Genome Biol Evol. 4:346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollocher H, Ting CT, Wu ML, Wu C-I. 1997. Incipient speciation by sexual isolation in Drosophila melanogaster: extensive genetic divergence without reinforcement. Genetics 147:1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollocher H, Ting C-T, Pollack F, Wu C-I. 1997. Incipient speciation by sexual isolation in Drosophila melanogaster: variation in mating preference and correlation between sexes. Evolution 51:1175–1181. [DOI] [PubMed] [Google Scholar]
- Hu TT, Eisen MB, Thornton KR, Andolfatto P. 2013. A second- generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Res. 23:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter S, Saminadin-Peter SS, Stephan W, Parsch J. 2008. Gene expression variation in African and European populations of Drosophila melanogaster. Genome Biol. 9:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayserili MA, Gerrard DT, Tomancak P, Kalinka AT. 2012. An excess of gene expression divergence on the X chromosome in Drosophila embryos: implications for the faster-X hypothesis. PLoS Genet. 8:e1003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res. 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, Weiss G, Lachmann M, Pääbo S. 2005. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309:1850–1854. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M, Hall DW. 2004. Male-biased mutation, sex linkage, and the rate of adaptive evolution. Evolution 58:437–440. [PubMed] [Google Scholar]
- Langley CH, Stevens K, Cardeno C, Lee YCG, Schrider DR, Pool JE, Langley SA, Suarez C, Corbett-Detig RB, Kolaczkowski B, et al. 2012. Genomic variation in natural populations of Drosophila melanogaster. Genetics 192:533–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte V, Charlesworth B. 2002. Effective population size and population subdivision in demographically structured populations. Genetics 162:501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopart A. 2012. The rapid evolution of X-linked male-biased gene expression and the large-X effect in Drosophila yakuba, D. santomea, and their hybrids. Mol Biol Evol. 29:3873–3886. [DOI] [PubMed] [Google Scholar]
- Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. 2012. The Drosophila melanogaster genetic reference panel. Nature 482:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Nam K, Ellegren H. 2010. Faster-Z evolution is predominantly due to genetic drift. Mol Biol Evol. 27:661–670. [DOI] [PubMed] [Google Scholar]
- Mank JE, Vicoso B, Berlin S, Charlesworth B. 2010. Effective population size and the Faster-X effect: empirical results and their interpretation. Evolution 64:663–674. [DOI] [PubMed] [Google Scholar]
- Marbach D, Roy S, Ay F, Meyer PE, Candeias R, Kahveci T, Bristow CA, Kellis M. 2012. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 22:1334–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massouras A, Waszak SM, Albarca-Aguilera M, Hens K, Holcombe W, Ayroles JF, Dermitzakis ET, Stone EA, Jensen JD, Mackay TFC, et al. 2012. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 8:e1003955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Coolon JD, Hartl DL, Wittkopp PJ. 2014. The roles of cis- and trans-regulation in the evolution of regulatory incompatibilities and sexually dimorphic gene expression. Genome Res. 24:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Connallon T. 2013. The faster-X effect: integrating theory and data. Trends Genet. 29:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. 2012. Faster-X evolution of gene expression in Drosophila. PLoS Genet. 8:e1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller L, Hutter S, Stamboliyska R, Saminadin-Peter SS, Stephan W, Parsch J. 2011. Population transcriptomics of Drosophila melanogaster females. BMC Genomics 12:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Nielsen R. 2008. The impact of founder events on chromosomal variability in multiply mating species. Mol Biol Evol. 25:1728–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC. 2008. Sex chromosomes and speciation in Drosophila. Trends Genet. 24:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackton TB, Corbett-Detig RB, Nagaraju J, Vaishna L, Arunkumar KP, Hartl DL. 2014. Positive selection drives faster-Z evolution in silkmoths. Evolution. 68:2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Pierre SE, Ponting L, Stefancsik R, McQuilton P. 2014. FlyBase 102—advanced approaches to interrogating FlyBase. Nucleic Acids Res. 42:D780–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks M, Dean R, Rogell B, Friberg U. 2015. Sex-specific trans-regulatory variation on the Drosophila melanogaster X chromosome. PLoS Genet. 11:e1005015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. 2013. R: a language and environment for statistical computing. Vienna (Austria): Available from: http://R-project.org/. [Google Scholar]
- Thornton K, Bachtrog D, Andolfatto P. 2006. X chromosomes and autosomes evolve at similar rates in Drosophila: no evidence for faster-X protein evolution. Genome Res. 16:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson DG, Singh RS. 2003. Sex-linked mammalian sperm proteins evolve faster than autosomal ones. Mol Biol Evol. 20:1705–1709. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. 2006. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet. 7:645–653. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. 2009a. Effective population size and the faster-X effect: an extended model. Evolution 63:2413–2426. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. 2009b. Recombination rates may affect the ratio of X to autosomal noncoding polymorphism in African populations of Drosophila melanogaster. Genetics 181:1699–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2004. Evolutionary changes in cis and trans gene regulation. Nature 430:85–88. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2008. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet. 40:346–350. [DOI] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. 2003. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 20:1377–1419. [DOI] [PubMed] [Google Scholar]
- Xu K, Oh S, Park T, Presgraves DC, Yi SV. 2012. Lineage-specific variation in slow- and fast-X evolution in primates. Evolution 66:1751–1761. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yukilevich R, Turner TL, Aoki F, Nuzhdin SV, True JR. 2010. Patterns and processes of genome-wide divergence between North American and African Drosophila melanogaster. Genetics 186:219–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvert G, Brem RB, Whittle J, Akey JM, Foss E, Smith EN, Mackelprang R, Kruglyak L. 2003. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet. 35:57–64. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. 2007. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 450:233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.