

Abstract
Multiple mechanisms exist for endocrine disruption; one nonreceptor-mediated mechanism is via effects on aromatase, an enzyme critical for maintaining the normal in vivo balance of androgens and estrogens. We adapted the AroER tri-screen 96-well assay to 1536-well format to identify potential aromatase inhibitors (AIs) in the U.S. Tox21 10K compound library. In this assay, screening with compound alone identifies estrogen receptor alpha (ERα) agonists, screening in the presence of testosterone (T) identifies AIs and/or ERα antagonists, and screening in the presence of 17β-estradiol (E2) identifies ERα antagonists. Screening the Tox-21 library in the presence of T resulted in finding 302 potential AIs. These compounds, along with 31 known AI actives and inactives, were rescreened using all 3 assay formats. Of the 333 compounds tested, 113 (34%; 63 actives, 50 marginal actives) were considered to be potential AIs independent of cytotoxicity and ER antagonism activity. Structure-activity analysis suggested the presence of both conventional (eg, 1, 2, 4, - triazole class) and novel AI structures. Due to their novel structures, 14 of the 63 potential AI actives, including both drugs and fungicides, were selected for confirmation in the biochemical tritiated water-release aromatase assay. Ten compounds were active in the assay; the remaining 4 were only active in high-throughput screen assay, but with low efficacy. To further characterize these 10 novel AIs, we investigated their binding characteristics. The AroER tri-screen, in high-throughput format, accurately and efficiently identified chemicals in a large and diverse chemical library that selectively interact with aromatase.
Keywords: AroER tri-screen, Tox21 10K library, environmental chemicals, quantitative high throughput screening, aromatase enzyme assay
A number of natural and synthetic chemicals in our environment have the potential to modulate endocrine signaling pathways and affect development and reproduction systems by mimicking natural hormones (Fowler et al., 2012; Nordkap et al., 2012). Additionally, many frequently used drugs have been shown to interfere with normal endocrine activity, resulting in significant attention from the Interagency Breast Cancer & Environmental Research Coordinating Committee (IBCERCC) (http://www.niehs.nih.gov/about/assets/docs/summary_of_recs_508.pdf) and the U.S. Food and Drug Administration (FDA) Center for Drug Evaluation and Research (CDER) (http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm369043.pdf).
To date, the preponderance of studies on endocrine disrupting chemicals (EDCs) has focused on their effects on steroid hormone receptors. However, considering the structural features of EDCs, it is likely that some of them could also alter normal physiological function through nonreceptor mechanisms by modifying the activity of enzymes responsible for the synthesis and metabolism of steroid hormones (Chen, 1998).
A goal of the U.S. Tox21 high-throughput screening program is to develop toxicity profiles for thousands of environmental chemicals using multiple cell-based assays targeting a variety of signaling pathways, including those that involve steroid hormone signaling (Tice et al., 2013). For assessing disruption of the hormone signaling pathways, many screens have been developed to identify compounds that directly interact with hormone receptors and have been adapted for use in the Tox21 program (Huang et al., 2011b, 2014). However, few test methods are available for identifying compounds that affect endocrine function through nonreceptor-mediated mechanisms, and none of these approaches were suitable for the quantitative high-throughput screening (qHTS) 1536-well plate format used by Tox21. Therefore, development of screening methods targeting nonreceptor-mediated mechanisms such as aromatase is needed to broaden the identification of potential environmental chemicals. Aromatase is a member of cytochrome P450 isozymes involving in the steroid biosynthesis pathway. A qHTS of chemicals interacting with 5 recombinant drug-metabolizing cytochrome P450 have been reported (Veith et al., 2009).
In the final step of the steroid biosynthesis pathway, aromatase converts testosterone (T) to estradiol (E2) or androstenedione to estrone. Aromatase plays a key role in maintaining the androgen/estrogen balance in many tissues throughout the body. Abnormal levels of aromatase enzyme activity have been linked to endocrine-related diseases (Jones et al., 2007). For example, abnormally low levels of aromatase adversely impact the development of reproductive organs (Braunstein, 1999), while excessively high enzyme activity promotes breast cancer cell proliferation (Chen, 1998). Prenatal suppression of aromatase was found to produce a subpopulation of male rats with same-sex preference (Olvera-Hernandez et al., 2015). Furthermore, while aromatase deficiency is extremely rare in humans, important information has been obtained from the examination of these patients (Chen et al., 2015). Affected women present with ambiguous genitalia at birth, elevated androgens and undetectable estrogens, primary amenorrhea, and failure of breast development at puberty. Men with aromatase deficiency usually present after puberty with continuing linear growth, tall stature, unfused epiphyses, delayed bone age, eunuchoid skeletal proportions, genu valgum, decreased bone mineral density, overweight or obesity, dyslipidemia, liver steatosis, insulin resistance, and impaired fertility. These observations support that abnormal reduction of aromatase would significantly affect normal development. In addition, unexpected suppression of aromatase during development could result in the accumulation of androgens. Androgens such as T are known to play a significant role in obesity, glucose homeostasis, and lipid metabolism (Saad and Gooren, 2011). Obesity and metabolic diseases have been shown to increase the incidence of breast cancer (Eliassen et al., 2006), increase aromatase expression in breast tissue (Morris et al., 2011), worsen the outcome of hormone-receptor-positive breast cancer (Sparano et al., 2010), and reduce responsiveness to endocrine therapy (Pfeiler et al., 2011). Also, it has been reported that obesity promotes breast cancer by modifying insulin and the insulin-like growth factor axis and by changing circulating levels of cytokines and adipokines (Roberts et al., 2010). While some of the pesticides and fungicides used in agriculture, as well as some nonagricultural chemicals, have been shown to inhibit aromatase (Cheshenko et al., 2008; Quignot et al., 2012; Sanderson, 2006; Vinggaard et al., 2000), it is expected that other compounds to which individuals are exposed, including some drugs, might also have the potential to inhibit aromatase activity.
The AroER tri-screen assay, a high-throughput screening system for detecting aromatase inhibiting chemicals, was developed by the stable transfection of an estrogen receptor (ER)-positive, aromatase-expressing human breast cancer cell line MCF-7 with an estrogen-responsive element (ERE)-driven luciferase reporter plasmid and validated against a small library of EDCs (Chen et al., 2014). In this study, the assay was used to screen the Tox21 10K compound library in an effort to detect environmental chemicals with heretofore unrecognized aromatase inhibiting activity.
MATERIALS AND METHODS
Tox21 10K compound library
The Tox21 compound library consists of ∼12 500 (∼8300 unique) compounds procured from commercial sources by the U.S. Environmental Protection Agency (EPA), the National Institute of Environmental Health Sciences (NIEHS)/National Toxicology Program (NTP), and the National Institutes of Health (NIH) Chemical Genomics Center (now part of the NIH National Center for Advancing Translational Sciences [NCATS] (Huang et al., 2011a). The library consists of a large variety of chemicals, including pesticides, industrial chemicals, natural food products, and drugs. The latter category includes failed drugs that did not make it to market, drugs that are no longer marketed and drugs that are marketed currently. The list of unique compound substances, including chemical names and Chemical Abstracts Service Registry Numbers (CASRN) as well as curated chemical structures and autogenerated structure identifiers (formula, systematic names, SMILES, desalted SMILES, InChI) can be downloaded from the EPA DSSTox website (http://www.epa.gov/ncct/dsstox/sdf_tox21s.html). Each substance was prepared as a stock solution (generally at 20 mM) in dimethyl sulfoxide (DMSO) and serially diluted in 1536-well microplates to yield 15 concentrations (11 concentrations in the aromatase follow-up screen) generally ranging from 1.1 nM to 92 μM (final concentrations in the assay wells).
AroER tri-screen
The AroER tri-screen assay was developed by stable transfection of ER-positive, aromatase-expressing human breast cancer cell line MCF-7 with an ERE-driven luciferase reporter plasmid (Chen et al., 2014). The AroER tri-screen can identify 3 different types of active compounds: (1) ERα agonists, which increase the luminescence signal in the absence of 17β-estradiol (E2); (2) ERα antagonists, which inhibit an E2-induced luminescence signal; and (3) aromatase inhibitors (AIs) and/or ERα antagonists, which suppress a T-induced luminescence signal. Only compounds that suppress T-induced luminescence signal independent of ER antagonism or cytotoxicity are classified as potential AIs. Briefly, the assay was first optimized in a 96-well plate format and then miniaturized into a 1536-well plate format for use in screening the Tox21 10K compound library. Because several Tox21 10K qHTS screens targeting the ER pathway have been conducted in the BG1 (full receptor) or Hek293 (partial receptor) cell lines (Huang et al., 2014), we screened the 10K library using the AroER tri-screen in the presence of T only. In addition, a cell viability counterscreen (CellTiter-Fluor; Promega Corp, Madison, Wisconsin) was conducted to discriminate the biological actives from cytotoxic compounds. The screening was performed 3 times and 333 substances were prioritized for follow-up testing in both T-stimulated and E2-stimulated versions of the AroER tri-screen assay to identify AIs that were independent of ER antagonism in the MCF-7 cell line. The initial 10K screen results can be found in PubChem (https://pubchem.ncbi.nlm.nih.gov/); the assay identification numbers are 743083 (primary screen), 743084 (cell viability counterscreen), and 743139 (summary).
Screen using testosterone (T)-stimulated mode
AroER tri-screen cells (Chen et al., 2014) were cultured in Minimum Essential Medium (MEM) medium (GE Healthcare Hyclone; Logan, Utah) supplemented with 10% fetal bovine serum (FBS), 20 µg/ml Hygromycin, 50 µg/ml G418 and 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen; Carlsbad, California). The cells were maintained at 37°C under a humidified atmosphere and 5% CO2. Two days prior to the assay, the cells were cultured in MEM phenol red free medium containing 10% charcoal-stripped FBS. The cells in the MEM phenol red free medium containing 10% charcoal-stripped FBS were dispensed at 1500 cells/4 µl/well in 1536-well white tissue cultured plates using a Thermo Scientific Multidrop Combi (Thermo Fisher Scientific Inc, Waltham, Massachussetts). After the assay plates were incubated at a 37°C/5% CO2 incubator for 5 h, 23 nl of compounds dissolved in DMSO, positive controls, or DMSO only was transferred to the assay plate by a pin tool (Kalypsys, San Diego, California), followed by the addition of T (Sigma-Aldrich, St. Louis, Missouri) (0.5 nM final concentration). The final compound concentrations in the 5 µl assay volume ranged from 5 nM to 92 µM. The positive control plate format was as follows: Column 1, concentration-response titration of letrozole (Tocris, St. Louis, Missouri) from 3.2 pM to 46 μM with 0.5 nM T; Column 2, 500 nM of letrozole with 0.5 nM T in top 16 wells and bottom 16 wells for 100 nM of letrozole with 0.5 nM T; Columns 3 and 4, DMSO only with 0.5 nM T in top 16 wells and bottom 16 wells for Columns 3 and 4 are 92 μM of tetraoctyl ammonium bromide (Sigma-Aldrich) with 0.5 nM T and DMSO only with assay buffer, respectively. The plates were incubated at 37°C for 24 h. For cell viability readout, after 23.5 h, 1 µl/well of CellTiter-Fluor reagent (Promega Co, Fitchburg, Wisconsin) was added into the assay plates using a Flying Reagent Dispenser (FRD; Aurora Discovery, Carlsbad, California). After 30 min incubation at 37°C, the fluorescence intensity in the plates was measured using a ViewLux plate reader (PerkinElmer, Shelton, Connecticut). This was followed by the addition of 4 μl of ONE-Glo Luciferase Assay reagent (Promega, Madison, Wisconsin) using an FRD (Aurora Discovery, Irvine, California). The plates were incubated at room temperature for 30 min, and luminescence intensity was measured by ViewLux plate-reader (Perkin Elmer, Waltham, Massachusetts). Data were expressed as relative luminescence units. The raw plate reads for each titration point were first normalized relative to the 0.5 nM T plus letrozole control (500 nM, 100%) and the 0.5 nM T plus DMSO-only wells (basal, 0%).
Screen using 17β-estradiol (E2)-stimulated mode and no chemical stimulation mode
The AroER tri-screen cells were cultured in MEM phenol red free medium containing 10% charcoal-stripped FBS for 2 days prior to the assay. The cells, suspended in the MEM phenol red free medium containing 10% charcoal-stripped FBS, were dispensed at 1500 cells/5 µl/well for the ER agonist assay or 1500 cells/4 µl/well for the ER antagonist assay in 1536-well white tissue cultured plates using a Thermo Scientific Multidrop Combi (Thermo Fisher Scientific Inc). After the assay plates were incubated in a 37°C/5% CO2 incubator for 5 h, 23 nl of compounds dissolved in DMSO, positive controls, or DMSO was transferred to the assay plate by a pin tool (Kalypsys) in the presence (ER antagonist screening) or absence (ER agonist screening) of 0.2 nM E2. The final compound concentration in the 5 µl assay volume ranged from 90 nM to 92 μM in 11 concentrations. The control plate format for ER agonist screening was as follows: Column 1, concentration-response titration of E2 from 0.32 fM to 4.6 nM; Column 2, 2 nM of E2 in the top 16 wells and 92 μM of tetraoctyl ammonium bromide in the bottom 16 wells; Column 3, 1 nM of E2 in the top 16 wells and DMSO only in the bottom 16 wells and Column 4, DMSO only. The control plate format for the ER antagonist screening was as follows: Column 1, concentration-response titration of 4-hydroxy tamoxifen (Sigma-Aldrich) from 3.2 pM to 46 μM with 0.2 nM E2; Column 2, concentration-response titration of ICI 182,780 (Tocris Bioscience, St. Louis, Missouri) from 3.2 pM to 46 μM with 0.2 nM E2; Column 3, top 12 wells with 1 μM of 4-hydroxy tamoxifen and 0.2 nM E2, the next 12 wells with 1 μM of ICI 182 780 and 0.2 nM E2, and the bottom 12 wells with 92 μM of tetraoctyl ammonium bromide and 0.2 nM E2; Column 4, DMSO only with 0.2 nM E2 in the top 16 wells and DMSO only with assay buffer in the bottom 16 wells. The plates were incubated at 37°C for 24 h. For cell viability readout, after 23.5 h, 1 µl/well of CellTiter-Fluor reagent was added into the assay plates using an FRD (Aurora Discovery). After 30 min incubation at 37°C, the fluorescence intensity in the plates was measured using a ViewLux plate reader (PerkinElmer). This was followed by the addition of 4 μl of ONE-Glo Luciferase Assay reagent (Promega) using a FRD (Aurora Discovery). The plates were then incubated at room temperature for 30 min, and luminescence intensity was measured using a ViewLux plate reader. Data were expressed as relative luminescence units. For ER agonist data analysis, raw plate reads for each titration point were first normalized relative to E2 control (2 nM, 100%) and DMSO-only wells (basal, 0%). For ER antagonist data analysis, raw plate reads for each titration point were first normalized relative to 0.2 nM E2 added 4-hydroxy tamoxifen control (1 μM, 100%) and 0.2 nM E2 added DMSO only wells (basal, 0%).
Tox21 qHTS data analysis
The raw plate reads for each titration point were first normalized relative to the positive control compound (E2, 100% [no chemical stimulation mode]; letrozole, −100% [T-stimulated mode]; 4-hydroxy tamoxifen, −100% [E2-stimulated mode]; tetra-n-octylammonium bromide, −100% [cell viability]) and DMSO-only wells (0%) as follows: % Activity = [(Vcompound − VDMSO)/(Vpos − VDMSO)] × 100, where Vcompound denotes the compound well values, Vpos denotes the median value of the positive control wells, and VDMSO denotes the median values of the DMSO-only wells. The data set was then corrected using the DMSO-only compound plates at the beginning and end of the compound plate stack by applying an in-house pattern correction algorithm (Huang et al., 2009).
After the initial screen, Curve Class (Huang et al., 2011b; Inglese et al., 2006) 3-stage approach (Shockley, 2012) and the weighted area under the curve (wAUC) approach (Hsieh et al., 2015) were used to prioritize the normalized concentration-response profiles into a candidate list for follow-up screens. A total of 333 substances were selected for follow-up screens in the T-stimulated and E2 stimulated AroER tri-screen assay. The data from the follow-up screens were analyzed by the wAUC approach, which can be used to estimate point-of-departure (POD) values as explained in greater detail later.
In the wAUC approach, Curvep (Hsieh et al., 2015; Sedykh et al., 2011), a noise filtering algorithm developed for qHTS data, was applied on the 3-run Tox21 concentration-response data. Based on the Curvep-processed curves, POD values were determined and used to calculate the wAUC values. The wAUC value quantifies the amount of activity across the tested concentration range, and the POD value is the concentration where the response exceeds the assay-dependent noise threshold. For each substance, the wAUC and POD values were summarized by the median value from the triplicates. Substances with summarized wAUC < 0 (wAUC > 0) suppress (enhance) luminescence signals. Both primary screens and cell viability counterscreens were processed. The actives are the substances that suppress luminescence signals independent of cytotoxicity (Supplementary Table S1). In summary, there are 4 activity outcomes in the initial screen (10 496 chemicals) and the follow-up screen (333 chemicals): actives (wAUC < −30), marginal active (wAUC < 0 and wAUC ≥ −30), inactive (wAUC = 0), and inconclusive (ie, cytotoxic). For the actives in the T-stimulated assay in the follow-up screen, the substances are further labeled whether the response is independent of ER antagonism (Supplementary Table S2). The AIs, independent of ER antagonism, are hierarchically clustered based on their chemical structural similarity defined by Leadscope structural fingerprints (Valerio et al., 2010) and are grouped using average linkage with similarity cutoff = 0.7.
Aromatase enzyme assay
To confirm if and how the selected compounds inhibited aromatase, we examined them for their response using an established aromatase inhibition assay (ie, tritiated water-release assay) (Grube et al., 2001). Tritiated water is produced when androst-4-ene-3,17-dione [1-β-3H(N)] (NEN-Dupont, Boston, Massachusetts) is converted to estrone by aromatase. In a serum-free condition, AroER tri-screen cells were incubated with 100 nM of tritiated androstenedione with or without varying concentrations of the selected compounds. After incubation, the unreacted 3H-androstenedione was absorbed by dextran-coated charcoal, and the tritiated water product was quantified by a liquid scintillation counter. Aromatase activity was expressed as picomole of tritiated water released per milligram protein per hour (pmol/mg/h).
Reversibility assay
AI-like chemicals were further examined for the reversibility of their inhibition of aromatase. Briefly, the compounds were incubated with AroER tri-screen cells for 3 h. After the incubation, the cells were washed with 100 μl of phosphate-buffered saline (PBS; GE Health Care Life Sciences, South Logan, Utah) 3 times to remove free/unbound inhibitors and assayed for aromatase activity using the tritiated water-release method.
Aromatase kinetics assay
Inhibition kinetic analysis was performed to determine how AI-like compounds inhibited aromatase. AroER tri-screen cells were incubated with androst-4-ene-3,17-dione [1-β-3H(N)] (5–100 nM) plus amoxapine (300 nM or 2 μM), amlodipine besylate (300 nM or 1 μM), trovafloxacin mesylate (200 nM or 1 μM), or erlotinib (10 μM) for 1 h. Tritiated water product in treated cells was quantified. Aromatase activity was expressed as picomole of tritiated water released per milligram protein per hour (pmol/mg/h). The double reciprocal Lineweaver Burk plot was generated to determine the type of inhibition by different compounds. The linear equation was used to analyze the data generated from tritiated water release assay.
RESULTS
The study design flowchart can be found in Figure 1.
FIG. 1.

Study design and outcome. Full color version available online.
qHTS Performance of AroER Triscreen
The Tox21 10K library was screened 3 times against the T-stimulated version of AroER tri-screen assay. Based on the results (Fig. 1), 333 substances (302 potential AIs along with 31 known actives and inactives) were selected to screen against the T-stimulated and E2-stimulated versions of the AroER tri-screen assay to identify AIs that are independent of ER antagonism. To rule out responses caused by compound cytotoxicity, a cell viability assay was conducted in the same well as the AroER tri-screen assay. Letrozole, the positive control in the T-stimulated mode, produced an IC50 value (ie, concentration calculated to inhibit a half maximal response with SD) of 6.9 ± 1.6 nM. The AroER tri-screen assay worked well in qHTS format as evaluated by average signal-to-background (S/B) ratios of 6.2, average coefficients of variation (CV) of 4.1, and average Z′ factors of 0.8. The cytotoxicity counterscreens also have acceptable performance with average S/B ratios of 4.1, average CV values of 7.4, and average Z′ factors of 0.77.
For the 10K library, the wAUC data reproducibility was evaluated by Pearson’s correlation (R) on the triplicate runs and the 88 duplicates intentionally plated on each plate for the T-stimulated AroER tri-screen assay and its cell viability counterscreen. For the 88 duplicates, the average R value is 0.97 and 0.97 with an average of 29% (∼25/88) and 32% (∼28/88) substances with significant responses for primary screen and counterscreen. For the triplicate runs, the average R value is 0.96 and 0.93 with an average of 19% (∼1994/10496) and 21% (∼2204/10 496) substances with significant responses for the primary screen and counterscreen. In follow-up rescreen, 333 substances were screened 3 times each in the T-stimulated and E2-stimulated versions of the AroER tri-screen assay. For the 3 runs of the T-stimulated AroER tri-screen assay, the average R value is 0.98 and 0.86 with an average of 80% (∼266/333) and 36% (∼120/333) substances with significant responses in the primary screen and counterscreen, respectively. For the 3 runs of the E2-stimulated AroER tri-screen assay, the average R value is 0.93 and 0.92 with an average of 49% (∼163/333) and 23% (∼77/333) substances with significant responses for the primary screen and counterscreen, respectively. We also compared the reproducibility of the wAUC data for the 333 substances in the two T-stimulated versions of the AroER tri-screen assay conducted at different times. As shown in Figure 2, the R value is ∼0.94 and the outlier - (Tox21_302369, raloxifene hydrochloride, x = −836; y = −579) presents the most potent POD (thus, highest wAUC) in the primary screen was less potent in the follow-up screen. The results demonstrate the high signal reliability of the AroER tri-screen assay even when conducted in 1536-well qHTS format.
FIG. 2.

Signal reproducibility between the initial screen and the follow-up screen of 333 substances. The Pearson’s correlation coefficient is ∼0.94. The magenta line represents the “active line.” Full color version available online.
Identification of AIs
After the initial screen (T-stimulated AroER tri-screen assay), 333 substances were selected for the follow-up screen using both T-stimulated and E2-stimulated versions of the AroER tri-screen assay to identify AIs independent of ER antagonism (Fig. 1).
Four activity calls defined by the wAUC pipeline (active, marginally active, inactive, inconclusive [e.g., cytotoxic]) were assigned to the 333 substances in the two T-stimulated AroER tri-screen assays conducted on 2 dates. In total, 278 compounds (of 333) were considered as hits (133 actives + 145 marginal actives) independent of cytotoxicity in the initial screen in the T-stimulated AroER tri-screen assay. In the follow-up screen, 227 compounds (out of 333) were considered as hits (127 actives + 100 marginal actives) independent of cytotoxicity in the T-stimulated AroER tri-screen assay. The activity call concordance between these 2 screens (initial vs follow-up) is provided in Table 1. Overall, more hits are identified in the initial screen than in the follow-up screen (278 vs 227). The major discordance observed was that ∼34% (50/145) of the marginal actives became inactive (33) or inconclusive (17) in the follow-up screen. The median value of the wAUC for these compounds was approximately 8.5 in the initial screen. The hit call concordance, a metric to show the chance an assigned hit to be remained as hit in the repeated assay, is defined as the average of the fraction of reproducible hits (actives + marginal actives, n = 216) within the total number of assigned hits in either the initial screen (n = 278) or the follow-up screen (n = 227). The hit call concordance was ∼0.86 between the initial screen and the follow-up screen. The overall concordance (matched categories/total number of substances) is ∼ 0.76
Table 1.
Activity Call Concordance
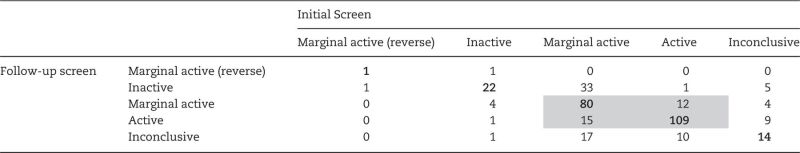 |
The bolded text represents the concordance results. The shaded text represents concordance hits.
To identify AIs independent of ER antagonism, the results from the E2-stimulated AroER tri-screen assay were incorporated. Using this approach, only 113 compounds (63 actives + 50 marginal actives) were considered to be AIs that were independent of ER antagonism; the filtered compounds (220, also see Fig. 1) could include either inactives, cytotoxic compounds, or ER antagonists.
Structure-Activity Relationship of AIs
To prioritize AIs identified in the AroER tri-screen assay for further mechanistic study, a structure-activity relationship (SAR) analysis was conducted on the 113 active compounds (Fig. 1). The compounds with similar chemical structures were grouped (hierarchical clustering using average linkage with a similarity cutoff of 0.7) based on Leadscope structural fingerprints (Valerio et al., 2010). In total, 17 clusters (number of members > 1) were formed, covering 71 substances; the remaining 42 substances were singletons. The POD distribution of the 18 clusters (singletons are placed in one cluster) is shown in Figure 3. The representative chemical structure in each cluster is shown in Supplementary Table S3. In total, 14 compounds, including both environmental chemicals and drugs, were selected for further confirmation assays and mechanistic studies (Table 2 and blue circle in Fig. 3) due to their novel structures; six of them are singletons.
FIG. 3.

POD distribution of AIs (111 substances) identified in the AroER tri-screen assay in qHTS format grouped by chemical structure. Substances that do not cluster with other substances based on defined similarity cutoff are labeled as singletons (Cluster ID = 0). The substances are annotated: the red or gray color is used to distinguish the high-efficacy (>70%, red) compounds from the low-efficacy compounds (≤ 70%, gray); the known AIs are labeled (hollow triangle); the 14 selected compounds for testing in the tritiated water-release assay are highlighted (blue hollow circle). Full color version available online.
Table 2.
The Summarized Results of the 14 Compounds Tested in the Tritiated Water Release Assay
| Assay Outcome |
Property |
T-Stimulated AroER tri-screen (Follow-Up) |
Tritiated Water Release | Reversibility and Kinetics | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tritiated Water Release | AroER tri-screen (Follow-Up) | CAS | LabelUsed in Tritiated Water Release | Chemical Name | Function | Cluster ID | POD | IC50 | Emax | IC50 | effect |
| Active | Active | 101-80-4 | H1 | 4,4'-Oxydianiline | Cross-linking agent for polymers | 6 | 8.71 | 42.66 | −90.87 | 7.863 | reversible |
| Active | Active | 17252-51-6 | C2 | 4,4'-Propane-1,3-diyldipyridine | Industrial compound | 0 | 5.33 | 11.48 | −87.19 | 1.450 | reversible |
| Active | Active | 111470-99-6 | A3 | Amlodipine besylate | Anti-hypertension, long acting calcium channel blocker | 12 | 11.08 | 19.50 | −98.24 | 0.577 | irreversible & competitive |
| Active | Active | 14028-44-5 | A1 | Amoxapine | Anti-depression | 0 | 5.76 | 19.95 | −117.9 | 0.767 | reversible |
| Inactive | Weak active | 134523-03-8 | D1 | Atorvastatin calcium | Statins,Anti-cholesterol drugs | 2 | 26.11 | 14.13 | −41.96 | N/A | N/A |
| Active | Active | 183321-74-6 | G1 | Erlotinib | Anti-cancer drugs | 0 | 7.77 | 15.85 | −87.44 | 3.391 | irreversible & noncompetition |
| Inactive | Active | 79241-46-6 | B2 | Fluazifop-P-butyl | Herbicides | 9 | 2.60 | 0.72 | −21.52 | N/A | N/A |
| Active | Active | 58594-72-2 | E1 | Imazalil sulfate | Fungicides | 1 | 0.31 | 0.71 | −100.99 | 0.005 | irreversible & noncompetition |
| Active | Active | 88671-89-0 | D2 | Myclobutanil | Fungicides | 1 | 1.92 | 5.62 | −105.98 | 0.217 | reversible |
| Inactive | Active | 147526-32-7 | F1 | Pitavastatin calcium | Statins,Anti-cholesterol drugs | 2 | 0.54 | 0.66 | −30.42 | N/A | N/A |
| Active | Active | 106134-32-1 | B3 | ro 22-9194 | Antiarrhythmic drug | 0 | 7.78 | 14.79 | −92.15 | 1.378 | reversible |
| Inactive | Active | 53123-88-9 | B1 | Sirolimus | Anti-cancer drug | 0 | 0.16 | 1.32 | −61.01 | N/A | N/A |
| Active | Active | 112281-77-3 | A2 | Tetraconazole | Pesticides | 1 | 4.74 | 8.51 | −98.95 | 1.058 | reversible |
| Active | Active | 147059-75-4 | C1 | Trovafloxacin mesylate | Antibiotic | 0 | 0.35 | 0.98 | −89.85 | 0.322 | irreversible & noncompetition |
Abbreviations: N/A, not available; CAS, Chemical Abstracts Services number; Emax, maximum efficacy in response; IC50, half maximum inhibitory response in µM; POD, point of departure in µM. cluster ID: 0, singleton
Confirmation of AI Activity Using the Aromatase Enzyme Assay
The aromatase inhibition activity of 14 compounds was verified by the aromatase tritiated water-release assay (Fig. 1). The results of these 14 compounds are shown in Table 2. Four of the 14 compounds (B1: sirolimus, D1: atorvastatin calcium, F1: pitavastatin calcium, and B2: fluazifop-P-butyl) were inactive in the aromatase enzyme assay (Table 2 and Fig. 4). The 10 active compounds have diverse structures (5 singletons; the remaining 5 compounds belong to 3 different clusters), and most of them do not belong to the traditional structural class of AIs (eg, triazole derivatives).
FIG. 4.

Dose-dependent inhibition of aromatase activity by each compound (10 nM to 10 μM) using the tritiated water-release assay. Letrozole (Let), a known AI, was used as a positive control. The assay was carried out in triplicate and data are expressed as the mean ± standard deviation (SD). The names of the chemicals are shown in Table 2.
To better understand the inhibition activity of these 10 active but structurally diverse compounds, we first examined the reversibility of their inhibitory activity (as indicated in Fig. 1). As reported previously, exemestane is an aromatase destabilizer that inhibits aromatase in an irreversible manner (Wang and Chen, 2006). Using exemestane as a reference control, reduction of aromatase activity by 4 of the 10 compounds was observed in an aromatase assay performed after a 3-h pretreatment of MCF-7aro cells with the compounds and the removal of unbound chemicals by washing the cells. Under the same treatment conditions, another AI letrozole was confirmed to inhibit aromatase in a reversible manner. The aromatase in cells pretreated with six of the 10 compounds was fully active after identical cell treatment. The 4 persistent AI chemicals were—trovafloxacin (C1), imazalil (E1), erlotinib (G1), and amlodipine besylate (A3) (Fig. 5A). The irreversible inhibition of aromatase by these compounds was concentration-dependent (Fig. 5B, Table 2). We can rule out that these compounds are cytotoxic because they are inactive in other assays such as ER antagonistic assay and cytotoxicity assay. Given this irreversible inhibition and thus the potential for long-lasting effects, these compounds may have significant physiological consequences (such as obesity) if exposure occurs at critical times during perinatal growth and development. The structures of these 4 compounds and their IC50 values for AI activity are shown in Supplementary Tables S1and S2, respectively. Three of the four compounds (erlotinib, imazalil sulfate, and trovafloxacin mesylate) are noncompetitive inhibitors with respect to the androgen substrate, whereas amlodipine besylate is a competitive inhibitor. The results of aromatase inhibition kinetic analysis of amoxapine, amlodipine besylate, trovafloxacin mesylate, and erlotinib are shown in Figure 6. As a competitive inhibitor, amlodipine besylate is thought to bind to the active site of the aromatase enzyme.
FIG. 5.
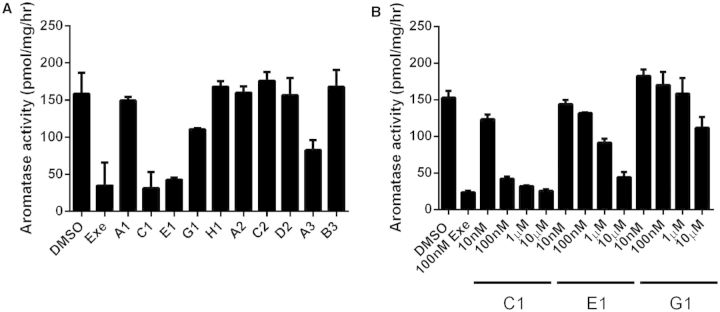
A, Inhibition of aromatase activity in MCF7aro cells by selected compounds. AI assays were carried out by incubating cells for 1 h with the androgen substrate and each compound at a single concentration (10 μM). Exemestane (EXE) (100 nM) was used as the positive control. The assay was carried out in triplicate and the data are expressed as the mean ± standard deviation (SD). The names of the chemicals are shown in Table 2. B, Confirmed inhibition of aromatase activity in MCF7aro cells by compounds trovafloxacin mesylate (C1), imazalil sulfate (E1), and Erlotinib (G1). Aromatase inhibition assays were carried out by incubation of cells for 1 h with the androgen substrate and each compound at various concentrations (10 nM–10 μM). Exemestane (100 nM) was used as a positive control. The assay was carried out in triplicate and the data are expressed as the mean ± standard deviation (SD).
FIG. 6.

Molecular basis of aromatase inhibition by amoxapine, amlodipine besylate, trovafloxacin mesylate, and erlotinib, examined by enzyme kinetic analysis. The double reciprocal Lineweaver Burk plot reveals that amlodipine besylate is a competitive inhibitor with respect to the androgen substrate. Amoxapine, erlotinib, and trovafloxacin mesylate inhibit aromatase in a noncompetitive manner. Full color version available online.
DISCUSSION
In this study, we used a 2-tiered qHTS approach, initially running just a T-stimulated AroER tri-screen assay on the full Tox21 10K library. In the second tier, a selected set of 333 compounds was tested in the complete AroER tri-screen assay (3 screening formats), to identify compounds selectively targeting aromatase. Overall, the 2-tiered qHTS screening data are robust, with good signal reproducibility observed in both the triplicate assay data and the Tox21 88 duplicate compounds (Pearson’s r > 0.9, except for the viability assay data in the tier-2 T-stimulated AroER tri-screen, where r = 0.86). Furthermore, the luminescence signals of the compounds in common between the tier-1 assay and the tier-2 assay, conducted at different times were also highly reproducible (Pearson’s r ∼0.94). Finally, the hit (i.e., actives independent of cytotoxicity) call concordance (0.86) and total call concordance (0.76) are acceptable. The major types of data discordance observed include the label switch from “marginally active” in the initial tier-1 screen to “inactive” in the follow-up tier-2 screen, and from “hit” in the initial screen to “assay interference” (ie, cytotoxicity) in the follow-up screen. Cytotoxicity was reported to be the major assay confounder in Tox21 qHTS inhibition-type assays (Hsieh et al., 2015). In the T-stimulated assays,∼35% of the inhibitory signals could have been confounded by cytotoxicity, and the PODs for the weaker signals tend be close to the dose region in which cytotoxicity is observed.
To prioritize novel structures for validation in an orthogonal assay, the tritiated water release assay, SAR analysis was conducted on the 113 potential AIs for which activity was independent of both cytotoxicity and ER antagonism. Most of the known AIs have POD values smaller than 1 µM (eg, 1, 2, 4-triazole class of AIs in cluster 13 and steroidal AIs in Cluster 4; see Figure 3 and Supplementary Table S3). As expected, the known AIs are significantly more potent than most of the novel AIs. In addition to the known AIs, fungicides/insecticides/herbicides are highly represented in the clusters (cluster 1, 3, 5, 8, 9). Some of them (eg, triflumizole and imazalil) have PODs comparable to the known AIs. Statins are also represented in 2 clusters (cluster 2 and 10). However, the efficacy values in the statin groups are low.
In total, 14 compounds were selected due to their novel structures for verification of AI using the tritiated water release aromatase assay. Four out of 14 compounds could not be confirmed in this alternative assay. All of the 4 compounds (atorvastatin calcium, pitavastatin calcium, sirolimus, and fluazifop-P-butyl) had reproducible concentration-response data in the high-throughput screen with good potency but with lower efficacy value (<70%). The molecular basis of the positive response of the 4 compounds in the high-throughput screen is currently not known. Therefore, an efficacy filter was applied to the 113 potential AIs and the resulting 50 potential AIs are presented in Supplementary Table S4.
The 10 compounds with AI activity confirmed in the tritiated water release assay were investigated further to assess reversibility of the effect; 4 were found to demonstrate irreversible inhibition of aromatase. Among these 4, amlodipine besylate is a long-acting dihydropyridine-type calcium channel blocker commonly used in the management of hypertension and coronary artery disease (Wang et al., 2014). Erlotinib is an epidermal growth factor receptor inhibitor, undergoing investigation in several tumor types as a single treatment or in combination chemotherapy (Yewale et al., 2013). Imazalil is registered for agricultural use in postharvest application and storage of various fruits, vegetables, forage, and grain crops. This chemical was reported previously to be able to inhibit aromatase (Sanderson et al., 2002; Vinggaard et al., 2000) as well as cortisol and aldosterone secretion (Ohlsson et al., 2010). Furthermore, imazalil has been reported to induce significant genetic damage (Sisman and Turkez, 2010; Turkez and Aydin, 2012). In mice, maternal exposure to imazalil was found to have an adverse impact on behavioral development in the F1-generation (Tanaka et al., 2013). Because aromatase plays an important role in estrogen action in the brain and has been shown to modulate sexual behavior (Charlier et al., 2010; Olvera-Hernandez et al., 2015), there might be a direct connection between imazalil’s potent (IC50 = 5 nM), irreversible anti-aromatase activity seen in our study and the adverse effects reported on brain function in mice. Trovafloxacin is a fluoroquinolone antibiotic. Because of its hepatic toxicity, trovafloxacin is only used for serious life- or limb-threatening infections (Andrade and Tulkens, 2011). Results from our study suggest that trovafloxacin is a potent, irreversible AI that may have adverse biological effects in addition to liver toxicity that should be considered during its use.
In addition to the 4 irreversible inhibitors of aromatase, the AroER tri-screen also identified amoxapine as a reversible inhibitor of aromatase. This compound is a tricyclic antidepressant (the structure is shown in Supplementary Table S1) that inhibits the reuptake of noradrenaline and acts as an antagonist at dopamine and serotonin receptors (Anderson, 2001; Kravetz de Srulijes et al., 1975). Considering the unique structure of amoxapine when compared with other AI inhibitors, kinetic analysis of its inhibitory activity was also performed. This revealed that it inhibits aromatase noncompetitively with respect to the androgen substrate, but in a reversible manner. Previously, we reported that paroxitine was a weak estrogenic chemical (Chen et al., 2014). Although FDA has approved the use of paroxitine for hot flashes, its weak estrogenic activity should not be ignored. In studies that used isolated human placentas, tricyclic antidepressants and selective serotonin reuptake inhibitors were shown to cross the placental barrier (Bourke et al., 2014). Therefore, identification of antidepressants as EDCs is a potentially important finding in our study. In view of the ability of these 5 structural classes to inhibit aromatase, exposure to such chemicals in the clinic or elsewhere can potentially affect the endocrine system and breast cancer development.
This study also confirmed 5 additional AI-like chemicals that inhibit aromatase in a reversible manner (Table 2). Myclobutanil and tetraconazole are triazole chemicals used as fungicides. Ro 22-9194 is a class I antiarrhythmic agent that inhibits thromboxane A2 synthase. There is no reported biological function for 4,4′-propane-1,3-diyldipyridine and 4,4′-oxydianiline; they are typically used as cross-linking agents for polymers (Murakami et al., 1996).
In summary, with the successful adaptation of AroER tri-screen to a 1536-well plate robotic platform, the U.S. Tox21 10K compound library was screened for chemicals having antiaromatase activity. Through this high-throughput screening approach, which included a confirmation assay for aromatase activity, 10 AI-like chemicals were identified. The analyses of the inhibition reversibility and enzyme inhibition kinetics provided key information regarding the mechanisms by which these compounds interact with aromatase, thereby potentially altering endocrine balance. Our findings suggest that endocrine health of human populations may be significantly impacted by unexpected or undetected exposures to these compounds. A prominent example is the irreversible, long-lasting, and potent (IC50 = 5 nM) anti-aromatase activity of imazalil, a fungicide widely in agriculture.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
Intramural Research Programs (Interagency agreement Y2-ES-7020-01) of the National Toxicology Program, NIEHS and the CBCRP grant (17UB-8701 to S.C.).
Supplementary Material
REFERENCES
- Anderson I. M. (2001). Meta-analytical studies on new antidepressants. Br. Med. Bull. 57, 161–178. [DOI] [PubMed] [Google Scholar]
- Andrade R. J., Tulkens P. M. (2011). Hepatic safety of antibiotics used in primary care. J. Antimicrob. Chemother. 66, 1431–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke C. H., Stowe Z. N., Owens M. J. (2014). Prenatal antidepressant exposure: Clinical and preclinical findings. Pharmacol. Rev. 66, 435–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein G. D. (1999). Aromatase and gynecomastia. Endocr. Relat. Cancer 6, 315–324. [DOI] [PubMed] [Google Scholar]
- Charlier T. D., Cornil C. A., Ball G. F., Balthazart J. (2010). Diversity of mechanisms involved in aromatase regulation and estrogen action in the brain. Biochim. Biophys. Acta 1800, 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. (1998). Aromatase and breast cancer. Front. Biosci. 3, d922–d933. [DOI] [PubMed] [Google Scholar]
- Chen S., Zhou D., Hsin L. Y., Kanaya N., Wong C., Yip R., Sakamuru S., Xia M., Yuan Y. C., Witt K., et al. (2014). AroER tri-screen is a biologically relevant assay for endocrine disrupting chemicals modulating the activity of aromatase and/or the estrogen receptor. Toxicol. Sci. 139, 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Wang O., Nie M., Elison K., Zhou D., Li M., Jiang Y., Xia W., Meng X., Chen S., et al. (2015). Aromatase deficiency in a Chinese adult man caused by novel compound heterozygous CYP19A1 mutations: effects of estrogen replacement therapy on the bone, lipid, liver and glucose metabolism. Mol. Cell. Endocrinol. 399, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshenko K., Pakdel F., Segner H., Kah O., Eggen R. I. (2008). Interference of endocrine disrupting chemicals with aromatase CYP19 expression or activity, and consequences for reproduction of teleost fish. Gen. Comp. Endocrinol. 155, 31–62. [DOI] [PubMed] [Google Scholar]
- Eliassen A. H., Colditz G. A., Rosner B., Willett W. C., Hankinson S. E. (2006). Adult weight change and risk of postmenopausal breast cancer. JAMA 296, 193–201. [DOI] [PubMed] [Google Scholar]
- Fowler P. A., Bellingham M., Sinclair K. D., Evans N. P., Pocar P., Fischer B., Schaedlich K., Schmidt J. S., Amezaga M. R., Bhattacharya S., et al. (2012). Impact of endocrine-disrupting compounds (EDCs) on female reproductive health. Mol. Cell. Endocrinol. 355, 231–239. [DOI] [PubMed] [Google Scholar]
- Grube B. J., Eng E. T., Kao Y. C., Kwon A., Chen S. (2001). White button mushroom phytochemicals inhibit aromatase activity and breast cancer cell proliferation. J. Nutr. 131, 3288–3293. [DOI] [PubMed] [Google Scholar]
- Hsieh J.-H., Sedykh A., Huang R., Xia M., Tice R. R. (2015). A data analysis pipeline accounting for artifacts in Tox21 quantitative high throughput screening assays. J. Biomol. Screen. 2015 Apr 22, 2015, pii: 1087057115581317 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R., Sakamuru S., Martin M. T., Reif D. M., Judson R. S., Houck K. A., Casey W., Hsieh J. H., Shockley K. R., Ceger P., et al. (2014). Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci. Rep. 4, 5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R., Southall N., Wang Y., Yasgar A., Shinn P., Jadhav A., Nguyen D. T., Austin C. P. (2011a). The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 3, 80ps16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R., Xia M., Cho M. H., Sakamuru S., Shinn P., Houck K. A., Dix D. J., Judson R. S., Witt K. L., Kavlock R. J., et al. (2011b). Chemical genomics profiling of environmental chemical modulation of human nuclear receptors. Environ. Health Perspect. 119, 1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. L., Southall N., Xia M. G., Cho M. H., Jadhav A., Nguyen D. T., Inglese J., Tice R. R., Austin C. P. (2009). Weighted feature significance: A simple, interpretable model of compound toxicity based on the statistical enrichment of structural features. Toxicol. Sci. 112, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J., Auld D. S., Jadhav A., Johnson R. L., Simeonov A., Yasgar A., Zheng W., Austin C. P. (2006). Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U.S.A. 103, 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. E., Boon W. C., McInnes K., Maffei L., Carani C., Simpson E. R. (2007). Recognizing rare disorders: Aromatase deficiency. Nat. Clin. Pract. Endocrinol. Metab. 3, 414–421. [DOI] [PubMed] [Google Scholar]
- Kravetz de Srulijes L., Israeli E., Barzilai D. (1975). RNA:DNA ratios in a developing fibrosarcoma and its lung metastases in C3H mice. J. Natl. Cancer Inst. 55, 659–663. [DOI] [PubMed] [Google Scholar]
- Morris P. G., Hudis C. A., Giri D., Morrow M., Falcone D. J., Zhou X. K., Du B., Brogi E., Crawford C. B., Kopelovich L., et al. (2011). Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prevent. Res. 4, 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M., Kinukawa M., Kanazawa T., Maruyama K., Miyagi M., Miyata H., Ujiie A. (1996). Effects of the new class I antiarrhythmic agent Ro 22-9194, (2R)-2-amino-N-(2,6-dimethylphenyl)-N-[3-(3-pyridyl)propyl]propionamide D-tartrate, on ischemia- and reperfusion-induced arrhythmias in dogs: involvement of thromboxane A2 synthase inhibitory activity. J. Pharmacol. Exp. Ther. 279, 877–883. [PubMed] [Google Scholar]
- Nordkap L., Joensen U. N., Blomberg Jensen M., Jorgensen N. (2012). Regional differences and temporal trends in male reproductive health disorders: Semen quality may be a sensitive marker of environmental exposures. Mol. Cell. Endocrinol. 355, 221–230. [DOI] [PubMed] [Google Scholar]
- Ohlsson A., Cedergreen N., Oskarsson A., Ulleras E. (2010). Mixture effects of imidazole fungicides on cortisol and aldosterone secretion in human adrenocortical H295R cells. Toxicology 275(1-3), 21–28. [DOI] [PubMed] [Google Scholar]
- Olvera-Hernandez S., Chavira R., Fernandez-Guasti A. (2015). Prenatal letrozole produces a subpopulation of male rats with same-sex preference and arousal as well as female sexual behavior. Physiol. Behav. 139, 403–411. [DOI] [PubMed] [Google Scholar]
- Pfeiler G., Konigsberg R., Fesl C., Mlineritsch B., Stoeger H., Singer C. F., Postlberger S., Steger G. G., Seifert M., Dubsky P., et al. (2011). Impact of body mass index on the efficacy of endocrine therapy in premenopausal patients with breast cancer: An analysis of the prospective ABCSG-12 trial. J. Clin. Oncol. 29, 2653–2659. [DOI] [PubMed] [Google Scholar]
- Quignot N., Desmots S., Barouki R., Lemazurier E. (2012). A comparison of two human cell lines and two rat gonadal cell primary cultures as in vitro screening tools for aromatase modulation. Toxicol. in vitro 26, 107–118. [DOI] [PubMed] [Google Scholar]
- Roberts D. L., Dive C., Renehan A. G. (2010). Biological mechanisms linking obesity and cancer risk: New perspectives. Annu. Rev. Med. 61, 301–316. [DOI] [PubMed] [Google Scholar]
- Saad F., Gooren L. J. (2011). The role of testosterone in the etiology and treatment of obesity, the metabolic syndrome, and diabetes mellitus type 2. J. Obes., 2011, 471584, doi:10.1155/2011/471584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson J. T. (2006). The steroid hormone biosynthesis pathway as a target for endocrine-disrupting chemicals. Toxicol. Sci. 94, 3–21. [DOI] [PubMed] [Google Scholar]
- Sanderson J. T., Boerma J., Lansbergen G. W., van den Berg M. (2002). Induction and inhibition of aromatase (CYP19) activity by various classes of pesticides in H295R human adrenocortical carcinoma cells. Toxicol. Appl. Pharmacol. 182, 44–54. [DOI] [PubMed] [Google Scholar]
- Sedykh A., Zhu H., Tang H., Zhang L., Richard A., Rusyn I., Tropsha A. (2011). Use of in vitro HTS-derived concentration-response data as biological descriptors improves the accuracy of QSAR models of in vivo toxicity. Environ. Health Perspect. 119, 364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shockley K. R. (2012). A three-stage algorithm to make toxicologically relevant activity calls from quantitative high throughput screening data. Environ. Health Perspect. 120, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisman T., Turkez H. (2010). Toxicologic evaluation of imazalil with particular reference to genotoxic and teratogenic potentials. Toxicol. Ind. Health 26, 641–648. [DOI] [PubMed] [Google Scholar]
- Sparano J. A., Wang M., Zhao F., Stearns V., Martino S., Ligibel J. A., Perez E. A., Saphner T., Wolff A. C., Sledge G. W., Jr, et al. (2010). Obesity at diagnosis is associated with inferior outcomes in hormone receptor-positive operable breast cancer. Cancer 118, 5937–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Ogata A., Inomata A., Nakae D. (2013). Effects of maternal exposure to imazalil on behavioral development in F(1)-generation mice. Birth Defects Res. B Dev. Reprod. Toxicol. 98, 334–342. [DOI] [PubMed] [Google Scholar]
- Tice R. R., Austin C. P., Kavlock R. J., Bucher J. R. (2013). Improving the human hazard characterization of chemicals: A tox21 update. Environ. Health Perspect. 121, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkez H., Aydin E. (2012). The protective role of ascorbic acid on imazalil-induced genetic damage assessed by the cytogenetic tests. Toxicol. Ind. Health 28, 648–654. [DOI] [PubMed] [Google Scholar]
- Valerio L. G., Yang C., Arvidson K. B., Kruhlak N. L. (2010). A structural feature-based computational approach for toxicology predictions. Expert Opin. Drug Metabol. Toxicol. 6, 505–518. [DOI] [PubMed] [Google Scholar]
- Veith H., Southall N., Huang R., James T., Fayne D., Artemenko N., Shen M., Inglese J., Austin C. P., Lloyd D. G., et al. (2009). Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 27, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinggaard A. M., Hnida C., Breinholt V., Larsen J. C. (2000). Screening of selected pesticides for inhibition of CYP19 aromatase activity in vitro. Toxicol. in vitro 14, 227–234. [DOI] [PubMed] [Google Scholar]
- Wang J. G., Yan P., Jeffers B. W. (2014). Effects of amlodipine and other classes of antihypertensive drugs on long-term blood pressure variability: Evidence from randomized controlled trials. J. Am. Soc. Hypertens., 8, 340–349. [DOI] [PubMed] [Google Scholar]
- Wang X., Chen S. (2006). Aromatase destabilizer: novel action of exemestane, a food and drug administration-approved aromatase inhibitor. Cancer Res. 66, 10281–10286. [DOI] [PubMed] [Google Scholar]
- Yewale C., Baradia D., Vhora I., Patil S., Misra A. (2013). Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 34, 8690–8707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.