

Abstract
Purpose
Discriminant markers for pancreatic cancer detection are needed. We sought to identify and validate methylated DNA markers for pancreatic cancer using next-generation sequencing unbiased by known targets.
Experimental Design
At a referral center, we conducted four sequential case-control studies: discovery, technical validation, biological validation, and clinical piloting. Candidate markers were identified using variance inflated logistic regression on reduced-representation bisulfite DNA sequencing results from matched pancreatic cancers, benign pancreas, and normal colon tissues. Markers were validated technically on replicate discovery study DNA and biologically on independent, matched, blinded tissues by methylation specific PCR. Clinical testing of 6 methylation candidates and mutant KRAS was performed on secretin-stimulated pancreatic juice samples from 61 pancreatic cancer patients, 22 with chronic pancreatitis and 19 with normal pancreas on endoscopic ultrasound. Areas under receiver operating characteristics curves (AUC) for markers were calculated.
Results
Sequencing identified >500 differentially hyper-methylated regions. On independent tissues, AUC on 19 selected markers ranged between 0.73 – 0.97. Pancreatic juice AUC values for CD1D, KCNK12, CLEC11A, NDRG4, IKZF1, PKRCB and KRAS were 0.92*, 0.88, 0.85, 0.85, 0.84, 0.83 and 0.75, respectively, for pancreatic cancer compared to normal pancreas and 0.92*, 0.73, 0.76, 0.85*, 0.73, 0.77 and 0.62 for pancreatic cancer compared to chronic pancreatitis (*p=0.001 vs KRAS).
Conclusion
We identified and validated novel DNA methylation markers strongly associated with pancreatic cancer. On pilot testing in pancreatic juice, best markers (especially CD1D) highly discriminated pancreatic cases from controls.
Keywords: DNA methylation, early detection of cancer, sensitivity and specificity, pancreatic neoplasms, pancreatic juice
INTRODUCTION
Incidence and mortality rates of pancreatic cancer continue to rise in the face of declining trends for other major cancers.(1) Some forecast that pancreatic cancer will become the second most fatal cancer in the United States before 2020.(2) Underscoring its extraordinarily high lethality, more than 46,000 Americans will be diagnosed with pancreatic cancer and nearly 40,000 will succumb this year.(1) Better approaches to pancreatic cancer control are urgently needed.
While population screening is currently not practiced, there are strong biological and clinical justifications to explore early detection. Recent studies on the molecular epidemiology of pancreatic carcinogenesis suggest slow rates of progression from premalignant neoplasms to cancer and from earliest stage cancer to metastatic disease.(3) Such long latency periods provide a window of opportunity for detection and curative treatment of presymptomatic precursor lesions or earliest stage pancreatic cancer. Indeed, incidentally discovered early-stage pancreatic cancers have the best reported cure rates.(4, 5) Precursor lesions, including pancreatic intraepithelial neoplasm (PanIN), intraductal papillary mucinous neoplasm (IPMN),(6) and pancreatic cancer are associated with molecular alterations(7–9) that could potentially serve as markers for early detection and screening.
Pancreatic neoplasms exfoliate cells and DNA into local effluent and ultimately stool. We and others have detected both genetic and epigenetic markers in pancreatic juice(10, 11) and stool(12–15) from patients with pancreatic cancer and precursor lesions. A limitation with mutation markers relates to the unwieldy process of their detection; typically, numerous mutations across several genes must be assayed separately to achieve high sensitivity. Additionally, some mutations common in pancreatic cancer may not be sufficiently specific; for example, mutant KRAS is frequently observed in chronic pancreatitis (CP).(16) Methylation of DNA at cytosine-phosphate-guanine (CpG) island sites provides marker candidates that are more broadly informative and sensitive than individual DNA mutations and may offer excellent specificity, as we have seen with stool DNA testing for colorectal cancer.(17, 18)
Identification of screening markers which are both highly sensitive and highly specific can be challenging. We have observed that methylation markers discriminant in primary tumor tissues often fail when assayed in an intended medium, such as stool.(15) Ideal candidate markers for pancreatic cancer screening would be universally present in pancreatic neoplasms, be absent in normal gastrointestinal mucosa, and have high signal strength.
Several methods are available to search for novel methylation markers. Micro-array based interrogation of CpG methylation is a high-throughput approach, but is biased towards known regions of interest, mainly the promotors of established tumor suppressor genes.(19) Alternative methods for genome-wide analysis of DNA methylation have been developed in the last decade.(20) Next-generation sequencing has provided important insights into the epigenetic regulation of gene expression in various cancers.(21–23) While whole-exome sequencing has been used to study mutations in pancreatic neoplasms,(24) we are unaware of any methylome-wide search for pancreatic cancer cancer screening markers using a next-generation sequencing approach.
We hypothesized that (1) a whole-methylome search by reduced representation bisulfite sequencing (RRBS)(25) would identify novel methylation markers which would discriminate pancreatic cancer from benign pancreatic tissues and have low background levels in other gastrointestinal epithelia and (2) discovered markers would accurately detect pancreatic cancer by assay of pancreatic juice.
METHODS
Study Overview
Four sequential case-control studies were conducted. In the first three tissue-based studies, we aimed to (1) discover novel and highly discriminant methylation markers for pancreatic cancer using RRBS; (2) technically confirm these findings using methylation-specific PCR (MSP), a more agile assay system; and (3) biologically validate top candidate markers in an independent, matched tissue sample set. In the fourth study, we clinically pilot-tested selected candidates on archival pancreatic juice samples using quantitative MSP (qMSP) and quantitative real-time allele-specific target and signal amplification (QuARTS) assay of mutant KRAS. All components of this investigation were approved by our Institutional Review Board.
Study Populations
Discovery
Tissue samples for the discovery selected from two existing institutional cancer registries at Mayo Clinic, Rochester Minnesota, and were reviewed by an expert gastrointestinal pathologist to confirm correct classification. All pancreatic tissues were collected by the Mayo Clinic SPORE in Pancreatic Cancer Patient Registry and Tissue Core, from patients enrolled between March 1998 and July 2011 (http://trp.cancer.gov/spores/abstracts/mayo_pancreatic.htm). Inclusion criteria for the registry were suspected pancreatic cancer and intent to perform a pancreaticoduodenectomy, distal pancreatectomy or total pancreatectomy. Pancreatic cancer case samples included pancreatic ductal adenocarcinoma tissues limited to early-stage disease (American Joint Committee on Cancer [AJCC] stage I and II),(26) of which there were approximately 600 in the registry. Patients having undergone neo-adjuvant therapy or those without matches to the control were excluded. Cases and both controls were matched by sex, age (in 5-year increments) and smoking status. There were two control groups studied. The first, termed “normal pancreas,” included histologically normal resection margins of low risk or focal pancreatic neoplasms (e.g. serous cystadenoma and neuroendocrine tumors) of which there were approximately 350 in the registry. The second control group included colonic epithelial tissues from patients confirmed to be free from pancreatic cancer or colonic neoplasm. Normal colon tissues were provided by the Biospecimens Linking Investigators and Clinicians to GIH Cell Signalling Research Clinical Core, which began recruitment on January 1, 2000. Normal colon samples were collected after informed consent from patients undergoing routine clinical colonoscopy. For both of the above tissue registries, all samples were procured at the time of surgery in the operating room by the Mayo Clinic Tissue Request Acquisition Group or at the time of endoscopic biopsy by trained study coordinators and immediately frozen to −80°C until utilized for research.
In a central core laboratory, DNA was extracted from micro-dissected tissues using a phenol-chloroform technique, yielding >500 ng of DNA per sample.
Technical Validation
Unblinded biological and technical replicate samples of pancreatic cancer and normal colon and technical replicates of normal pancreas were studied to ensure that the sites of differential methylation percentage identified by the RRBS data filtration, would be reflected in qMSP, where the unit of analysis was the copies per sample of the target sequence, corrected by the concentration of DNA in each sample, measured prior to bisulfite treatment.
Biological Validation
Top technically validated candidates were assayed by qMSP in independent pancreatic cancer, benign pancreas and normal colon samples from the same registries, above, which were matched, blinded and randomly allocated.
Clinical Pilot Testing
Selected methylated candidates and mutant KRAS were assayed by qMSP and QuARTS, respectively, on DNA extracted from blinded pancreatic juice samples, collected via simple duodenal luminal aspiration following a 16 microgram intravenous dose of secretin (ChiRhoClin, Burtonsville MD), as previously described.(27) Pancreatic juice samples were prospectively collected at Mayo Clinic, Jacksonville Florida, from March 1, 2012 to November 1, 2012. Patients were enrolled prospectively at the time of routine endoscopic ultrasound (EUS) or esophagogastroduodenoscopy (EGD) into one of three groups: those with pain suggestive of pancreatic disease; those suspected of having pancreatic cancer; and, those undergoing diagnostic EGD without suspicion of pancreatic disease or cancer. The latter group received EUS for research purposes. Patients were excluded if they could not provide informed consent or for prior gastric, pancreatic or duodenal resection. Pancreatic cancer or main-duct IPMN diagnoses were confirmed by histopathology; chronic pancreatitis and normal-appearing pancreas diagnoses were confirmed by magnetic resonance imaging and EUS. Juice was rapidly placed in 2 mL vials, immediately snap-frozen in liquid nitrogen, and stored at −80°C.
Reduced Representation Bisulfite Sequencing
Library preparation
(25) Genomic DNA (300 ng) was fragmented by digestion with 10 Units of MspI, a methylation-specific restriction enzyme which recognizes CpG-containing motifs, to enrich sample CpG content and eliminates redundant areas of the genome. Digested fragments were end-repaired and A-tailed with 5 Units of Klenow fragment (3’-5’ exo-), and ligated overnight to methylated TruSeq adapters (Illumina, San Diego CA) containing barcode sequences (to link each fragment to its sample ID.) Size selection of 160–340bp fragments (40–220 bp inserts) was performed using Agencourt AMPure XP SPRI beads/buffer (Beckman Coulter, Brea CA). Buffer cutoffs were 0.7X - 1.1X sample volumes of beads/buffer. Final elution volume was 22 uL (EB buffer – Qiagen, Germantown MD); qPCR was used to gauge ligation efficiency and fragment quality on a small sample aliquot. Samples then underwent bisulfite conversion (twice) using a modified EpiTect protocol (Qiagen). qPCR and conventional PCR (PfuTurbo Cx hotstart – Agilent, Santa Clara CA) followed by Bioanalyzer 2100 (Agilent) assessment on converted sample aliquots determined the optimal PCR cycle number prior to final library amplification. The following conditions were used for final PCR: 1.) each 50uL reaction contained 5uL of 10X buffer, 1.25uL of 10 mM each deoxyribonucleotide triphosphate (dNTP), 5uL primer cocktail (~5uM), 15uL template (sample), 1uL PfuTurbo Cx hotstart and 22.75 water; temperatures and times were 95C-5min; 98C-30sec; 16 cycles of 98C-10sec, 65C-30sec, 72C-30sec, 72C-5min and 4C hold, respectively. Samples were combined (equimolar) into 4-plex libraries based on the randomization scheme and tested with the bioanalyzer for final size verification, and with qPCR using phiX standards and adaptor-specific primers.
Sequencing and Bioinformatics
Samples were loaded onto flow cells according to a randomized lane assignment with additional lanes reserved for internal assay controls. Sequencing was performed by the Next Generation Sequencing Core at the Mayo Clinic Medical Genome Facility on the Illumina HiSeq 2000. Reads were unidirectional for 101 cycles. Each flow cell lane generated 100–120 million reads, sufficient for a median coverage of 30–50 fold sequencing depth (read number per CpG) for aligned sequences. Standard Illumina pipeline software called bases and sequenced read generation in the fastq format. As described previously, (28) SAAP-RRBS, a streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing, was used for sequence alignment and methylation extraction.
MSP Primer design
Primers for each marker were designed to target the bisulfite-modified methylated sequences of each target gene (IDT, Coralville IA) and a region without CpG sites in the β-actin gene, a reference of bisulfite treatment and DNA input. The design was done by either Methprimer software (University of California, San Francisco CA) or by semi-manual methods. Assays were tested and optimized by qPCR with SYBR Green (Life Technologies, Grand Island NY) dyes on dilutions of universally methylated and unmethylated genomic DNA controls.
Methylation specific PCR
Quantitative MSP reactions were performed on tissue-extracted DNA as previously described.(15) Additional specifications are provided in the Supplemental Methods.
Quantitative allele-specific real time target and signal amplification
QuARTS assays were used for KRAS assays as previously published.(29) Briefly, KRAS was PCR amplified with primers flanking codons 12/13 using 10 μl captured KRAS DNA templates. QuARTS assays then evaluated seven mutations at codons 12 and 13. Each QuARTS reaction incorporated primers, detection probes, an invasive oligo, FAM (Hologic, Madison WI), Yellow (Hologic), Quasar® 670 (BioSearch Technologies, Novato CA) fluorescence resonance energy transfer reporter cassettes (FRETs), Cleavase® 2.0 (Hologic), GoTaq® DNA polymerase (Promega, Madison WI), MOPS buffer, MgCl2, and deoxyribonucleotide triphosphate (dNTP). Plates contained standards made of engineered plasmids, +/− controls, and water blanks, and were run in a LightCycler 480 (Roche).
Both methylated candidates and mutant KRAS copy numbers per sample were calculated in reference to standard curves. In the qMSP and QuARTS reactions any sample for which at least 50 copies of β-actin were measured was included in the analysis. Any PCR product which amplified in reactions with primers and probes directed at the methylated or mutant target sequence was quantified by fluorescence values in relationship to the 1:5 serially-diluted reference standards which reproducibly amplify at 5000, 1000, 200, 40, 8 and 1.6 copies per well, respectively. For values below the analytical threshold, the copies per sample were assigned a value of 1 copy in order to normalize results for all samples by β-actin copy number or concentration, respectively.
Statistical Analysis
Overall approach
Candidate CpGs were filtered by a priori read-depth and variance criteria, significance of differential methylation percentages between cases and controls and discrimination of cases from controls based on area under the receiver operating characteristics curve (AUC) and target to background ratio. For the RRBS discovery phase, the primary comparison of interest was the methylation difference between cases, pancreatic controls and colon controls at each mapped CpG. CpG islands are biochemically defined by an observed to expected CpG ratio >0.6.(30) However, for this model, tiled units of CpG analysis “differentially methylated region (DMR)” were created based on distance between CpG site locations for each chromosome. Islands with only single CpGs were excluded. Individual CpG sites were considered for differential analysis only if the total depth of coverage per disease group was ≥200 reads (an average of 10 reads/subject) and the variance of %-methylation was >0 (non-informative CpGs were excluded). To estimate the sample size required per group for DNA sequencing, we assumed a minimum read depth of 10 reads per sample and that the primary comparison is between normal tissues (pancreatic & colon) and cancer tissues. The highest background for the average normal tissue methylation was assumed to be 5% and a three-fold increase in the odd ratio was deemed as the minimum effect difference that is biologically relevant. At the minimum depth of cover of 10 reads, a minimum of 18 samples per group was required to achieve 80% power with a 2-sided test at a significance level of 5% and assuming binomial variance inflation factor of 1. As the estimated variance inflation factor increases, the power drops with only 18 subjects per group. However, we accepted sites with a minimum read depth of 20 to maintain sufficient power, across all inflation factors, for the given sample size under these assumptions.
Statistical significance was determined by logistic regression of the methylation percentage per DMR, based on read counts. To account for varying read depths across individual subjects, an over-dispersed logistic regression model was used, where dispersion parameter was estimated using the Pearson Chi-square statistic of the residuals from fitted model. DMRs, ranked according to their significance level, were further considered if -methylation in benign pancreas and colon controls, combined, was ≤1% but ≥10% in pancreatic cancer.
For the validation and feasibility studies, the primary outcome was the area under the receiver operating characteristics curve (AUC) for each marker, as calculated from the concentration-corrected copies per sample of each marker with pancreatic cancer in comparison to normal pancreas and normal colon. For the technical and biological validation phases, 17 patients per group provided 80% power to distinguish an area under the curve of 0.85 from a null hypothesis of 0.50 in a 1-sided test at the 0.05 level of significance. After technical validation confirmed AUC >0.85, a quantitative difference in median values of candidate copies per sample between cases and controls of at least 10-fold was used to select markers for biological validation and pancreatic juice testing.
Pancreatic juices were convenience samples from an existing archive from which all samples were analyzed. AUC values of the each methylation marker in juice were compared to that of mutant KRAS in the same samples. The method of DeLong, DeLong and Clarke-Pearson (31) was used to compare AUCs and measure significance of differences. A Bonferroni correction was used to avoid bias from multiple comparisons, establishing a significance threshold p-value of <0.008. Samples included 61 cancers and two control groups of approximately 20. Using the normal approximation of the AUC, the sample number per group was used to determine the variance of the statistic. With this approximation and assuming a one-sided significance level of 0.05 and 80% power, the minimum detectable AUC for the feasibility study was 0.70 in comparisons to the null hypothesis of 0.5. When comparing any two markers the paired variance was estimated assuming a low correlation between markers of 0.3 and a moderate correlation of 0.6. Assuming a one-sided significance level of 0.008 with 80% power, the minimum detectable difference between any paired markers was 0.32 (0.60 vs 0.92) for a correlation of 0.3 between markers and 0.29 (0.60 vs 0.89) for a correlation of 0.6 between markers. Regression models also tested the influence of age, sex, clinical tumor stage (T1&2 vs T3&4, determined by endoscopic ultrasound) and tumor location (head vs body/ tail) on the strength of association between marker levels and case or control status.
The point-value, in copies per sample, for each marker was identified at the false positive rate-based cut-offs of 5% and 10% among normal pancreas controls and used to estimate marker sensitivity and 95% confidence intervals for pancreas cancer in separate comparisons to normal pancreas and chronic pancreatitis.
RESULTS
RRBS marker discovery
DNA extracts from 54 tissue samples (18 pancreatic cancer tumors, 18 benign pancreatic control tissues and 18 normal colonic epithelial) were sequenced by RRBS (Figure 1). Median age was 61 (interquartile range 52 – 65), 61 % were women, and 44% were current or former smokers. A total of 6,101,049 CpG sites were captured in any of the samples with at least 10X coverage. After selecting CpG sites where group coverage and variance criteria were met, a total of 1,217,523 CpG sites were further analyzed. Approximately 500 DMRs met significance criteria. Among these, we identified 87 candidate regions with sufficient methylation signatures for MSP primer design. Methylation signatures ranged from 3–52 neighboring CpGs. Methylation levels in pancreatic cancer samples were typically below 25%, reflecting the common contamination by stromal cells. The degree of stromal cell contamination could be quantified indirectly by KRAS testing; among pancreatic cancer specimens which harbored a heterozygous KRAS base change, the frequency of the mutant allele was generally 4 times less than the corresponding wild-type allele (Supplemental Figure 1).
Figure 1.

Study flow diagram of four sequential case control studies for marker discovery and validation
Technical validation
After primer design, MSP assayed the 87 candidates in samples of DNA from an additional 20 unblinded pancreatic cancer lesions, 10 additional normal colonic epithelial samples (biologic replicates) as well as remaining DNA samples from the 18 sequenced pancreatic cancer lesions,15 of the sequenced benign pancreatic tissues and 10 of the sequenced normal colon samples (technical replicates). β-actin amplified in all samples. With either first or second-pass MSP, 38 of 87 candidate markers had an AUC > 0.85 (Figure 2, Supplemental Table 1). RRBS-identified candidates were compared to two published reports of pancreatic cancer methylation measured by microarray.(8, 32) RRBS candidate pool was corroborated and comparably informative; however, 10 of the 38 top candidates were novel genes, not identified by hybridization array methods.
Figure 2.

Quantitative methylation specific PCR validates candidate methylated gene regions identified by reduced representation bisulfite sequencing. Areas under receiver operating characteristics curves (AUC) measure strength of association with pancreatic cancer compared to controls. Controls were combined benign pancreas and normal colon samples. Confidence intervals for the AUC estimates are provided in Supplemental Table 1.
Biological validation
Based on the magnitude of difference in median copies per sample between cases and controls for each candidate marker, ABCB1, ADCY1, AK055957, BMP3, C13ORF18, CACNA1C, CD1D, CLEC11A, ELMO1, FOXP2, GRIN2D, IKZF1, KCNK12, KCNN2, NDRG4, PRKCB, RSPO3, SCARF2, SHH, SLC38A3, TWIST1, VWC3 and WT1 were selected for validation in independent, matched, blinded, randomly allocated DNA from 72 tissue samples. These included 18 pancreatic cancers, 18 benign pancreas tissues and 36 normal colon epithelia. The median age of this subset was 60 (interquartile range 54 – 64). The majority (55%) of samples came from men and 61% were current or former smokers. β-actin amplified in all samples. As shown (Figure 3), candidates were strongly associated with pancreatic cancer in comparison to benign pancreatic and colonic controls, combined. The individual AUC values (and 95% confidence intervals) for AK055957, WT1, GRIN2D, CACNA1C, ELMO1, ABCB1, KCNN2, CD1D, TWIST1, C13ORF18, and CLEC11A were outstanding at 0.97 (0.92–1), 0.97 (0.93–1), 0.97 (0.93–1), 0.95 (0.91–1), 0.95 (0.9–1), 0.94 (0.88–1), 0.94 (0.86–1), 0.94 (0.86–1), 0.93 (0.83–1), 0.93 (0.84–1), and 0.93 (0.84–1), respectively. Excellent association was seen with 9 other candidates with AUC values for RSPO3, PRKCB, KCNK12, SLC38A3, SHH, VWC2, SCARF2 and ADCY1of 0.92 (0.85–0.98), 0.91 (0.81–1), 0.91 (0.83–98), 0.89 (0.78–1), 0.88 (0.77–0.99), 0.87 (0.73–1), 0.86 (0.74–0.98) and 0.85 (0.69–1).
Figure 3.

Area under the receiver operating characteristics curve (AUC) plotted against the log ratio of median case to control copy numbers per sample of each marker shows both accuracy and signal strength. Control samples combined benign pancreas and normal colon. Confidence intervals for the AUC estimates and the signal to noise ratios are provided in Supplemental Table 2.
The majority of novel candidates showed excellent signal to noise ratios. Specifically, for 10 candidate markers, methylated copy numbers were more than 30-fold higher among cases, compared to controls. For AK055957, KCNK12, ADCY1, ELMO1 and PRKCB, copy numbers of methylated candidates were more than 100-fold greater in cases, compared to controls (Figure 3, Supplemental Table 2). The biologically validated DMRs were compared to an open-access published data set from the International Cancer Genome Consortium (ICGC). This set included 167 pancreas cancer and 29 control tissues in which DNA methylation was interrogated by Infinium Human Methylation 450K BeadChips (Illumina, San Diego, CA).(32) All RRBS-derived biologically validated genes were corroborated by the ICGC results. The published sequences of the CpG probe sets for each annotated gene were compared to the coordinates and sequences for the RRBS-derived DMRs (Supplemental Table 2). Of 19 RRBS-derived DMRs, 13 had no sequence overlaps with the 450K probes. Of the remaining 6, the RRBS-derived DMRs had at least one novel CpG, not contained in the list of significant probes reported for the hybridization array method.
Pilot testing in pancreatic juice
At the time of the pancreatic juice pilot, the full biological validation analysis had not been completed. Six candidate methylation markers reflecting a range of AUC values and signal to noise ratios of at least 10 were chosen for feasibility testing in pancreatic juice. All 102 pancreatic juice samples from a pre-existing freezer archive were tested and included 61 patients with pancreatic cancer, 22 with chronic pancreatitis and 19 with normal pancreas (Table 1). β-actin amplified in all samples.
Table 1.
Patients who submitted pancreatic juice for clinical validation
| Pancreatic cancer (n=61)* | Chronic pancreatitis (n=22) | Normal pancreas (n=19) | p-value | |
|---|---|---|---|---|
| Age, median (IQR), years | 67 (61 – 76) | 64 (53 – 72) | 60 (49 – 70) | 0.02 |
| Men (%) | 34 (58) | 15 (68) | 4 (21) | 0.007 |
| Smoking (%) | 0.03 | |||
| Current | 11 (18) | 10 (45) | 2 (11) | |
| Former | 19 (32) | 5 (23) | 4 (21) | |
| Never | 30 (50) | 7 (32) | 13 (68) | |
| Diabetic (%) | 15 (25) | 6 (27) | 1 (5) | 0.4 |
| Tumor location | ||||
| Head (%) | (71) | -- | -- | -- |
| Body (%) | (8) | -- | -- | -- |
| Tail (%) | (21) | -- | -- | -- |
| EUS tumor stage, T1 or 2, % | 16 (26) | |||
Smoking history, diabetes diagnosis and endoscopic ultrasound (EUS) stage were missing on a single pancreatic cancer patient
Samples were available on only 3 patients with main duct IPMN; due to insufficient power, these were not included in regression analyses. Median age (range) for pancreatic cancer patients was 67 (IQR, 61 – 76), slightly older than those for chronic pancreatitis and normal pancreas patients at 64 (IQR 53 – 72) and 60 (IQR 49 – 70), respectively (p = 0.02). While the majority of pancreatic cancer and chronic pancreatitis patients were men (58% and 68%, respectively), most normal pancreas patients (79%) were women (p=0.007). A higher percentage of normal pancreas patients (68%) were never smokers, compared to pancreatic cancer (50%) and chronic pancreatitis (32%) groups (p=0.04). Of pancreatic cancer cases, 16 (26%) were EUS T-stage 1&2 and 43 (71%) were located in the head of the pancreas.
For the detection of pancreatic cancer in comparison to normal pancreas, the AUC values for CD1D, KCNK12, CLEC11A, NDRG4, IKZF1, PKRCB and KRAS were 0.92 (0.86–0.98), 0.88 (0.80–0.95), 0.85 (0.76–0.95), 0.85 (0.77–0.94), 0.84 (0.75–0.93), 0.83 (0.74–0.92) and 0.75 (0.64–0.86), respectively. Sensitivity at 90% specificity is shown (Table 2). Two of 3 patients with main duct IPMN had methylated CD1D levels exceeding the 90% specificity threshold (not shown).
Table 2.
Discrimination of DNA markers in pancreatic juice for pancreatic cancer
| Marker | Area under ROC curve (95% CI) | Sensitivity at 90% Specificity (95% CI) | ||
|---|---|---|---|---|
|
| ||||
| Pancreatic cancer vs Normal pancreas | Pancreatic cancer vs Chronic pancreatitis | Pancreatic cancer vs Normal pancreas | Pancreatic cancer vs Chronic pancreatitis | |
| Methylated | ||||
| CD1D | 0.92 (0.86–0.98)* | 0.92 (0.85–0.98)* | 0.79 (0.67–0.87) | 0.84 (0.72–0.91) |
| KCNK12 | 0.88 (0.80–0.95)† | 0.73 (0.61–0.86) | 0.79 (0.67–0.87) | 0.46 (0.34–0.58) |
| CLEC11A | 0.85 (0.76–0.95) | 0.76 (0.64–0.87)‡ | 0.67 (0.55–0.78) | 0.53 (0.40–0.65) |
| NDRG4 | 0.85 (0.77–0.94) | 0.85 (0.76–0.94)* | 0.72 (0.6–0.82) | 0.67 (0.55–0.78) |
| IKZF1 | 0.84 (0.75–0.93) | 0.73 (0.61–0.86) | 0.62 (0.5–0.73) | 0.54 (0.42–0.66) |
| PRKCB | 0.83 (0.74–0.92) | 0.77 (0.65–0.89)† | 0.67 (0.55–0.78) | 0.38 (0.27–0.50) |
| Mutant | ||||
| KRAS | 0.75 (0.64–0.86) | 0.62 (0.49–0.74) | 0.56 (0.44–0.68) | 0.39 (0.28–0.52) |
p=0.001 vs KRAS
p=0.03 vs KRAS
p=0.06 vs KRAS
ROC, receiver operating characteristics curve; CI, confidence interval;
For the detection of pancreatic cancer in comparison to chronic pancreatitis, the AUC values for CD1D, KCNK12, CLEC11A, NDRG4, IKZF1, PKRCB and KRAS were 0.92 (0.85–0.98), 0.73 (0.61–0.86), 0.76 (0.64–0.87), 0.85 (0.76–0.94), 0.73 (0.61–0.86), 0.77 (0.65–0.89) and 0.62 (0.49–0.74), respectively. CD1D was the most discriminant individual marker for detection of pancreatic cancer in comparison to normal pancreas or chronic pancreatitis (Figure 4, Supplemental Figures 2–7) and was significantly more discriminant than mutant KRAS (p=0.001). From specificity cut-offs determined in normal pancreas patients, CD1D detected 75% of pancreatic cancer at 95% specificity, while falsely positive in only 9% of chronic pancreatitis patients (p<0.0001, Fisher exact). In contrast mutant KRAS was only positive in 55% of pancreatic cancer samples and falsely positive in 41% of chronic pancreatitis (p=0.3). At 100% specificity CD1D falsely detected only 5% chronic pancreatitis patients (p<0.0001), whereas KRAS was false positive in 32% of chronic pancreatitis (p=0.4).
Figure 4.
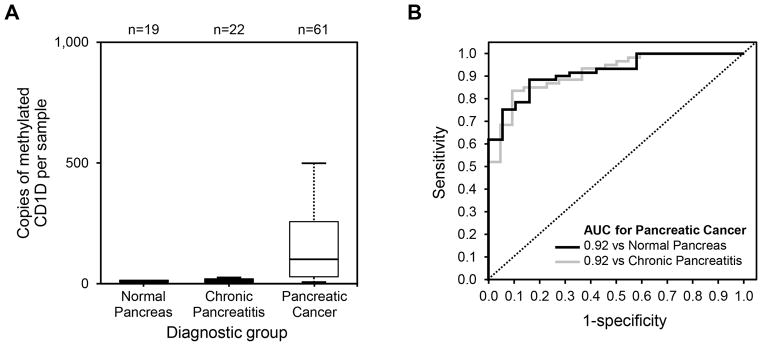
(A) Copy numbers per sample of methylated CD1D, assayed from pancreatic juice samples of patients with normal pancreas, chronic pancreatitis, and pancreatic cancer were used to calculate (B) receiver operating characteristics curves of methylated CD1D for the detection of pancreatic in comparison to normal pancreas (black) and chronic pancreatitis (grey).
Age, sex or current smoking did not significantly influence the strength of association between methylated marker levels and pancreatic cancer. There were no significant differences when patients were stratified for T-stage 1&2 compared to T3&4 or for tumor location in the head of the pancreas compared to body & tail.
DISCUSSION
Methylome sequencing, without a priori bias to known CpG islands, yielded novel highly discriminant methylation markers for pancreatic cancer. Importantly, these findings were confirmed using an independent sample set of tumor and control tissues, showing that the RRBS process can successfully identify pancreatic cancer markers with low background levels in normal pancreatic parenchyma and colonic epithelial tissues. Many of the markers with the strongest association to pancreatic cancer also showed greater than 30-fold increases in the median copies per sample compared to controls; this observation is critical to the application of these markers in diagnostic test development where assays must detect tumor signal against the background biological milieu. Novel candidates identified by this method were clinically piloted by assay from pancreatic juice, demonstrating utility for the detection of pancreatic cancer in blinded comparisons, even to diseased controls with chronic pancreatitis.
In the present study, a single marker, methylated CD1D was sensitive and specific for pancreatic cancer. Candidate marker performance was superior to mutant KRAS, which was poorly specific in patients with chronic pancreatitis. The methylation marker levels in pancreatic juice were unaffected by age, gender, cancer stage or site, similar to our observations of methylated DNA in pancreatic cancer when assayed from stool.(15)
Some of the methylated DNA markers that we found to be highly discriminant for pancreatic neoplasia have been previously identified in array-based studies.(8),(32, 33) Several of our RRBS-discovered markers were found on genes known to be important generally in tumorigenesis, cell signaling, and epithelial-to-mesenchymal transition (Supplemental Table 3), while others have no apparent or reported tumor-related role. Some of the identified markers may prove to be organ-specific. Because DNA methylation is a highly conserved regulator of tissue development,(34) the identification of unreported candidates raises optimism for the existence of DNA methylation events potentially unique to tumor type and site. Indeed, our preliminary observations suggest site specificity of various methylated DNA tumor markers.(35) To our knowledge 10 of the top technically validated markers have not been previously described and are novel to pancreatic cancer. Among 19 biologically validated markers, all contain CpG sites which are not captured by the Infinium 450K hybridization chip. These comparisons demonstrate the value of genome-wide scanning without bias to known DMRs or established gene promotors.
Our results also add significantly to the emerging body of data on next-generation sequencing in human cancer biology. Studies directly comparing these discovery techniques are limited but several recent reports highlight important differences. Among several genome-wide DNA methylation technologies, we selected RRBS for comparatively deeper genomic coverage than methylated DNA immunoprecipitation sequencing (MeDIP-seq) and methylated DNA capture by affinity purification (MethylCap-seq), though the latter two approaches may cover a wider range of genomic CpGs.(19) While MethylCap-seq may identify a greater number of hyper-methylated DMRs,(21) RRBS data output has the advantage of single-nucleotide sequence resolution, which permits optimal design of secondary clinical assay platforms. As the study of genome-wide DNA methylation mapping progresses, modifications to sequencing based platforms are likely to further improve marker yield and accuracy.(22) However, with any discovery strategy, DMRs must be found in a sufficient majority of samples to permit tests of statistical and clinical significance.(23)
There are several limitations to the present study. First, the sample size for RRBS is small but was determined by the power needed to detect a region with at least a 10% differential methylation rate in cases from among controls with the lowest background. With 18 subjects in each group, the overall sample size was similar to or larger than other available genome-wide studies.(8, 21–24) Statistical power was further augmented by analysis of only CpGs with sufficient read depth and group coverage. Samples in the RRBS and the biological validation experiments were tightly matched and randomly allocated to blinded flow cell lane assignment and well assignments, respectively. Tissues from patients with chronic pancreatitis were deliberately excluded from the marker discovery process to ensure the greatest homogeneity of observations within groups(36) and also to exclude possible field effects(37) from undiagnosed pancreatic cancer or precursor lesions. Furthermore, the inclusion of samples from individuals with chronic pancreatitis in clinical piloting controlled for the exclusion of this group in the discovery process. Inclusion of chronic pancreatitis controls may also partially explain why not all markers discriminant in tissues performed equally as well in pancreatic juice. When markers were selected for the pancreatic juice pilot, the full analysis of the biological validation was not yet completed. In the full and completed biological validation, several candidates emerged that might have superior performance. At this time, the DNA from the pancreatic juice analysis had been exhausted, prohibiting testing of those candidates; however, these markers will be of great interest in analysis of new samples, to be collected in a planned prospective clinical trial. Second, samples for the pilot study were from a prospectively enrolled convenience sample and were not matched. This resulted in several significant differences in baseline variables across groups, notably in age, sex and smoking history. However, adjusted analyses did not demonstrate any significant influence of those clinical variables on DNA markers. Greater statistical power may also facilitate the study of marker combinations for improved discrimination. Third, the pancreatic juice sample collection method was also designed to study protein markers (27) and may not have been optimal for DNA recovery. Despite the inclusion of a protease inhibitor in the sample preparation and the use of non-optimized first-pass primer designs, we were able to recover and assay sufficient marker DNA to make highly significant observations. Additionally, the use of secretin stimulation in the collection protocol minimized potential for background contamination during duodenal luminal sampling and avoided the risks of pancreatic duct cannulation, as reviewed by Mastsubayashi and colleagues.(10) Limited by total pancreatic juice DNA quantity, not all biologically validated markers were assessed in pancreatic juice; it is therefore likely that additional highly discriminant markers remain in the initial dataset and deserve further analysis.
Two of 3 patients with IPMNs containing high grade dysplasia had substantially elevated marker levels in pancreatic juice. While corroboration in larger sample size studies are clearly needed, this interesting finding suggests a potential future role for pancreatic juice testing to help guide management of cystic pancreatic lesions.
In this translational investigation from discovery to clinical application, our genome-wide search with RRBS identified novel DNA methylation markers which highly discriminated early stage pancreatic from normal tissue. Initial pilot studies on pancreatic juice both validate the biological discrimination of these new markers and demonstrate their clinical feasibility for use in minimally invasive biological media, such as pancreatic juice, blood, stool or urine. Moving forward, we are compelled to corroborate these findings in expanded patient populations, validate additional candidates, and further assess tumor site-specificity of DNA methylation markers.
Supplementary Material
Statement of translational relevance.
Pancreatic cancer mortality is rising; screening tests are urgently needed. Assay of molecular markers in distant media such as pancreatic juice, stool, urine or blood, is a rational but nascent approach to early detection. Next-generation sequencing, an unbiased marker discovery technique, is largely unexplored in pancreatic cancer. From >6 million CpGs genome-wide, top markers achieved high discrimination in pancreatic cancer tissues and were validated in independent samples. In pancreatic juice samples, methylated DNA markers were highly sensitive and specific, even against chronic pancreatitis controls, and superior to mutant KRAS. Known tumor suppressors were among methylated genes discovered but, more importantly, RRBS revealed novel candidates without previously reported roles in cancer biology. Methylated DNA markers hold promise in noninvasive tools for pancreatic cancer detection from stool or blood. Assay of these markers from pancreatic juice by duodenal aspiration at esophagogastroduodenoscopy could complement imaging in evaluation of pancreatic masses or cystic neoplasms.
Acknowledgments
Funding: This was made possible by grants (to J.B.K.) from the Jack and Maxine Zarrow Family Foundation of Tulsa Oklahoma and the Paul Calabresi Program in Clinical-Translational Research (NCI CA90628). Additional partial support was provided by the Carol M. Gatton endowment for Digestive Diseases Research. Biospecimens were provided by support from the Mayo Clinic SPORE in Pancreatic Cancer (P50 CA102701), the Lustgarten Foundation for Pancreatic Cancer Research and the Clinical Core of the Mayo Clinic Center for Cell Signalling in Gastroenterology (P30DK084567). Reagents for QuARTS assays were provided by Exact Sciences (Madison WI).
Footnotes
Disclosures:
Mayo Clinic is a minor equity investor in and has licensed intellectual property to Exact Sciences (Madison WI). Consistent with Mayo Clinic policy, Drs. Kisiel and Ahlquist, Ms Yab and Messrs. Taylor and Mahoney could share in potential future equity or royalties.
Presented in part at Digestive Disease Week, May 2013, Orlando FL, and Digestive Disease Week, May 2014, Chicago IL
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer Statistics, 2014. CA: Cancer J Clin. 2014 doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Matrisian LM, Aizenberg R, Rosenzweig A. The alarming rise of pancreatic cancer deaths in the United states: Why we need to stem the tide today. 2012 [cited June 12, 2013]; Available from: http://www.pancan.org/section_research/reports/pdf/incidence_report_2012.pdf.
- 3.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 467:1114–7. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeo CJ, Cameron JL, Lillemoe KD, Sitzmann JV, Hruban RH, Goodman SN, et al. Pancreaticoduodenectomy for cancer of the head of the pancreas. 201 patients. Ann Surg. 1995;221:721–31. doi: 10.1097/00000658-199506000-00011. discussion 31–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cleary SP, Gryfe R, Guindi M, Greig P, Smith L, Mackenzie R, et al. Prognostic factors in resected pancreatic adenocarcinoma: analysis of actual 5-year survivors. J Am Coll Surg. 2004;198:722–31. doi: 10.1016/j.jamcollsurg.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-del Castillo C, Adsay NV. Intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology. 139:708–13. 13e1–2. doi: 10.1053/j.gastro.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 7.Sato N, Fukushima N, Hruban RH, Goggins M. CpG island methylation profile of pancreatic intraepithelial neoplasia. Mod Pathol. 2008;21:238–44. doi: 10.1038/modpathol.3800991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vincent A, Omura N, Hong SM, Jaffe A, Eshleman J, Goggins M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin Can Res. 2011;17:4341–54. doi: 10.1158/1078-0432.CCR-10-3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waraya M, Yamashita K, Katoh H, Ooki A, Kawamata H, Nishimiya H, et al. Cancer specific promoter CpG Islands hypermethylation of HOP homeobox (HOPX) gene and its potential tumor suppressive role in pancreatic carcinogenesis. BMC Cancer. 2012;12:397. doi: 10.1186/1471-2407-12-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsubayashi H, Canto M, Sato N, Klein A, Abe T, Yamashita K, et al. DNA methylation alterations in the pancreatic juice of patients with suspected pancreatic disease. Cancer Res. 2006;66:1208–17. doi: 10.1158/0008-5472.CAN-05-2664. [DOI] [PubMed] [Google Scholar]
- 11.Kato N, Yamamoto H, Adachi Y, Ohashi H, Taniguchi H, Suzuki H, et al. Cancer detection by ubiquitin carboxyl-terminal esterase L1 methylation in pancreatobiliary fluids. World J Gastroenterol. 2013;19:1718–27. doi: 10.3748/wjg.v19.i11.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern SE. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer research. 1994;54:3568–73. [PubMed] [Google Scholar]
- 13.Berndt C, Haubold K, Wenger F, Brux B, Muller J, Bendzko P, et al. K-ras mutations in stools and tissue samples from patients with malignant and nonmalignant pancreatic diseases. Clin Chem. 1998;44:2103–7. [PubMed] [Google Scholar]
- 14.Zou H, Harrington JJ, Taylor WR, Devens ME, Cao X, Heigh RI, et al. T2036 Pan-Detection of Gastrointestinal Neoplasms By Stool DNA Testing: Establishment of Feasibility. Gastroenterology. 2009;136:A-625. [Google Scholar]
- 15.Kisiel JB, Yab TC, Taylor WR, Chari ST, Petersen GM, Mahoney DW, et al. Stool DNA testing for the detection of pancreatic cancer: assessment of methylation marker candidates. Cancer. 2012;118:2623–31. doi: 10.1002/cncr.26558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Queneau PE, Adessi GL, Thibault P, Cleau D, Heyd B, Mantion G, et al. Early detection of pancreatic cancer in patients with chronic pancreatitis: diagnostic utility of a K-ras point mutation in the pancreatic juice. Am J Gastroenterol. 2001;96:700–4. doi: 10.1111/j.1572-0241.2001.03608.x. [DOI] [PubMed] [Google Scholar]
- 17.Lidgard GP, Domanico MJ, Bruinsma JJ, Light J, Gagrat ZD, Oldham-Haltom RL, et al. Clinical performance of an automated stool DNA assay for detection of colorectal neoplasia. Clin Gastroenterol Hepatol. 2013;11:1313–8. doi: 10.1016/j.cgh.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 18.Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, et al. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N Engl J Med. 2014;370:1287–97. doi: 10.1056/NEJMoa1311194. [DOI] [PubMed] [Google Scholar]
- 19.Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nature Biotechnol. 2010;28:1106–14. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 21.Simmer F, Brinkman AB, Assenov Y, Matarese F, Kaan A, Sabatino L, et al. Comparative genome-wide DNA methylation analysis of colorectal tumor and matched normal tissues. Epigenetics. 2012;7:1355–67. doi: 10.4161/epi.22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS genetics. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012;7:567–78. doi: 10.4161/epi.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011;108:21188–93. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu H, Bock C, Mikkelsen TS, Jager N, Smith ZD, Tomazou E, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods. 2010;7:133–6. doi: 10.1038/nmeth.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edge SBB, DR, Compton CC, Fritz AG, Greene FL, Trotti A, editors. AJCC Cancer Staging Manual. 7. Springer; New York: 2010. [Google Scholar]
- 27.Noh KW, Pungpapong S, Wallace MB, Woodward TA, Raimondo M. Do cytokine concentrations in pancreatic juice predict the presence of pancreatic diseases? Clin Gastroenterol Hepatol. 2006;4:782–9. doi: 10.1016/j.cgh.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 28.Sun Z, Baheti S, Middha S, Kanwar R, Zhang Y, Li X, et al. SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics. 2012;28:2180–1. doi: 10.1093/bioinformatics/bts337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahlquist DA, Zou H, Domanico M, Mahoney DW, Yab TC, Taylor WR, et al. Next-generation stool DNA test accurately detects colorectal cancer and large adenomas. Gastroenterology. 2012;142:248–56. doi: 10.1053/j.gastro.2011.10.031. quiz e25–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–82. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 31.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–45. [PubMed] [Google Scholar]
- 32.Nones K, Waddell N, Song S, Patch AM, Miller D, Johns A, et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014;135:1110–8. doi: 10.1002/ijc.28765. [DOI] [PubMed] [Google Scholar]
- 33.Omura N, Li CP, Li A, Hong SM, Walter K, Jimeno A, et al. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer biology & therapy. 2008;7:1146–56. doi: 10.4161/cbt.7.7.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song F, Smith JF, Kimura MT, Morrow AD, Matsuyama T, Nagase H, et al. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc Natl Acad Sci U S A. 2005;102:3336–41. doi: 10.1073/pnas.0408436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kisiel JB, Taylor WR, Yab TC, Mahoney DW, Sun Z, Middha S, et al. 769 Novel Methylated DNA Markers Predict Site of Gastrointestinal Cancer. Gastroenterology. 2013;144:S-84. [Google Scholar]
- 36.Bock C. Epigenetic biomarker development. Epigenomics. 2009;1:99–110. doi: 10.2217/epi.09.6. [DOI] [PubMed] [Google Scholar]
- 37.Kisiel JB, Garrity-Park M, Taylor WR, Smyrk TC, Ahlquist DA. Methylated Eya4 Gene in Non-Neoplastic Mucosa of Ulcerative Colitis Patients With Colorectal Cancer: Evidence for a Field Effect. Gastroenterology. 2011;140:S-348-S-9. doi: 10.1097/MIB.0b013e31829b3f4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.