

Abstract
Background
FGFR1 copy number gain (CNG) occurs in head and neck squamous cell cancers (HNSCC) and is used for patient selection in FGFR-specific inhibitor clinical trials. This study explores FGFR1 mRNA and protein levels in HNSCC cell lines, primary tumors and patient-derived xenografts (PDXs) as predictors of sensitivity to the FGFR inhibitor, NVP-BGJ398.
Methods
FGFR1 status, expression levels and BGJ398 sensitive growth were measured in 12 HNSCC cell lines. Primary HNSCCs (n=353) were assessed for FGFR1 CNG and mRNA levels and HNSCC TCGA data were interrogated as an independent sample set. HNSCC PDXs (n=39) were submitted to FGFR1 copy number detection and mRNA assays to identify putative FGFR1-dependent tumors.
Results
Cell line sensitivity to BGJ398 is associated with FGFR1 mRNA and protein levels, not FGFR1 CNG. 31% of primary HNSCC tumors expressed FGFR1 mRNA, 18% exhibited FGFR1 CNG, 35% of amplified tumors were also positive for FGFR1 mRNA. This relationship was confirmed with the TCGA dataset. Using high FGFR1 mRNA for selection, 2 HNSCC PDXs were identified, one of which also exhibited FGFR1 CNG. The non-amplified tumor with high mRNA levels exhibited in vivo sensitivity to BGJ398.
Conclusion
FGFR1 expression associates with BGJ398 sensitivity in HNSCC cell lines and predicts TKI sensitivity in PDXs. Our results support FGFR1 mRNA or protein expression, rather than FGFR1 CNG as a predictive biomarker for the response to FGFR inhibitors in a subset of patients suffering from HNSCC.
Keywords: FGFR1, HNSCC, BGJ398, small molecule inhibitor, targeted therapy
Introduction
Head and neck squamous cell carcinoma (HNSCC) is a common and lethal cancer with a 40-50% 5-year survival rate (1). The risk factors for HNSCC include tobacco and alcohol use, human papilloma virus (HPV) infection and inherited disorders such as Fanconi anemia (1-3). With the exception of the anti-EGFR therapeutic cetuximab, HNSCC has evaded the modern advances of personalized medicine involving deployment of targeted therapeutics that block distinct driver oncogenes present in individual tumors. Recently, a wealth of genomic information has become available on HNSCC with the expectation that driver oncogenes susceptible to targeted therapies would be identified. In contrast to other solid tumors such as lung adenocarcinoma and melanoma where discrete subsets of these cancers are defined by actionable driver oncogenes, a similar picture in HNSCC has not emerged so far (4-7). Rather, sporadic mutations in RAS family members and PIK3CA were observed with the majority of tumors lacking an obvious driver oncogene (4).
The FGFR family of receptor tyrosine kinases (RTKs) is encoded by 4 distinct genes and are established as bona fide oncogenes in diverse human cancers through somatic mutation, gene rearrangements encoding activated fusion proteins, gene amplification and by ligand-dependent activation through paracrine and autocrine FGFs (8-11). Our group and others have reported FGFR1 amplification in HNSCC at a frequency of 15% (9, 12), consistent with the frequency of FGFR1 amplification observed in lung squamous cell carcinomas (13, 14). In fact, the association of FGFR1 amplification in lung cancer cell lines with sensitivity to FGFR-specific TKIs (13, 14) provides the rationale for ongoing trials of two FGFR inhibitors, NVP-BGJ398 (15) and AZD4547 (16), in human cancers where FGFR1 amplification status as well as FGFR2 amplification and FGFR3 mutation serve as a biomarker for patient enrollment. BGJ398 is an orally bioavailable, small molecule pan FGFR kinase inhibitor, predominantly active on FGFR1-3 (17). BGJ398 is currently being tested in ten clinical trials (Phase I and II), out of which three are on solid tumors (NCT01004224, NCT01928459, NCT02160041). Gain-of-function mutations in FGFR2 and FGFR3 have recently been detected in HPV positive HNSCC, invoking an oncogenic role for these FGFRs distinct from FGFR1 that is largely amplified in HPV negative HNSCC (6).
Distinct from the aforementioned mechanisms of FGFR pathway activation, our previous study demonstrated a requirement for autocrine FGF2 in the growth of a subset of HNSCC cell lines (18). It is noteworthy that ligand-mediated paracrine and autocrine activation mechanisms of FGFR cannot be detected by the genomic landscape projects, such as The Cancer Genome Project (TCGA), as no mutations or amplifications are required. In fact, increased expression levels of FGFs and FGFRs would be predicted to serve as markers of autocrine/paracrine FGFR activation. In this regard, the aims of our present study were to rigorously define the ability of FGFR1 amplification to predict FGFR inhibitor sensitivity in HNSCC cell lines and patient-derived xenografts (PDXs) relative to FGFR1 mRNA expression levels. In addition, we defined the prevalence of increased FGFR1 mRNA expression in primary HNSCC and determined the degree of overlap of a FGFR1 copy number gain (CNG) with increased mRNA expression. The findings support a view that FGFR1 mRNA expression may serve as the more accurate and comprehensive biomarker of FGFR1 dependent HNSCC.
Methods
HNSCC patient cohort
The patient cohort was described in a previous publication (9). In brief, we assessed 452 primary tumor tissues where 353 were measurable for both FGFR1 copy number by FISH and mRNA levels by in situ hybridization (see below). Sites of available primary tumor tissue origin were distributed as follows: hypopharynx (n=56), oropharynx (n=142), oral cavity (n=111), larynx (n=143). Clinico-pathological data was available for all patients. In a previous study, all patient samples were tested for p16 positivity. P16-positive cases were then further analyzed for HPV expression (9). The study was approved by the institutional review board of the University Hospital of Bonn (#148/11).
mRNA in situ hybridization (ISH) assay
The FGFR1 mRNA expression status of 452 primary HNSCC patients was examined using the RNA Scope technology for mRNA in situ hybridization (Advanced Cell Diagnostics, Hayward, CA). FGFR1 mRNA molecules were detected with single copy detection sensitivity. All tumor microarray (TMA) slides were digitized using a Zeiss MIRAX MIDI scanner. Staining intensity was evaluated by two independent observers (F.G., A.F.) and scored as described in the legend to Fig. S5. In cases of discrepant results, samples were reassessed by an independent evaluator (S.P.) to obtain consensus.
HNSCC cell lines
The HNSCC cell lines in Tab. 1 were obtained from the collection at the University of Colorado Anschutz Medical Campus or the Leibniz Institute DSMZ (for cell line HN). All were submitted to genomic DNA fingerprinting to verify authenticity and cultured in Dulbecco’s Modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) except for 584-A2 cells (RPMI1640 containing 10% FBS) or SCC9 cells (DMEM/F12 containing 10% FBS and 0.4 μg/ml hydrocortisone).
Table 1.
Characteristics of FGFR1-3 and BGJ398-sensitivity in HNSCC cell lines
| Cell Line | Histology | BGJ398 IC50, [nM] | FGFR1 (protein) |
FGFR1 (mRNA) |
FGFR1
(FISH) |
FGF2 (mRNA) |
FGFR2 (mRNA) |
FGFR3 (mRNA) |
|
|---|---|---|---|---|---|---|---|---|---|
| CyQUANT | Clono- genic |
||||||||
| CCL30 | Nasal Septum |
8 | 0.670 | 0.128 | 0.97 | 2.76 | 0.34 | 0.10 | |
| 584-A2 | Larynx | 14 | 1.4 | 1.000 | 1.000 | 0.47 | 1.86 | 0.15 | 0.01 |
| SCC9 | Tongue | 78 | 69 | 0.580 | 0.104 | 0.97 | 1.35 | 0.69 | 0.02 |
| Cal27 | Tongue | 502 | 0.058 | 0.004 | 0.93 | 0.70 | 6.04 | 0.11 | |
| UMSCC25 | Larynx | 574 | 956 | 0.026 | 0.036 | 0.96 | 0.02 | 1.63 | 0.00 |
| HN6 | Tongue | 611 | 0.039 | 0.005 | 0.96 | 2.78 | 2.87 | 0.02 | |
| UMSCC8 | Alveloar Ridge |
733 | 0.079 | 0.038 | 0.82 | 1.00 | 1.00 | 1.00 | |
| JHU011 | Larynx | 761 | 0.035 | 0.005 | 0.53 | 0.58 | 3.44 | 0.04 | |
| HN12 | Hypopharynx | 822 | 0.057 | 0.006 | 0.67 | 0.56 | 6.81 | 0.01 | |
| UMSCC1 | Floor of Mouth |
860 | 956 | 0.039 | 0.003 | 0.94 | 1.23 | 1.51 | 0.04 |
| HN | Lymph Node Met |
>1000 | 0.068 | 0.017 | 2.50 | 1.34 | - | - | |
| FaDu | Hypopharynx | >1000 | 0.031 | 0.004 | 0.98 | 0.24 | 2.16 | 0.08 | |
| Pearson correlation coefficient |
0.875 | 0.59 | 0.365 | 0.525 | 0.473 | 0.175 | |||
| P value | 0.00019 | 0.040 | n.s. | n.s. | n.s. | n.s. | |||
The IC50s for BGJ398 derived from inhibition of cell proliferation assessed by the CyQUANT assay and/or by clonogenic/anchorage-independent growth assays are shown. FGFR1 protein levels were quantified by densitometry of immunoblots and normalized to the level of the α-subunit of the NaK-ATPase measured following stripping and reprobing the membrane. Likewise FGFR1 mRNA was measured by RT-QPCR and normalized to GAPDH mRNA levels measured in replicate cDNA samples. For both FGFR1 mRNA and protein, the normalized expression values were adjusted relative to the levels in 584-A2 which was assigned a value of 1. For FGFR1 copy number status the target and reference probe signals were quantified in 100 nuclei for each cell line. The ratio presented is target signals/ reference signals. FGF2, FGFR2 and FGFR3 mRNA levels were measured by RT-QPCR and normalized to GAPDH mRNA levels and adjusted relative to the levels in UMSCC8 cells. The association of the different measurements with the sensitivity to BGJ398 was determined by Pearson correlation with the coefficients and P values tabulated (p>0.05, not significant).
HNSCC patient-derived xenograft (PDX) studies
The collection of 39 HNSCC PDX models (Tab. S1) developed and maintained at the University of Colorado Hospital and Anschutz Medical Campus in accordance with an approved protocol (COMIRB #08-0552) has been previously described (19). For analysis of FGFR1 mRNA and gene copy number, a TMA was stained as described above. Studies involving propagation of the PDXs in female nu/nu mice and treatment of xenograft bearing mice with BGJ398 were approved by the Institutional Animal Care and Use Committee Office of the University of Colorado Anschutz Medical Campus. PDXs (passage 2 to 3) were placed in collecting medium consisting of RPMI supplemented with 10% FBS, 200 units/mL penicillin, and 200 μg/mL streptomycin, and cut into ~10 mm3 pieces. The PDX pieces (CUNH015, CUHN072) were implanted in both flanks of twenty mice (40 tumors total/PDX model) and treatment by daily oral gavage of BGJ398 (30 mg/kg) or diluent was initiated when the tumors reached 100 mm3. Tumor volume was measured twice per week with calipers and individual mice were continued on treatment until signs of morbidity or maximal permitted tumor size at which point they were euthanized.
Fluorescence in situ hybridization (FISH) assay for FGFR1 copy number status in HNSCC cell lines
The FGFR1 copy number status of all cell lines was measured with a FISH assay as previously described (20). An FGFR1 target probe (BAC RP11-148D21 spanning the FGFR1 gene locus 8p11.23 to 8p11.22; Invitrogen, Darmstadt, Germany) and a commercially available chromosome 8 centromeric (CEP8) reference probe (Metasystems, Altussheim, Germany) were used for examination. Nuclei were annotated as high-level amplified if the number of red target signals was at least nine greater than the number of green reference signals. Samples with fewer than nine but greater than two excess red target signals relative to the number of green reference signals were annotated as low-level amplified. Polyploidy was defined as an equal number of red target and green reference signals (ratio 1:1) with both probes showing ≥ 4 signals. Amplification and polyploidy was classified as copy number gain (CNG). All cases were evaluated by two independent observers (F.G., A.F.).
Cell proliferation assay
Cell lines were plated at 100 to 500 cells per well in 96 well tissue culture plates and treated with NVP-BGJ398 (Novartis, Nuernberg, Germany) at various doses in triplicate (DMSO diluent, 0.01, 0.03, 0.1, 0.3, and 1.0 µM). Cells were fed with fresh medium containing drugs after 3 days and allowed to proliferate for a total of 7 days. Relative cell number was assessed using a CyQUANT Direct Cell Proliferation Assay (Invitrogen, Darmstadt, Germany) according to instructions provided by the manufacturer.
Clonogenic or anchorage-independent growth assays
To measure the effect of NVP-BGJ398 on single cell colony formation in a clonogenic assay, UMSCC1 and UMSCC25 cells were seeded at 200 cells/well in 6-well plates in full media. Twenty-four hours later, cells were treated with BGJ398 at various doses (DMSO, 0.01, 0.03, 0.1, 0.3, 1.0 µM) and allowed to grow for 10-14 days with feeding once a week. Wells were then rinsed with 1X PBS and stained with 0.5% crystal violet in 6% gluteraldehyde solution. For measurement of anchorage-independent growth of 584-A2 and SCC9 cell in soft agar, 20,000 cells were suspended in 1.5mL media and 0.35% noble agar and overlaid on base layers containing 1.5mL media and 0.5% noble agar in 6-well plates. Wells were fed once a week and allowed to grow for 14 to 21 days. Viable colonies were stained for 24 hrs with 250µL 1mg/mL nitroblue tetrazolium. Digital photographs of both clonogenic and soft agar wells were used to quantify total colony area by Metamorph imaging software.
Silencing of FGFR1 mRNA for testing growth dependency on FGFR1
HNSCC cell lines were transduced with a lentivirus encoding a shRNA targeting green fluorescent protein (GFP) as a negative control for RNAi or lentiviruses encoding two independent FGFR1-targeting shRNAs. The cells were selected for growth in puromycin-containing medium for 14 days and the viable cells were stained with crystal violet. The percent of clonogenic outgrowth relative to the GFP shRNA was determined by Metamorph quantification.
Immunoblot assays
Extracts were prepared as previously described (18) and SDS-PAGE was performed using 10% (ERK1/2) or 8% (FGFR1) polyacrylamide gels. Following electrophoretic transfer, the filters were blocked in Tris-buffered saline containing 0.1% Tween-20 and 3% bovine serum albumin and then incubated overnight in the same solution containing antibodies. Antibodies for analysis of downstream targets were purchased from Cell Signaling Technologies (Danvers, MA); phospho-p44/42 MAPK (ERK1/2) (Thr 202/Tyr 204) (D13.14.4E) XP® Rabbit mAb (#4370); p44/42 MAPK (ERK1/2) (137F5) Rabbit mAb (#4695). Antibodies used for immunoblot analysis of FGFR1 were a rabbit monoclonal antibody (D8E4, Cell Signaling Technologies) and mouse monoclonal antibody (M2F12, internal epitope from Santa Cruz Biotechnology, Santa Cruz, CA). Detection of the α-subunit of NaK-ATPase with a mouse monoclonal antibody (Santa Cruz Biotechnology) or β-actin (#4967, Cell Signaling Technology) was performed as a loading control. FGFR1 protein levels detected with the C-terminal antibody were assessed from densitometry of the immunoblot and normalized to the α-subunit of NaK-ATPase; the resulting value for 584-A2 cells was designated as 1.0.
Real-time quantitative PCR assays
Total RNA was purified from cells using RNeasyTM mini kits (QIAGEN, Valencia, CA) and aliquots (5 μg) were reverse transcribed in a 20 μl volume using random hexamers and MMLV reverse transcriptase. Aliquots (5 μl) of the reverse transcription reactions were submitted to PCR reactions with SYBR® green Jumpstart Taq Readymix (Sigma-Aldrich, St. Louis, MO) using an I Cycle (BioRad, Hercules, CA). Primers used for FGF2, FGFR1-3 QPCR assays are as previously described (18). Expression of the different mRNAs in samples was normalized to GAPDH mRNA levels measured by quantitative RT-PCR in replicate samples. Data are presented as “Relative Expression”.
Ki-67 and cleaved caspase 3 staining
Cytospins of 584-A2 HNSCC cells (~1×105 cells/slide) were generated with a Shandon Cytospin 4 centrifuge (Thermo Electron Corporation) and fixed with paraformaldehyde overnight. Subsequently, immunohistochemistry was performed using the Ventana Discovery automated immunostaining system (Ventana Medical Systems, Tuscon, AZ) and Ventana reagents. Antibodies used were rabbit monoclonal Ki67 (30-9; 1:100; Ventana Discovery, Roche, Mannheim, Germany) and rabbit polyclonal cleaved caspase 3 (1:100; DCS Innovative Diagnostic Systems).
Results
FGFR1 expression and copy number status in HNSCC cell lines
We explored the relationship between FGFR1 CNG, FGFR1 mRNA and protein expression and the expression of FGFR1-3 and FGF2 mRNA in a panel of twelve HNSCC cell lines, all of which proved to be HPV-negative (Tab. 1). Among the cell lines, FGFR1 mRNA was exclusively expressed in CCL30, 584-A2 and SCC9 cells. Immunoblot analysis of cell extracts confirmed the mRNA measurement with FGFR1 protein expression restricted to these three cell lines (Fig. 1). FGFRs are subject to extensive alternative mRNA splicing (see (8-11)) that results in the multiple polypeptides detected by the antibody. Note that the FGFR1 polypeptide expressed by CCL30 cells is not detected by an antibody directed against the C-terminus, but is detectable with an antibody directed against an epitope within the N-terminal region of the protein. FGFR1 copy number status was measured in the HNSCC cell lines by FISH (Fig. S1). Among the twelve HNSCC cell lines, only HN cells exhibit evidence for FGFR1 CNG and UMSCC8 cell exhibit high-level polysomy for FGFR1. Thus, the findings demonstrate that neither expression of FGFR1 mRNA nor protein is associated with FGFR1 CNG in HNSCC cells. It is noteworthy that CCL30, 584-A2 and SCC9 cells express abundant FGF2 mRNA (Tab. 1). These three cell lines also express low FGFR2 and FGFR3 mRNA (Tab. 1) relative to many of the HNSCC cell lines that lack FGFR1 expression.
Figure 1. Expression of FGFR1 protein in HNSCC cell lines.
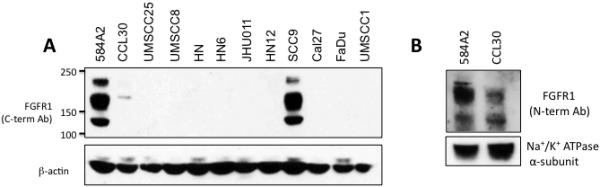
A, Extracts from the indicated HNSCC cell lines were submitted to SDS-PAGE and immunoblotted for FGFR1 with an antibody against the C-terminus and the α-subunit of NaK-ATPase as a loading control. B, Extracts from 584-A2 and CCL30 were submitted to immunoblot analysis using an antibody recognizing an epitope within the extracellular domain. Note that the FGFR1 polypeptide in CCL30 cells is not detected by the C-terminal-directed antibody, suggesting that the cells express an alternatively spliced variant.
Sensitivity of HNSCC cells to BGJ398 is associated with FGFR1 mRNA and protein expression, not FGFR1 CNG
The panel of HNSCC cell lines was tested for growth sensitivity with CyQUANT assays (Fig. 2A) or anchorage-independent/clonogenic growth assays (Fig. 2B) in the presence of increasing concentrations of the FGFR-specific TKI, BGJ398 (15). The calculated IC50 values are graphed in Fig. 2C and presented in Tab. 1. Combined, the data demonstrate high sensitivity of 584-A2, CCL30 and SCC9 cells to BGJ398 with IC50 values less than 100 nM. As a confirmatory measurement, analysis of the Ki67 proliferation index also showed a dose dependent reduction in proliferative activity after 48 hours of BGJ398-treatment in 584-A2 cells (Fig. S2). The remaining HNSCC cells we examined for growth inhibition exhibited IC50s that are greater than 500 nM, a concentration that is outside the therapeutic spectrum of BGJ398 for FGFRs (15, 17). Increased levels of cleaved caspase 3 in BGJ398-treated 584-A2 cells is indicative of an increased apoptosis rate and suggests FGFR pathway addiction in this cell line (Fig. S2B). In addition to inhibition of multiple measures of growth and apoptosis in HNSCC cell lines expressing FGFR1, BGJ398 also reduced the basal levels of the activated downstream target phospho-ERK1/2 in 584-A2, CCL30 and SCC9 cells in a dosage-dependent manner (Fig. S2C). By contrast, there was no effect of BGJ398 on pERK levels in HN, UMSCC25 or UMSCC1 cells that lack FGFR1 expression (Fig. S2C) or on the pAKT status in any of these cell lines (data not shown).
Figure 2. Growth inhibition of HNSCC cell lines by BGJ398 and association of sensitivity with FGFR1 expression.

The indicated cell lines were submitted to proliferation assays (A) or clonogenic/anchorage-independent growth assays (B) in the presence of BGJ398 over a concentration range of 0 to 1 μM. The cell numbers in the proliferation experiments were quantified by the CyQUANT method. In C, dose response data from the CyQUANT assays were used to calculate the IC50 values using the Prism software program which were plotted from lowest to highest. The relationship of the BGJ398 IC50 values with FGFR1 mRNA or protein levels are graphed. A full analysis of FGFR1-3 mRNA, FGFR1 protein, FGFR1 GCN and FGF2 mRNA versus BGJ398 sensitivity is presented in Fig. S3.
The full panel of HNSCC cell lines ranking from the most to the least sensitivity to BGJ398 as determined by inhibition of cell proliferation is shown in Fig. 2C and reveals a correlation of TKI sensitivity with the expression of FGFR1 mRNA (p=0.04) and protein p=0.0002). While FGF2 mRNA expression was weakly associated (p=0.079) with the BGJ398 IC50 values, FGFR2 and FGFR3 mRNA levels were not significantly associated and generally low in the BGJ398-sensitive cell lines (Tab. 1, Fig. S3). In addition, the FGFR1 GCN status did not associate with BGJ398 IC50 values or with FGFR1 mRNA or protein expression (Tab. 1, Fig. S3). In fact, the only HNSCC cell line bearing FGFR1 CNG (HN) failed to express FGFR1 mRNA or protein and showed no sensitivity to BGJ398. In summary, analysis of a panel of twelve HNSCC cell lines reveals three cell lines that are BGJ398 sensitive and demonstrates an association with FGFR1 mRNA and protein expression, but not FGFR1 CNG. In conclusion, the predictive biomarker for FGFR1 TKI sensitivity in HNSCC is FGFR1 mRNA and protein expression.
To confirm FGFR1 dependency regarding treatment with BGJ398, the BGJ398-sensitive cells 584A2 and CCL30 as well as BGJ398-insensitive UMSCC8 cells were transduced with lentiviral vectors encoding two independent shRNAs targeting FGFR1. As shown in Fig. S4, clonogenic growth of 584A2 and CCL30 cells was inhibited by FGFR1 shRNAs relative to the GFP control. This correlated with reduction of FGFR1 mRNA levels. UMSCC8 cells which lack FGFR1 expression (Fig. 1) and are insensitive to BGJ398 (Fig. 2) were unaffected by transduction with shRNAs targeting FGFR1. Thus, the findings reveal that a subset of HNSCC cell lines highly express and are dependent on FGFR1 for growth as assessed by both molecular and pharmacological means.
Partial overlap of FGFR1 mRNA expression with FGFR1 CNG in primary HNSCC
We employed in situ hybridization (ISH) for detection of FGFR1 mRNA in primary HNSCC specimens (Fig. 3 and Fig. S5). Expression scores were binned into no expression (0), low expression (1-2) and medium/high expression (3-4). Overall, 29% (111/385 cases) displayed elevated levels (score 3-4), 56% (214/385) displayed a low expression pattern (1-2) and 16% (60/385) showed no FGFR1 mRNA transcripts (Fig. 4A). Next, we correlated FGFR1 mRNA levels with FGFR1 status in 353 cases of formalin-fixed paraffin-embedded (FFPE) patient samples for which both FGFR1 mRNA and copy number status were available. Among the 63 FGFR1 amplified/polyploid cases (41 low level amplification, 13 high level amplification and 9 polyploid), 35% (22/63 cases) also displayed elevated levels of FGFR1 mRNA (score 3-4+), whereas 65% (41/63) of cases harbouring CNG expressed FGFR1 mRNA at low (0-2+) levels (Fig. 4A). Furthermore, of the 290 patient samples without FGFR1 CNG, 31% (89/290) exhibited increased FGFR1 mRNA levels (3-4+), whereas 69% (201/290) of diploid/deleted cases had no or low FGFR1 mRNA levels (0-2) (Fig. 4A). Staining patterns of specific tumors exhibiting FGFR1 mRNA and/or CNG are shown in Fig. 3. To independently validate this modest degree of overlap between FGFR1 mRNA expression and CNG, we queried RNAseq and gene copy number data derived from 279 HNSCC tumors that are deposited in The Cancer Genome Atlas (TCGA) (Fig. 4B). Using FGFR1 mRNA levels ≥ mean of the 279 tumors as the threshold for positivity, 34% (94/279) of tumors were positive for FGFR1 mRNA and 9% (24/279) showed FGFR1 CNG. Among the tumors with FGFR1 CNG, 58% were also positive for FGFR1 mRNA (Fig. 4B). Thus, the overall frequency of increased FGFR1 mRNA is similar in the University Hospital of Bonn cohort and TCGA tumor collection (29% and 34%, respectively) with degrees of overlap between mRNA positivity and CNG of 35% and 15%, respectively. While increased FGFR1 GCN has been observed to be enriched in HPV-negative HNSCC tumors (6), elevated FGFR1 mRNA expression was detected at similar frequencies in both HPV positive and negative cases (Fig. S6).
Figure 3. Representative HNSCC tumors positive and negative for FGFR1 GCN and mRNA levels.

Representative ISH staining (A1-A4) and FISH staining (B1-B4) of HNSCC tumors exhibiting increased FGFR1 mRNA, but not GCN (A1, B1), increased FGFR1 mRNA and GCN (A2, B2), increased FGFR1 GCN, but not mRNA (A3, B3) and a tumor with diploid FGFR1 GCN and negative for mRNA (A4, B4).
Figure 4. Overlap of FGFR1 mRNA positivity and increased GCN in primary HNSCC.

A, Primary HNSCC (n=353) in the University Hospital of Bonn cohort evaluable for both FGFR1 mRNA (ISH) and GCN (FISH, see (9)). B, As a validation set, The Cancer Genome Atlas HNSCC dataset containing RNAseq and GCN measurements in 279 HNSCC tumors was queried for positivity for FGFR1 mRNA (expression level ≥ mean) and increased GCN. Also, positivity for FGF2 and FGF9 mRNA expression as well as mutations in FGFR2, FGFR3, PIK3CA and TP53 are indicated.
FGFR1 mRNA as a predictive biomarker for BGJ398 sensitivity in a panel of HNSCC patient-derived xenografts (PDXs)
To independently test the utility of FGFR1 mRNA and gene copy number status as predictive biomarkers of FGFR inhibitor response, a collection of 39 HNSCC PDXs was assayed for FGFR1 mRNA and gene copy number status (Tab. S1). Among these, 7/39 (18%) expressed FGFR1 mRNA at 3-4+; two PDX cases exhibiting 4+ mRNA staining were selected (CUHN015 and CUHN072). Only 1/39 PDXs (CUHN072) exhibited high-level FGFR1 CNG while CUHN015 showed a FGFR1/CEP8 signal ratio of 1.6. Importantly, RNAseq analysis of a subset of the PDX models revealed FGFR1 expression patterns that were in good agreement with the in situ hybridization-based assay (Tab. S1, p=0.0147). As an FGFR1 mRNA and CNG negative control, CUHN036 was selected (ISH score = 0). All three cases are HPV-negative. FGFR1 protein levels were assessed by immunoblot analysis of tumor extracts (Fig. 5A), revealing high FGFR1 protein levels in both of the mRNA positive tumors. CUHN015 and CUHN072 were propagated as flank xenografts in nu/nu mice and treated by daily gavage with BGJ398 (30 mg/kg) or with diluent control. CUHN015 xenografts exhibited significant growth inhibition with BGJ398 relative to diluent control-treated tumors (Fig. 5B), demonstrating that a non-CNG HNSCC tumor can respond to an FGFR inhibitor. Surprisingly, and despite amplification and high expression of FGFR1, CUHN072 xenografts exhibited no significant response to BGJ398 (Fig. 5C). The CUHN072 tumors expressed FGF2 protein at levels equivalent to cultured 584-A2 cells (65.5 and 55.8 pg/μg protein, respectively), suggesting that the failure to respond to BGJ398 was not due to inadequate ligand expression. The mechanism for the intrinsic resistance of CUHN072 to BGJ398 is presently under further investigation and preliminary evidence supports strong activation of the MTOR pathway as a candidate explanation.
Figure 5. Sensitivity of HNSCC PDXs to BGJ398.

A, The FGFR1 protein expression levels in extracts from a PDX model negative for FGFR1 mRNA and diploid for GCN (CUHN036), a PDX showing high expression of FGFR1 protein and diploid for GCN (CUHN015) and a PDX exhibiting high FGFR1 mRNA and increased GCN (CUHN072) was measured by immunoblot analysis using the C-terminal antibody (Epitomics) with the α-subunit of the NaK-ATPase as a loading control. The effect of daily oral gavage with BGJ398 (30 mg/kg) on flank xenograft growth of CUHN015 (B), and CUHN072 (C) is shown. The experiment was performed as described in the Materials and Methods.
Discussion
Recurrent genetic alterations associated with tumorigenesis in lung and other common sites of epithelial malignancies such as HNSCC have become a focus of attention in hopes that such alterations will represent opportunities for the development of targeted therapies (21). The frequent amplification of FGFR1 in both squamous lung tumors and HNSCC (9, 12-14, 22-24), while not associated with gain-of-function mutations, highlights this RTK as a target for already available anti-FGFR small molecule inhibitors in these cancers. Therefore, clinical trials for two FGFR inhibitors in human cancers have recruited patients based on the presence of FGFR1 CNG, alone. While FGFR1 amplification has been put forth as a relevant indicator of FGFR1 pathway dependence in lung cancers and serves as precedent for its use as a predictive biomarkers in the aforementioned clinical trials (13, 14), our findings in HNSCC cell lines, patient-derived HNSCC xenografts and primary HNSCC tumors reveal only modest overlap of FGFR1 expression at the mRNA or protein level with FGFR1 CNG. Thus, reliance on FGFR1 CNG, alone, will significantly underestimate the full extent of FGFR1-driven HNSCC and also introduce a significant number of HNSCC patients whose tumors exhibit FGFR1 CNG, but fail to express the gene products. As sensitivity to BGJ398 is dependent on mRNA expression and not gene copy number status in cell line experiments, we recommend redefining selection criteria for the enrollment of patients with HNSCC into clinical trials. Moreover, the results of this study provide precedent for a further exploration of the best predictive biomarker for selection of patients bearing other solid tumors such as lung and breast cancer to FGFR inhibitor trials.
Similar to our findings with FGFR1 mRNA and gene copy number status in HNSCC, FGFR1 protein levels showed no correlation to FGFR1 amplification status (12). Others and we have also observed a lack of correlation between FGFR1 mRNA levels and gene copy number status in lung cancer (25-27). Our findings suggest additional mechanisms regulate the transcription of FGFR1 beyond simple increases in gene copy number. Also, the discrepant prognostic associations assigned to FGFR1 amplification in lung squamous cancers (28, 29) and HNSCC (9) may, in fact, be linked to neighboring genes in the amplicon such as WHSC1L1. Moreover, alternative mRNA splicing may occur at exons encoding the extreme C-terminus of FGFR1 (see Fig. 1) similar to what has been described for FGFR2 (30). To date, regulatory epigenetic mechanisms leading to FGFR1 mRNA and protein up-regulation are poorly defined and should be a subject of additional investigation.
FGFR inhibitors such as BGJ398 are not specific for FGFR1, but also inhibit FGFR2 and FGFR3 (15). FGFR2 and FGFR3 mRNA levels are low in all 3 BGJ398-sensitive cell lines (CCL30, SCC9, 584-A2), as compared to insensitive cells (see Tab. 1, Fig. S3). Thus, we propose that FGFR2 and FGFR3 play little or no role in driving growth of these cell lines and that the response of the cells to BGJ398 is therefore likely due to an inhibition of FGFR1 activity. FGF2 levels proved to be relatively high in TKI-sensitive cells, pointing to an activation of FGFR1 through autocrine signaling as previously proposed. The reduction of basal phospho-ERK levels in 584-A2, CCL30 and SCC9 cells is consistent with the proposed autocrine activity of FGF2 in these HNSCC cells as detected with TKIs and an FGF ligand trap (18). The degree of co-expression of FGF2 and FGF9 with FGFR1 mRNA in TCGA HNSCC tumors (Fig. 4B) is consistent with this notion. It is noteworthy that the FGFR1 mRNA positive tumors likely represent a distinct subset from HNSCCs bearing FGFR2 or FGFR3 mutations (Fig. 4B). The two FGFR2/3 mutation positive tumors within the FGFR1 mRNA positive subset do not express these genes at the mRNA level (data not shown). This observation is consistent with a recent study demonstrating that HNSCCs bearing FGFR2 and FGFR3 mutations largely represent HPV-positive tumors relative to FGFR1 CNG, a marker enriched in HPV-negative HNSCC (6). In our previous study we also reported FGFR1 amplification and HPV-infection to occur mutually exclusive (11). By contrast, in our present study, we observe increased FGFR1 mRNA levels being in HPV-positive as well as in HPV-negative tumor samples. Additionally, our analysis of HNSCC TCGA data indicates that FGFR1 mRNA positivity overlaps broadly with PIK3CA and TP53 mutations, suggesting that presence of these mutations will not exclude sensitivity to FGFR inhibitors.
Finally, this study provides important precedent for therapeutic advancement on an oncogene driver pathway that cannot be identified in human tumors through simple assessment of somatic mutation or gene amplification events. Our previous studies demonstrated the importance of autocrine FGF2 for the growth of both lung cancer and HNSCC cell lines (18, 31). In fact, an autocrine role for FGFs in cancer is consistent with the long-established findings that specific FGF genes are transforming genomic integration sites for MMTV in murine breast cancer (32). Thus, in addition to the documented role somatic mutations in FGFR family members play in human solid cancers (6, 33) and the transforming role of FGFR oncogene fusions in hematological cancers and solid tumors including lung adenocarcinoma (34), we propose that FGFR1 will function as an actionable oncogene driver in many cancers including HNSCC through autocrine activation by co-expressed FGFs. It is important to note that co-expression of wild-type FGF2 and FGFR1 will not be identified by the completed and ongoing cancer genome projects, but can be readily assessed through straightforward measurement of FGFR1 protein or mRNA expression. If true, FGFR1 may function as a targetable oncogene driver in as many as 25 to 30% of HNSCC patients.
The IC50s for BGJ398 derived from inhibition of cell proliferation assessed by the CyQUANT assay and/or by clonogenic/anchorage-independent growth assays are shown. FGFR1 protein levels were quantified by densitometry of immunoblots and normalized to the level of the α-subunit of the NaK-ATPase measured following stripping and reprobing the membrane. Likewise FGFR1 mRNA was measured by RT-QPCR and normalized to GAPDH mRNA levels measured in replicate cDNA samples. For both FGFR1 mRNA and protein, the normalized expression values were adjusted relative to the levels in 584-A2 which was assigned a value of 1. For FGFR1 copy number status the target and reference probe signals were quantified in 100 nuclei for each cell line. The ratio presented is target signals/ reference signals. FGF2, FGFR2 and FGFR3 mRNA levels were measured by RT-QPCR and normalized to GAPDH mRNA levels and adjusted relative to the levels in UMSCC8 cells. The association of the different measurements with the sensitivity to BGJ398 was determined by Pearson correlation with the coefficients and P values tabulated (p>0.05, not significant).
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
FGFR1 has emerged as a therapeutic target in a number of human solid cancers including lung squamous cell carcinomas and breast cancers. Herein, we demonstrate that growth inhibition of HNSCC cell lines and patient-derived xenografts is statistically associated with FGFR1 mRNA and protein expression, but not copy number gain in HNSCC. Importantly, as a single biomarker, FGFR1 amplification greatly underestimates the potential prevalence of FGFR1-driven HNSCC and only partially overlaps with FGFR1 mRNA expression. Thus, a biomarker encompassing FGFR1 expression, either mRNA or protein, should be considered for more accurate and comprehensive enrollment of HNSCC patients to clinical trials with FGFR-specific tyrosine kinase inhibitors.
Acknowledgements
NVP-BGJ398 was kindly provided by Novartis. The results in Figure 5B are derived completely from data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
We would like to thank Z. Auberpferd for exceptional technical support.
Grant Support
This study was supported by a grant of the Rudolph-Becker-Foundation (SP), by VA Merit grant 1 BX001994-01A1 (LEH), NIH R21DE019712 (AJ), and by NIH grant P50 CA15187 (LEH, FRH).
Footnotes
Author’s Contributions
Conception and design: F. Göke, S. Perner, L. E. Heasley, A. Franzen, T. K. Hinz,
Acquisition of data: T. K. Hinz, P. Yoon, L. A. Marek, E. K. Kleczko, J. R. Eagles, F. R. Hirsch, A. Franzen, A. Schroeck, T. Van Bremen, A. Göke, R. Sharma, A. von Maessenhausen, M. Bode, C. Golletz, D.Boehm, W. Vogel, J. Kim
Analysis and interpretation of data: F. Göke, S. Perner, L. E. Heasley, A.C. Tan, A. Franzen, R. Kirsten
Writing, review, and/or revision of manuscript: F. Göke, S. Perner, L. E. Heasley, A. Jimeno, A. Franzen, F. Bootz, B. Lankat-Buttgereit
Providing tissue: G. Kristiansen
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
REFERENCES
- 1.Rothenberg SM, Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. The Journal of clinical investigation. 2012;122:1951–7. doi: 10.1172/JCI59889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, et al. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Archives of otolaryngology--head & neck surgery. 2003;129:106–12. doi: 10.1001/archotol.129.1.106. [DOI] [PubMed] [Google Scholar]
- 3.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nature reviews Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 4.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Wawrose JS, Gooding WE, Garraway LA, Lui VW, Peyser ND, et al. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: a rational approach to preclinical model selection. Molecular cancer research : MCR. 2014;12:571–82. doi: 10.1158/1541-7786.MCR-13-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker TP, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 doi: 10.1158/1078-0432.CCR-13-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature reviews Cancer. 2010;10:116–29. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 9.Goke F, Bode M, Franzen A, Kirsten R, Goltz D, Goke A, et al. Fibroblast growth factor receptor 1 amplification is a common event in squamous cell carcinoma of the head and neck. Mod Pathol. 2013;26:1298–306. doi: 10.1038/modpathol.2013.58. [DOI] [PubMed] [Google Scholar]
- 10.Goke F, Goke A, von Massenhausen A, Franzen A, Sharma R, Kirsten R, et al. Fibroblast growth factor receptor 1 as a putative therapy target in colorectal cancer. Digestion. 2013;88:172–81. doi: 10.1159/000355018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehnen NC, von Massenhausen A, Kalthoff H, Zhou H, Glowka T, Schutte U, et al. Fibroblast growth factor receptor 1 gene amplification in pancreatic ductal adenocarcinoma. Histopathology. 2013;63:157–66. doi: 10.1111/his.12115. [DOI] [PubMed] [Google Scholar]
- 12.Freier K, Schwaenen C, Sticht C, Flechtenmacher C, Muhling J, Hofele C, et al. Recurrent FGFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC) Oral oncology. 2007;43:60–6. doi: 10.1016/j.oraloncology.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PloS one. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. Journal of medicinal chemistry. 2011;54:7066–83. doi: 10.1021/jm2006222. [DOI] [PubMed] [Google Scholar]
- 16.Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer research. 2012;72:2045–56. doi: 10.1158/0008-5472.CAN-11-3034. [DOI] [PubMed] [Google Scholar]
- 17.Guagnano V, Kauffmann A, Wohrle S, Stamm C, Ito M, Barys L, et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012;2:1118–33. doi: 10.1158/2159-8290.CD-12-0210. [DOI] [PubMed] [Google Scholar]
- 18.Marshall ME, Hinz TK, Kono SA, Singleton KR, Bichon B, Ware KE, et al. Fibroblast growth factor receptors are components of autocrine signaling networks in head and neck squamous cell carcinoma cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:5016–25. doi: 10.1158/1078-0432.CCR-11-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keysar SB, Astling DP, Anderson RT, Vogler BW, Bowles DW, Morton JJ, et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Molecular oncology. 2013;7:776–90. doi: 10.1016/j.molonc.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heist RS, Mino-Kenudson M, Sequist LV, Tammireddy S, Morrissey L, Christiani DC, et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol. 2012;7:1775–80. doi: 10.1097/JTO.0b013e31826aed28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HR, Kim DJ, Kang DR, Lee JG, Lim SM, Lee CY, et al. Fibroblast growth factor receptor 1 gene amplification is associated with poor survival and cigarette smoking dosage in patients with resected squamous cell lung cancer. J Clin Oncol. 2013;31:731–7. doi: 10.1200/JCO.2012.43.8622. [DOI] [PubMed] [Google Scholar]
- 24.Young RJ, Lim AM, Angel C, Collins M, Deb S, Corry J, et al. Frequency of fibroblast growth factor receptor 1 gene amplification in oral tongue squamous cell carcinomas and associations with clinical features and patient outcome. Oral oncology. 2013;49:576–81. doi: 10.1016/j.oraloncology.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Network CGAR Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wynes MW, Hinz TK, Gao D, Martini M, Marek LA, Ware KE, et al. FGFR1 mRNA and Protein Expression, not Gene Copy Number, Predict FGFR TKI Sensitivity across All Lung Cancer Histologies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:3299–309. doi: 10.1158/1078-0432.CCR-13-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pros E, Lantuejoul S, Sanchez-Verde L, Castillo SD, Bonastre E, Suarez-Gauthier A, et al. Determining the profiles and parameters for gene amplification testing of growth factor receptors in lung cancer. Int J Cancer. 2013;133:898–907. doi: 10.1002/ijc.28090. [DOI] [PubMed] [Google Scholar]
- 28.Cihoric N, Savic S, Schneider S, Ackermann I, Bichsel-Naef M, Schmid RA, et al. Prognostic role of FGFR1 amplification in early-stage non-small cell lung cancer. British journal of cancer. 2014;110:2914–22. doi: 10.1038/bjc.2014.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craddock KJ, Ludkovski O, Sykes J, Shepherd FA, Tsao MS. Prognostic value of fibroblast growth factor receptor 1 gene locus amplification in resected lung squamous cell carcinoma. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2013;8:1371–7. doi: 10.1097/JTO.0b013e3182a46fe9. [DOI] [PubMed] [Google Scholar]
- 30.Moffa AB, Tannheimer SL, Ethier SP. Transforming potential of alternatively spliced variants of fibroblast growth factor receptor 2 in human mammary epithelial cells. Molecular cancer research : MCR. 2004;2:643–52. [PubMed] [Google Scholar]
- 31.Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol. 2009;75:196–207. doi: 10.1124/mol.108.049544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J, et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet. 2007;39:759–69. doi: 10.1038/ng2034. [DOI] [PubMed] [Google Scholar]
- 33.Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3:264–79. doi: 10.1158/2159-8290.CD-12-0362. [DOI] [PubMed] [Google Scholar]
- 34.Wang R, Wang L, Li Y, Hu H, Shen L, Shen X, et al. FGFR1/3 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:4107–14. doi: 10.1158/1078-0432.CCR-14-0284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.