

Abstract
Acute lymphoblastic leukemia (ALL), the most common malignancy of childhood, is a genetically complex entity that remains a major cause of childhood cancer-related mortality. Major advances in genomic and epigenomic profiling during the past decade have appreciably enhanced knowledge of the biology of de novo and relapsed ALL and have facilitated more precise risk stratification of patients. These achievements have also provided critical insights regarding potentially targetable lesions for development of new therapeutic approaches in the era of precision medicine. This review delineates the current genetic landscape of childhood ALL with emphasis upon patient outcomes with contemporary treatment regimens, as well as therapeutic implications of newly identified genomic alterations in specific subsets of ALL.
Keywords: acute lymphoblastic leukemia, cytogenetics, genomics, pediatrics, therapy
Introduction
Substantial advances have been made in the past five decades in the treatment of patients with acute lymphoblastic leukemia (ALL), the most common malignancy of childhood, which was universally fatal fifty years ago. Approximately 3,000 children are diagnosed with ALL each year in the United States, and long-term survival rates approach 85-90% with contemporary therapy.1, 2 Improved cure rates have been largely attributable to recognition of the critical importance of central nervous system (CNS)-directed anti-leukemia therapy, implementation of multi-agent chemotherapy cycles with a prolonged maintenance phase, and improved supportive care measures. A key feature of contemporary ALL treatment regimens is stratification of patients into different risk groups based upon clinical and biological characteristics of the patient and the leukemia, as well as early treatment response. Therapies of different intensity are applied to the different risk groups with more intensive therapy needed to maximize the chance of cure for higher risk patients. This approach has provided a paradigm for the treatment of many pediatric and adult cancers.
Patient age and initial white blood cell (WBC) count are consistent predictors of outcome, as patients of older age and/or higher WBC counts fare less well than younger patients and/or those with lower WBC counts.3 These continuous variables were dichotomized by the National Cancer Institute (NCI)-Rome criteria to define standard-risk (SR; age 1-9.99 years and WBC <50,000/microliter) and high-risk (HR; age ≥10 years and/or WBC ≥50,000/microliter) subgroups.4 NCI-Rome classification criteria are used by many consortia for risk stratification of children with B-ALL, but have limited predictive power in T-ALL.
Improved understanding of the biologic heterogeneity of ALL and development of sensitive polymerase chain reaction- or flow cytometry-based minimal residual disease (MRD) response monitoring techniques have facilitated modern risk stratification for childhood ALL.5-7 Implementation of risk-adapted algorithms with application of appropriately intensive therapy in subsets of patients with ALL has helped to improve relapse-free and overall survival (OS) and to minimize toxicities, although such algorithms remain imperfect. Approximately 75% of B-ALL cases have somatic aneuploidy or recurrent chromosomal translocations, many of which have prognostic significance. Conversely, the clinical significance of most recurrent genomic alterations in T-ALL is less clear, and current clinical trials stratify patients with T-ALL based primarily upon MRD responses to induction and/or post-induction chemotherapy. Despite excellent survival for most pediatric patients with ALL, relapse occurs in 15-20% of children and remains a significant source of childhood-cancer morbidity and mortality.8
Substantial genomic sequencing efforts now underway will better characterize B-ALL and T-ALL, define new prognostic factors, and identify genomic lesions and pathways suitable for molecularly-targeted therapies. As most children with de novo ALL diagnosed in North America and Western Europe are treated in large cooperative group clinical trials, correlation of leukemia genomics data with well-annotated clinical trial response and outcome data will continue to inform the development and prioritization of new therapeutic approaches for appropriate patient subsets. In this review, we discuss current knowledge concerning the genomic landscape of ALL and its relevance to treatment of children and adolescents/young adults (AYAs) with ALL.
B-Cell Acute Lymphoblastic Leukemia (B-All)
B-ALL comprises approximately 85% of pediatric ALL. The majority of childhood B-ALL cases are currently classified based upon the presence of specific recurrent genetic lesions delineated by the World Health Organization 2008 criteria (Figure 1).9 Rare bone marrow failure and other constitutional leukemia predisposition syndromes, such as trisomy 21 and TP53 mutations in Li-Fraumeni syndrome, are associated with higher incidence of childhood ALL and are discussed in detail elsewhere. Recent studies have also identified genomic polymorphisms in genes including ARID5B, CEBPE, GATA3, and IKZF1 that increase the risk of developing ALL, and rare germline mutations in PAX5 and ETV6 have been linked to familial ALL occurrence.10-15
Figure 1. Frequency of cytogenetic alterations in childhood B-ALL and T-ALL.
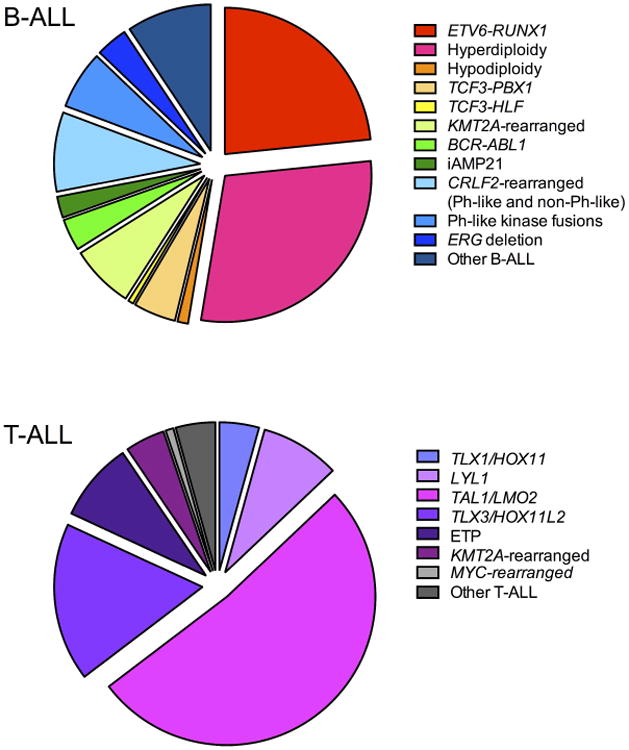
Data regarding submicroscopic genetic alterations are not included.
Aneuploidy
Leukemia-associated whole chromosome gains and losses are readily identified by conventional cytogenetics and/or fluorescence in situ hybridization (FISH), and many numerical alterations have prognostic and therapeutic significance (Table 1).16 High hyperdiploidy with greater than 50 chromosomes per leukemia cell occurs in approximately 25% of childhood ALL, is most frequent in younger children (particularly those with SR ALL), and is associated with favorable chemotherapy responses and 5-year OS exceeding 90-95%.17 Children with high hyperdiploid SR ALL who are rapid early responders to induction chemotherapy comprise a low-risk subgroup; therapy reduction for such patients can minimize therapy-associated toxicities without compromising cure rates.18, 19
Table 1. Common genetic alterations in childhood ALL and associated clinical outcomes.
| Genetic subtype | Common alterations | Frequency in ALL | Prognosis | Comment |
|---|---|---|---|---|
| B-ALL | ||||
| Aneuploidy | ||||
| Hyperdiploidy (>50 chromosomes) | 25% | Favorable | ||
| Hypodiploidy (<44 chromosomes) | Near-haploidy (24-31 chromosomes), low-hypodiploidy (32-39 chromosomes) | 1-2% | Unfavorable | Association with TP53 mutations, IKZF2 and IKZF3 deletions, and Ras and PI3K pathway mutations |
| Chromosomal Translocations | ||||
| t(12;21)(p13;q22) | ETV6-RUNX1 (TEL-AML1) | 20% | Favorable | |
| t(1;19)(q23;p13.1) | TCF3-PBX1 (E2A-PBX1) | 4% | Intermediate | |
| t(17;19)(q22;p13) | TCF3-HLF | <0.5% | Unfavorable | |
| KMT2A (MLL) rearrangements | 5-6% | Unfavorable (infants), intermediate (non-infants) | Highest frequency in younger infants (80%); associated with FLT3 overexpression and epigenetic dysregulation | |
| t(1;11)(q21;q23) | KMT2A-MLLT11 | Less unfavorable | Rare | |
| t(4;11)(q21;q23) | KMT2A-AFF1(AF4) | Particularly unfavorable | Comprises 50% of infant KMT2A-rearranged ALL | |
| t(9;11)(p22;q23) | KMT2A-MLLT3(AF9) | Comprises 15% of infant KMT2A-rearranged ALL | ||
| t(10;11)(p12;q23) | KMT2A-AF10 | Comprises 5% of infant KMT2A-rearranged ALL | ||
| t(11;19)(q23;p13.3) | KMT2A-ENL | Comprises 20-25% of infant KMT2A-rearranged ALL | ||
| Other fusion partners | ||||
| t(9;22)(q34;q11.2) | BCR-ABL1 | 3-5% | Unfavorable prior to TKI therapy, intermediate with TKI therapy? | Associated with IKZF1 deletions |
| Other | ||||
| iAMP21 | Multiple copies of RUNX1 | 2% | Unfavorable | Rare rob(15;21)(q10;q10)c associated with greatly increased risk of iAMP21 ALL |
| Trisomy 21-associated ALL | P2RY8-CRLF2, JAK2 mutations | Intermediate | ||
| Philadelphia chromosome-like (Ph-like) | IGH-CRLF2, P2RY8-CRLF2 | 7-8% | Unfavorable | 50% of Ph-like; associated with JAK1 and JAK2 mutations, CDKN2A/B deletions, IKZF1 deletions; increasing incidence with older age; possibly targetable with TKIs |
| ABL1, ABL2, CSF1R, PDGFRB rearrangements | 5-6% | Unfavorable | 10-20% of Ph-like; potentially targetable with TKIs | |
| EPOR, JAK2 rearrangements | 2% | Unfavorable | 10% of Ph-like; potentially targetable with TKIs | |
| ERG deletion | 3% | Favorable | Associated with IKZF1 deletions and aberrant CD2 expression | |
|
| ||||
| T-ALL* | ||||
| Transcription factor oncogenes | ||||
| t(10;14)(q24;q11) | TLX1 (HOX11) fusions | 5-10% of T-ALL | Favorable | Associated with PHF6 mutations |
| t(7;19)(q34;p13) | LYL1 fusions | 10% of T-ALL | Unfavorable | |
| t(1;14)(p32;q11), t(1;7)(p32;q34), t(11;14)(p15;q11), t(11;14)(p13;q11) | TAL1, LMO1, LMO2 fusions | 50-60% of T-ALL | Unfavorable | Associated with PHF6 mutations |
| t(11;14)(p15;q11), t(5;14)(q35;q32) | TLX3 (HOX11L2) fusions | 20-25% of T-ALL | Unfavorable (some studies), intermediate (some studies), favorable (some studies) | Associated with PHF6 mutations |
| 7p15 translocations | HOXA10, HOXA9 overexpression | 3% of T-ALL | Unfavorable | |
| KMT2A rearrangements | ||||
| KMT2A-AFF1, KMT2A-MLLT1 | 5% of T-ALL | Possibly favorable | ||
| PICALM-MLLT10 | 5-10% of T-ALL | Unfavorable (some studies), intermediate (other studies) | Associated with EZH2 alterations | |
| Other | ||||
| t(8;14)(q24;q11) | TRA-MYC, TRC-MYC | 1% of T-ALL | Likely unfavorable | Associated with MYC activation and aggressive phenotype |
| ETP | 10-15% of T-ALL | Unfavorable (some studies), intermediate (other studies) | Associated with Ras pathway mutations; characteristic immunophenotype (CD1a-, CD8-, CD5- or CD5-dim with co-expression of myeloid or stem cell markers) | |
| NOTCH1 mutations | 50-60% of T-ALL | Favorable | Associated with CDKN2A and FBXW7 deletions | |
| FBXW7 mutation | 15% of T-ALL | Associated with NOTCH1 activation via impairment of proteasomal degradation of NOTCH1 | ||
| t(9;14)(q34;q32) | NUP214-ABL1 | 5-15% of T-ALL | Unfavorable (some studies), intermediate (other studies) | Associated with HOX11 and HOX11L2 overexpression |
| Other T-ALL | 20% of T-ALL | |||
|
| ||||
| Relapsed ALL | Associated with chemotherapy resistance | |||
| CREBBP mutation | 20% of relapsed ALL | Likely confers resistance to glucocorticoids | ||
| NT5C2 mutation | 20% of relapsed ALL | Likely confers resistance to anti-metabolite drugs | ||
| MSH6 deletion | ||||
| NR3C1 deletion | ||||
| SETD2 mutation | 12% of relapsed ALL | |||
| KDM6 mutation | ||||
| MLL2 mutation | ||||
| Ras pathway mutations | 30-50% of relapsed ALL | |||
Frequency of alterations in T-ALL exceeds sum of 100% due to co-occurrence of lesions.
TKI = tyrosine kinase inhibitor
Conversely, hypodiploidy with fewer than 44 chromosomes accounts for 1-2% of childhood ALL and is associated with poor outcomes.20, 21 A recent report from the Children's Oncology Group (COG) found that children with hypodiploid ALL had a 5-year event-free survival (EFS) rate of 47% versus 85% for children with non-hypodiploid ALL.22 MRD response was highly predictive of outcome among the hypodiploid patients, and those with end-Induction MRD ≥0.01% had a 5-year EFS of <30%.22 Based on these poor outcomes, hypodiploidy is often considered to be an indication for allogeneic hematopoietic stem cell transplant (HSCT) in first remission.23-26
Hypodiploid ALL can be further classified based upon the degree of hypodiploidy, such as low-hypodiploidy (32-39 chromosomes) and near-haploidy (24-31 chromosomes), which may correlate with incrementally inferior clinical outcomes.24 The genomics of near-haploid and low-hypodiploid ALL are quite different. Remarkably, over 90% of pediatric low-hypodiploid ALL cases harbor TP53 mutations, which are germline events indicative of Li-Fraumeni syndrome in half of patients.27 The pattern of mutations present in hypodiploid ALL suggests that there may be clinical utility of phosphatidylinositol-3-kinase (PI3K) or mitogen/extracellular signal-regulated kinase (MEK) inhibitors in this high-risk ALL subset.27
Sentinel Chromosomal Translocations
Nearly half of childhood B-ALL cases harbor somatic chromosomal translocations, many of which may be cryptic on conventional cytogenetic analyses, but readily detectable by FISH or reverse-transcriptase polymerase chain reaction (RT-PCR) amplification of fusion genes created by these translocations. The most common is t(12;21)(p13;q22) resulting in ETV6-RUNX1 (TEL-AML1) fusion that occurs in 20-25% of childhood NCI SR B-ALL. ETV6-RUNX1 alterations are often accompanied by other submicroscopic alterations in lymphoid development and tumor suppressor genes.28 Children with NCI SR ETV6-RUNX1 ALL have an outstanding prognosis with >95% OS, and therapy de-escalation trials with lower-intensity chemotherapy regimens for this patient population have been conducted by some cooperative groups.18, 19 However, MRD response remains a critical predictor of outcome in childhood ALL, including ETV6-RUNX1 ALL, and children with elevated end-Induction MRD remain at higher risk of treatment failure and/or relapse regardless of underlying leukemia-associated alterations.5, 7 ETV6-RUNX1 translocations have been detected at low levels in preserved neonatal blood spot specimens (Guthrie cards) from children who subsequently develop ALL, implicating a prenatal origin of this subtype of childhood ALL.29 However, ETV6-RUNX1 fusions are also detectable in blood spots from children who do not subsequently develop ALL, suggesting that additional cooperating mutations are necessary for leukemogenesis.30
Other recurrent chromosomal translocations in B-ALL include t(1;19)(q23;p13.3) resulting in TCF3-PBX1 (E2A-PBX1) fusion, rearrangement of KMT2A (formerly MLL; 11q23), and t(9;22)(q34;q11.2) (the Philadelphia chromosome; Ph+) resulting in BCR-ABL1 fusion. While TCF3-PBX1 ALL was previously associated with an intermediate or unfavorable prognosis, modern therapeutic regimens have improved outcome, and TCF3-PBX1 fusion is no longer considered for risk stratification.31-33 Children with TCF3-PBX1 ALL appear to have higher risk of CNS relapse and may merit intensification of CNS-directed therapy.34 TCF3-HLF fusion resulting from t(17;19)(q22;p13.3) occurs in less than 0.5% of patients with B-ALL and, despite its rarity, has been associated with extremely poor outcomes.35, 36
KMT2A is a promiscuous oncogene with rearrangements involving >75 fusion partners, and the incidence of KMT2A rearrangements in childhood B-ALL differs markedly by age.37, 38 Approximately 75% of infants less than 1 year old with ALL harbor somatic KMT2A rearrangements, and these patients have high rates of hyperleukocytosis and CNS involvement with leukemia. The near-universal concordance rate of KMT2A-rearranged ALL in monozygotic twin infants suggests prenatal origin of leukemogenesis in this subtype of ALL.39, 40 Infants with KMT2A-rearranged ALL have poor OS (<50% at 4 years) despite intensive multi-agent chemotherapy.41, 42 Major adverse risk factors in infant ALL include age less than 90 days and WBC ≥300,000/microliter.41, 43 Given concomitant overexpression of the fms-related tyrosine kinase 3 receptor (FLT3) in KMT2A-rearranged infant ALL and promising preclinical data with FLT3 inhibition,44-46 clinical testing of FLT3 inhibitors (e.g., lestaurtinib, quizartinib) with chemotherapy in infants with KMT2A-rearranged ALL is ongoing to determine if combination therapy can diminish relapse risk and/or improve EFS (www.clinicaltrials.gov NCT00557193 and NCT0141126747). Similarly, given frequent epigenetic dysregulation reported in KMT2A-rearranged infant ALL, demethylating/hypomethylating agents (e.g., decitabine, 5-azacytidine, DOT1L inhibitors) and histone deacetylation inhibitors (e.g., vorinostat, panobinostat, bromodomain inhibitors) are also under preclinical and early clinical evaluation.48 The genomics of KMT2A-rearranged ALL differ between infants and older children. In infants, KMT2A rearrangement is accompanied by a remarkable paucity of other somatic mutations.49, 50 Many mutations occur in genes associated with activated Ras or PI3K pathway signaling, although were often detected in subclonal populations and/or were diminished or lost at relapse. These data suggest that such alterations may be passenger mutations, and a potential role for PI3K pathway or MEK inhibition in infant ALL is not clear.50 KMT2A-rearranged leukemias in older children and AYAs have a higher number of somatic mutations including mutations in epigenetic regulators in about half of cases.50 The frequency of KMT2A-rearranged ALL diminishes markedly with increased age, occurring in approximately 5% of children with B-ALL older than one year.43
BCR-ABL1-rearranged (Ph+) ALL occurs in 3-5% of children with B-ALL and conferred a particularly dismal prognosis in the pre-tyrosine kinase inhibitor (TKI) treatment era with 35% 3 year EFS.51, 52 Sentinel studies conducted in the early 2000s demonstrated that ABL1-targeting TKIs (e.g., imatinib; now also dasatinib, nilotinib, and ponatinib) could induce potent leukemia cytotoxicity in Ph+ ALL.51, 53 Mature clinical trial data now demonstrate that combination therapy with imatinib and intensive cytotoxic chemotherapy has substantially improved EFS and OS in children with Ph+ ALL and has minimized need for HSCT in first remission, one of the true precision medicine successes in pediatric oncology.54, 55 As in chronic myelogenous leukemia, development of ABL1 “gatekeeper” kinase domain mutations in patients treated chronically with TKIs remains a potential mechanism of drug resistance, although these mutations have not appeared to occur at high frequency in children with Ph+ ALL to date.25, 56 Most Ph+ ALL cases also have deletions in the transcription factor IKZF1, which has been associated with poor prognosis in Ph+ and other subtypes of ALL.57-60
Intrachromosomal Amplification of Chromosome 21
Intrachromosomal amplification of chromosome 21 (iAMP21) is a high-risk subset comprising approximately 2% of childhood B-ALL. This subtype was initially discovered via detection of multiple RUNX1 copies on routine ETV6-RUNX1 FISH testing. iAMP21 ALL generally occurs in older children with a median age of 9 years old. Although previously associated with poor outcomes, recognition of patients with iAMP21 ALL as high-risk/very high-risk with appropriate therapy intensification has improved their survival substantially without HSCT.61-63 Although extremely rare, the constitutional Robertsonian translocation rob(15;21)(q10;q10)c is associated with a dramatically increased risk of developing ALL with iAMP21.64
Trisomy 21-associated ALL
Children with trisomy 21 (Down Syndrome) have increased risk of developing B-ALL, although the role of germline trisomy 21 in leukemogenesis remains incompletely understood.65 Common childhood ALL-associated translocations occur less frequently in Down Syndrome-associated ALL (DS-ALL), and outcomes of children with DS-ALL are inferior to those without DS.66-68 However, 50-60% of children with DS-ALL have rearrangements in the CRLF2 (cytokine receptor-like factor 2) gene located in the pseudoautosomal region (PAR1) of chromosomes X and Y that most commonly result from interstitial PAR1 deletion causing P2RY8-CRLF2 fusion.69, 70 Concomitant JAK mutations (generally missense point mutations in JAK2) occur in half of CRLF2-rearranged DS-ALL cases, which demonstrate activated JAK/STAT and other cytokine receptor pathway signaling.71, 72 Investigation of small molecule JAK inhibitors is thus of potential therapeutic interest given the mutational profile and potential for toxicity in patients with DS-ALL, but has not yet been studied in the clinic.
Philadelphia Chromosome-like ALL
Philadelphia chromosome-like (Ph-like) or BCR-ABL1-like ALL is a recently recognized subset of B-ALL defined by a gene expression profile similar to that of BCR-ABL1+ ALL and suggestive of activated tyrosine kinase signaling.73 Deletions of IKZF1 and other genes encoding transcription factors that regulate B-cell differentiation commonly occur in Ph-like ALL.74-76 IKZF1-deleted ALL has been associated with a high rate of treatment failure and/or relapse in HR patients, suggesting that new treatment approaches may be necessary to improve outcomes in some patients.57-60, 74, 75, 77 Clinical outcomes of patients with Ph-like ALL are generally poor, although one small study reported better outcomes with MRD-based application of HSCT in first remission.78-81 The prevalence of Ph-like ALL increases with age from approximately 10% in NCI SR ALL to 13% of NCI HR ALL, 21% of adolescents, and 27% of young adults with ALL.82
The genomics of Ph-like ALL are complex, but frequently cause activated kinase and cytokine receptor signaling.73, 83 CRLF2 rearrangements resulting in overexpression are present in approximately 50% of Ph-like ALL cases in the United States (US) and include P2RY8-CRLF2 fusions and (often cryptic) IGH-CRLF2 translocations.69, 70 Interestingly, IGH-CRLF2 translocations are more common in older adolescents and young adults and linked to Hispanic ethnicity and Native American genetic ancestry in the US, which may partially explain the difference in frequency of this abnormality between US and European reports.84-86 Mutations of JAK1 and particularly JAK2 occur in half of CRLF2-rearranged ALL cases and are rarely detected in ALL cases without CRLF2 rearrangements.69, 70, 84
Among the remaining half of Ph-like ALL cases, about one-third harbor often cryptic genomic rearrangements involving the ABL1, ABL2, CSF1R, or PDGFRB kinase genes.82, 83 These rearrangements involve a diverse variety of translocation partners and encode fusion proteins (ABL-class fusions) that join the intact kinase domain of the carboxy terminal ABL-class kinase in frame to the amino terminus of the partner gene. The resultant fusion proteins phenocopy BCR-ABL1 in terms of their ability to confer growth factor independence in murine cell lines and, importantly, to respond to ABL-class TKIs imatinib and dasatinib.82, 83
A second major subgroup accounting for about 20% of CRLF2-intact Ph-like ALL is comprised of cases with genomic rearrangements that produce JAK2 fusion genes analogous to ABL-class fusions or rearrangements targeting the erythropoietin receptor (EPOR). These lesions are also transforming in preclinical model systems and are highly sensitive to JAK2 inhibitors, including ruxolitinib.81, 83, 87
The observed in vitro sensitivity of ABL-class fusions to imatinib and dasatinib and JAK2 and EPOR fusions to ruxolitinib strongly suggests that these TKIs might have clinical efficacy in genomically-defined subsets of Ph-like ALL. Numerous anecdotes have been reported of patients with ABL1-class fusions who had poor early responses to chemotherapy, then subsequently dramatic responses following addition of imatinib or dasatinib to chemotherapy.81, 88, 89 It is now clear that PDGFRB fusions are present in a significant percentage of patients who fail to enter remission after one month of induction chemotherapy.81 Because PDGFRB rearrangements can be detected readily via FISH with commercially available probes, such testing should be performed on any cases of ALL with overt induction failure (>25% marrow blasts).90
Based upon these and other very promising preclinical and early clinical data, gene expression and sequencing assays for rapid identification of patients with Ph-like ALL and characterization of the driver genomic lesions are in development.91 Such assays will facilitate incorporation of appropriate TKIs into treatment of subsets of patients with newly diagnosed Ph-like ALL.
Intragenic Deletion of ETS-Related Gene
Very recently, recurrent intragenic deletions of the transcription factor ERG (ETS-related gene) have been described in approximately 3% of children with B-ALL.73, 92, 93 ERG-deleted ALL occurs in older children (median age 7 years), is associated with aberrant CD2 surface expression, and frequent IKZF1 deletions. Despite the generally unfavorable prognosis of IKZF1-deleted ALL, patients with ERG-deleted ALL (even those with IKZF1 deletions) appear to have excellent outcomes with standard therapy with >85% 8-year EFS and >95% 8-year OS in one study.93
T-Cell Acute Lymphoblastic Leukemia (T-All)
T-ALL comprises 10-15% of pediatric ALL and is associated with older age at diagnosis and greater incidence in males and in African Americans.94 While outcomes of children with T-ALL were historically inferior to those with B-ALL, therapy intensification has recently resulted in excellent outcomes with >85% 5-year EFS, although T-cell immunophenotype remains an adverse risk factor in multivariate analyses of clinical trials.2, 19, 95 Age and WBC, which are very powerful factors in B-ALL, have little prognostic impact in T-ALL.96
Genomic alterations in T-ALL are often cytogenetically cryptic, and the prognostic impact of the various T-ALL-associated lesions identified to date remain poorly understood (Figure 1). Consequently, genomic alterations are not currently used for risk stratification of T-ALL patients, which instead relies primarily upon CNS status at diagnosis and MRD response. Treatment of patients with relapsed T-ALL with the nucleoside analogue nelarabine demonstrated impressive early activity, but was associated with significant neurotoxicity.97, 98 This toxicity delayed subsequent frontline testing efforts, but the COG has now shown that nelarabine can be safely added to treatment regimens for newly-diagnosed T-ALL patients.99 A recently completed phase 3 clinical trial will determine, once outcome data are mature, whether addition of nelarabine improves outcomes for children and AYAs with T-ALL.100 Clinical testing of γ-secretase inhibitors (GSIs) and other NOTCH1-targeted molecular therapeutic agents (discussed below) is also in progress.
Chromosomal Translocations
Approximately 50% of T-ALL cases harbor chromosomal translocations that most commonly involve fusion of T-cell receptor genes to various oncogenes (e.g., TLX1-TCRδ) or interstitial deletions resulting in juxtaposition of two genes (e.g., STIL-TAL1). Gene expression profiling studies have classified T-ALL into four molecular subtypes based upon underlying genetic alterations and resulting oncogenic signaling pathway activation: (1) TLX1/HOX11, (2) LYL1, (3) TAL1/LMO2, and (4) TLX3/HOX11L2.101-104 The prognostic significance of these alterations in childhood T-ALL remains largely unknown, although patients with TLX1 alterations appear to have more favorable outcomes (Table 1).103, 105
Mutations in X Chromosome Genes
The TLX1/HOX11, TAL1/LMO2, and TLX3/HOX11L2 subsets are associated with inactivating mutations and deletions in PHF6 (plant homeodomain finger 6) located on the X chromosome, which may partially help to explain the greater frequency of T-ALL in males.106 PHF6 is hypothesized to function as a tumor suppressor given its role as an RNA-interacting protein and component of the nucleosome remodeling and deacetylation (NuRD) complex.106 Somatic loss-of-function mutations in the X-linked histone H3K27me3 demethylase ubiquitously transcribed X (UTX) chromosome also occur frequently in T-ALL cases, are enriched in males, and appear sensitive in vitro to histone methyltransferase EZH2 inhibition.107
NOTCH1 Mutations
Somatic mutations in NOTCH1, which encodes a transmembrane receptor responsible for T-cell lineage commitment and survival, result in constitutive PI3K pathway signaling activation. NOTCH1 mutations occur in greater than 50% of T-ALL cases and are generally associated with more favorable therapeutic responses and outcomes.108, 109 Common cooperating lesions such as deletion of tumor suppressor CDKN2A or mutation in ubiquitin protein ligase FBXW7 are also hypothesized to enhance aberrant PI3K signaling via attenuation of NOTCH1 protein degradation. Various NOTCH1-directed strategies are under preclinical and clinical study, such as anti-NOTCH1 antibodies and small molecule GSIs to block NOTCH1 pathway signaling and proteolytic degradation of NOTCH1, respectively. To date, first-generation GSIs have not been efficacious in patients with T-ALL at least in part due to on target/off tumor gastrointestinal toxicity.110 Development of newer GSIs with more acceptable toxicity profiles is in progress.
Early Thymic Precursor ALL
A fifth subtype of T-ALL called early thymic precursor or early T-cell precursor (ETP) accounts for 10-15% of T-ALL and is diagnosed by its characteristic immature immunophenotype (CD1a-, CD8-, CD5- or CD5dim with co-expression of myeloid or stem cell markers).111 Patients with ETP ALL tend to respond more slowly to induction chemotherapy, although most patients appear to achieve molecular remission with negative MRD by the end of consolidation therapy.95 An initial study of patients with ETP ALL reported extremely poor outcomes with high rates of primary chemotherapy-refractory disease and/or relapse and 19% 10-year EFS.111 More recent data from the United Kingdom Medical Research Council and the COG suggest that outcomes of children with ETP ALL are comparable to those of patients with non-ETP ALL when stratified by MRD responses with overall 80-85% 5-year EFS.95, 112
Whole-genome sequencing of ETP ALL cases demonstrated high frequency of activating mutations in Ras pathway and cytokine receptor signaling (e.g., FLT3, IL7RA, JAK1 and JAK3, KRAS and NRAS, SH2B3) in comparison to non-ETP ALL cases.113 Inactivating mutations in hematopoiesis development and histone modification genes also were common in ETP ALL. These mutational profiles appear similar to those of myeloid leukemias, suggesting that ETP ALL may be part of a spectrum of leukemias derived from very early progenitor cells and/or hematopoietic stem cells. Recent preclinical studies also suggest potential therapeutic efficacy of molecularly-targeted agents such as JAK inhibitors to target aberrant cytokine receptor signaling.114-116
Other recurrent alterations in T-ALL include KMT2A rearrangements (10-15% of T-ALL), t(9;14)(q34;q32) resulting in NUP214-ABL1 fusion (10%), and t(8;14)(q24;q11) resulting in TRA-MYC or TRD-MYC fusion (1%).103 The PICALM-MLLT10 rearrangement in particular has been associated with very poor outcomes, although more recent studies suggest intermediate outcomes with intensive therapy.103, 117
Relapsed All
Prognostic factors are imperfect, and relapse occurs across the genomic spectrum of childhood ALL. Some subtypes are associated with particularly high risk of relapse, such as Ph+ ALL in the pre-TKI era, hypodiploid ALL, and the newly-recognized Ph-like ALL subset. Relapsed ALL is often associated with greater resistance to chemotherapy, which may result from emergence of a pre-existent resistant subclone and/or from mutation acquisition during chemotherapy exposure that promote drug resistance (Figure 2).118, 119 Genomic profiling of matched diagnosis, remission, and relapsed ALL specimens has helped to elucidate various features of leukemia clonal evolution, mechanisms of chemoresistance, and emergence of new mutations.119-122 The majority of relapsed ALL cases maintain key genetic features from diagnosis, particularly sentinel chromosome translocations which are almost always retained and are likely very early or even initiating events in leukemogenesis. Almost all relapsed ALL cases also exhibit new genetic alterations, suggesting dynamic evolution of leukemogenesis.120, 121 Interestingly, the bulk leukemia clone present at diagnosis is typically eradicated, while rare subclones persist and acquire new mutations to become the predominant clone at relapse.121 Genome-wide association studies have also recently identified specific germline single nucleotide polymorphisms that occur more frequently in patients with relapsed ALL.123
Figure 2. Impact of genetic features of ALL upon therapy response.
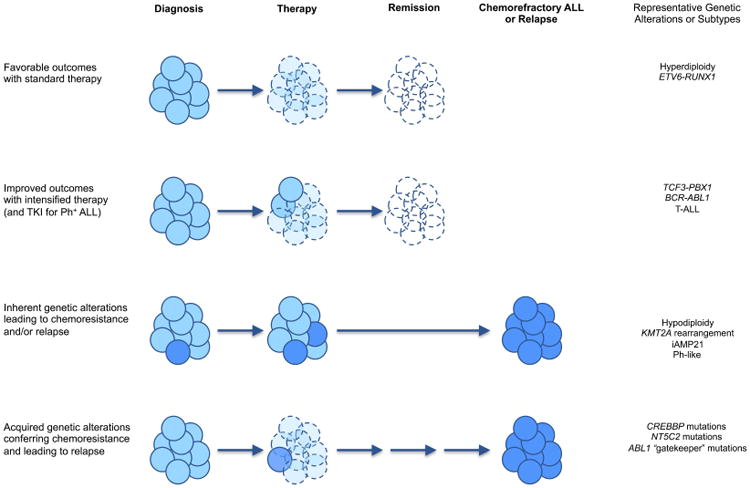
Chemosensitive ALL cells (pale blue) respond to appropriately intensive chemotherapy based upon underlying leukemic genetics and risk stratification, often allowing patients to achieve sustained remission. Chemoresistant ALL cells (dark blue) may result from inherently drug-resistant subclones present at diagnosis or from selective pressure during chemotherapy that leads to development of acquired mutations and facilitates relapse. Very rarely, secondary malignancies may arise from genetically distinct subclones than those present at diagnosis.
CREBBP (CREB-binding protein) mutations have been reported in approximately 20% of patients with relapsed ALL.124, 125 The CREBBP (also known as CBP) protein is involved in glucocorticoid-mediated transcription and histone deacetylation; preclinical studies demonstrate activity of histone deacetylase inhibition and other small molecule inhibitor treatment of steroid-resistant ALL, which may merit further study in the clinic.124, 126 Co-occurrence of CREBBP and KRAS mutations was also recently reported in a large proportion of early relapse ALL cases, providing additional rationale for exploration of small molecule inhibitor therapies (e.g., MEK inhibitors, PI3K pathway inhibitors) in this subset.125 More recently, gain-of-function NT5C2 mutations were identified in nearly 20% of relapsed B- and T-ALL cases.127, 128 NT5C2 is a 5′-nucleotidase catalytic enzyme that metabolizes and inactivates nucleoside analogues, such as mercaptopurine and thioguanine, which are critical components of anti-ALL therapy. Acquisition of NT5C2 mutations thus likely confers resistance to anti-metabolite drugs, and NT5C2 mutations occur predominantly in patients that relapse during therapy, particularly during maintenance chemotherapy.126, 127 Other recurrent somatic mutations identified in relapsed ALL include deletions in the DNA mismatch repair gene MSH6 and the glucocorticoid receptor NR3C1 and mutations in the H3K36 trimethyltransferase SETD2, the lysine-specific demethylase KDM6, and the epigenetic regulator MLL2. 121, 129-131 Activating somatic mutations in Ras pathway-associated genes (e.g., KRAS, NRAS, FLT3, and PTPN11) have also been identified in one-third to one-half of patients with relapsed ALL, and inhibition of leukemia proliferation with MEK inhibitor treatment of Ras pathway-mutant human ALL xenograft models was recently reported.130, 132
Summary and Clinical Implications
Despite similar morphological features, ALL is a genetically heterogeneous group of diseases. To date, therapeutic success for children and AYAs with ALL has largely been driven by implementation of biology- and response-based risk stratification with appropriate chemotherapy intensification for high-risk patients. Depth and timing of leukemia remission after initial chemotherapy, as measured by MRD testing, remain critical determinants of long-term outcomes.133-135 Advances in next-generation sequencing technologies have facilitated the identification of additional high-risk subtypes of de novo ALL, as well as alterations enriched at relapse. Efforts are ongoing to integrate genomic features with biochemically- and epigenetically-informed molecular treatment approaches. TKI addition to cytotoxic chemotherapy for children with Ph+ ALL has dramatically improved EFS and OS, and implementation of similar strategies for patients with other high-risk ALL genetic subgroups may also improve outcomes. However, testing of precision medicine therapies is currently in its early stages for most potentially “targetable” subtypes of ALL, and the long-term success of such approaches remains unknown. In addition, presence of inherited leukemia predisposition gene variants and detection of chemoresistance-associated mutations in sizable proportions of patients who relapse demonstrates the dynamic biology of ALL that remains incompletely understood.
In vitro and in vivo evaluation of new agents in ALL model systems with swift bench-to-bedside translation of preclinically promising therapies remains a lengthy and often imperfect system. Single-agent biologic activity in murine models does not always translate into clinical activity in patients, and improper drug sequencing can result in untoward clinical outcomes.136 Access to biologically relevant new agents for children with ALL and other cancers often appreciably lags behind that of adult hematology and oncology. Initial evaluation of new anti-cancer drugs in children generally occurs in the multiply relapsed/chemorefractory setting where lack of response may not necessarily mirror activity in newly-diagnosed patients or predict activity when combined with chemotherapy. Distinct drug dose intensity and tolerability factors for children also exist that must be carefully considered, particularly as new drugs are incorporated into multi-agent regimens.137 Drug development may be halted when deleterious side effects occur in patients that were not previously observed or predicted by preclinical modeling, as well as for economic reasons despite evidence of anti-cancer activity in patients.137 It is becoming increasingly apparent that combination of two or more targeted agents (e.g., growth factor receptor inhibitors, TKIs) for some cancers will be necessary to evoke robust anti-tumor efficacy while minimizing development of resistance mutations or upregulation of “escape” pathways. Improved collaboration among cancer researchers, clinical oncologists, government agencies, and pharmaceutical industry partners is thus likely necessary to accelerate the pipeline of preclinical drug discovery and early clinical testing of promising new drugs. Clinical trial design for evaluation of new agents in increasingly smaller “boutique” subsets of genomically-defined patients also presents new challenges, and development of robust biomarkers of response (pharmacokinetic, pharmacodynamic, others) will be necessary to define clinical activity and to prioritize further drug development for children with ALL.
Treatment of children and AYAs with years of multi-agent chemotherapy is well-associated with untoward short- and long-term sequelae, and it is likely that a proportion of children with favorable-risk ALL (e.g., rapid early responders with ETV6-RUNX1 translocation or with hyperdiploidy) can achieve similarly outstanding cure rates with less intensive therapy. Therapy reduction for strictly defined low-risk subsets of ALL has been investigated or is under current evaluation by several cooperative groups, and several studies have demonstrated excellent feasibility of this approach.18, 19, 116, 138 The major challenges for the next decade are to improve cure rates through improved risk stratification and more widespread application of molecularly-targeted therapies, including TKIs and antibody- and T-cell-based immunotherapies, as well as to minimize late effects by identifying the least toxic regimens that can cure low-risk patients.
Acknowledgments
This work was supported by the National Cancer Institute (K08CA184418 to SKT), the Alex's Lemonade Stand Foundation (SKT and MLL), the Rally Foundation for Childhood Cancer Research (SKT), and the St. Baldrick's Foundation (MLL and SPH). SKT was an Alex's Lemonade Stand Foundation Scholar in Developmental Therapeutics. MLL is the Deborah and Arthur Ablin Chair in Molecular Pediatric Oncology. We apologize in advance that space limitations made it impossible to reference many important publications.
Footnotes
Financial Disclosures and Conflicts of Interest: The authors have no relevant financial disclosures or conflicts of interest.
Authorship Contributions: SKT, MLL, and SPH wrote, edited, and approved the final manuscript.
References
- 1.Pui CH. Recent advances in acute lymphoblastic leukemia. Oncology. 2011;25:341, 346–347. [PubMed] [Google Scholar]
- 2.Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pui CH, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120:1165–1174. doi: 10.1182/blood-2012-05-378943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1996;14:18–24. doi: 10.1200/JCO.1996.14.1.18. [DOI] [PubMed] [Google Scholar]
- 5.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111:5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunger SP, Loh ML, Whitlock JA, et al. Children's Oncology Group's 2013 blueprint for research: acute lymphoblastic leukemia. Pediatric blood & cancer. 2013;60:957–963. doi: 10.1002/pbc.24420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pui CH, Pei D, Coustan-Smith E, et al. Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: a prospective study. Lancet Oncol. 2015;16:465–474. doi: 10.1016/S1470-2045(15)70082-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children's Oncology Group study. Leukemia. 2008;22:2142–2150. doi: 10.1038/leu.2008.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 10.Perez-Andreu V, Xu H, Yang JJ. The novel susceptibility variants for childhood acute lymphoblastic leukemia. J Natl Cancer Inst. 2013;105:1512–1513. doi: 10.1093/jnci/djt230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, Yang W, Perez-Andreu V, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105:733–742. doi: 10.1093/jnci/djt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah S, Schrader KA, Waanders E, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45:1226–1231. doi: 10.1038/ng.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez-Andreu V, Roberts KG, Xu H, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood. 2015;125:680–686. doi: 10.1182/blood-2014-09-595744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47:180–185. doi: 10.1038/ng.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015 doi: 10.1038/ng.3253. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison CJ, Haas O, Harbott J, et al. Detection of prognostically relevant genetic abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: recommendations from the Biology and Diagnosis Committee of the International Berlin-Frankfurt-Munster study group. Br J Haematol. 2010;151:132–142. doi: 10.1111/j.1365-2141.2010.08314.x. [DOI] [PubMed] [Google Scholar]
- 17.Moorman AV, Chilton L, Wilkinson J, Ensor HM, Bown N, Proctor SJ. A population-based cytogenetic study of adults with acute lymphoblastic leukemia. Blood. 2010;115:206–214. doi: 10.1182/blood-2009-07-232124. [DOI] [PubMed] [Google Scholar]
- 18.Moricke A, Reiter A, Zimmermann M, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood. 2008;111:4477–4489. doi: 10.1182/blood-2007-09-112920. [DOI] [PubMed] [Google Scholar]
- 19.Vora A, Goulden N, Wade R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14:199–209. doi: 10.1016/S1470-2045(12)70600-9. [DOI] [PubMed] [Google Scholar]
- 20.Pui CH, Williams DL, Raimondi SC, et al. Hypodiploidy is associated with a poor prognosis in childhood acute lymphoblastic leukemia. Blood. 1987;70:247–253. [PubMed] [Google Scholar]
- 21.Heerema NA, Nachman JB, Sather HN, et al. Hypodiploidy with less than 45 chromosomes confers adverse risk in childhood acute lymphoblastic leukemia: a report from the children's cancer group. Blood. 1999;94:4036–4045. [PubMed] [Google Scholar]
- 22.Devidas M, Raetz EA, Loh ML, et al. Outcome For Children With Hypodiploid Acute Lymphoblastic Leukemia (ALL) on Contemporary Children's Oncology Group (COG) Clinical Trials. Pediatric blood & cancer. 2013;60 abstract #O0036. [Google Scholar]
- 23.Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112–1115. doi: 10.1182/blood-2006-07-038299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrison CJ, Moorman AV, Broadfield ZJ, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125:552–559. doi: 10.1111/j.1365-2141.2004.04948.x. [DOI] [PubMed] [Google Scholar]
- 25.Soverini S, De Benedittis C, Papayannidis C, et al. Drug resistance and BCR-ABL kinase domain mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia from the imatinib to the second-generation tyrosine kinase inhibitor era: The main changes are in the type of mutations, but not in the frequency of mutation involvement. Cancer. 2014;120:1002–1009. doi: 10.1002/cncr.28522. [DOI] [PubMed] [Google Scholar]
- 26.Mehta PA, Eapen M, Zhang MJ, et al. Transplant Outcomes for Children with Hypodiploid Acute Lymphoblastic Leukemia: the Cibmtr Experience. Biol Blood Marrow Transplant. 2014;20:S87. doi: 10.1016/j.bbmt.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker H, An Q, Barber K, et al. The complex genomic profile of ETV6-RUNX1 positive acute lymphoblastic leukemia highlights a recurrent deletion of TBL1XR1. Genes Chromosomes Cancer. 2008;47:1118–1125. doi: 10.1002/gcc.20613. [DOI] [PubMed] [Google Scholar]
- 29.Mori H, Colman SM, Xiao Z, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002;99:8242–8247. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greaves M. Darwin and evolutionary tales in leukemia. The Ham-Wasserman Lecture. Hematology Am Soc Hematol Educ Program. 2009:3–12. doi: 10.1182/asheducation-2009.1.3. [DOI] [PubMed] [Google Scholar]
- 31.Raimondi SC, Behm FG, Roberson PK, et al. Cytogenetics of pre-B-cell acute lymphoblastic leukemia with emphasis on prognostic implications of the t(1;19) J Clin Oncol. 1990;8:1380–1388. doi: 10.1200/JCO.1990.8.8.1380. [DOI] [PubMed] [Google Scholar]
- 32.Pui CH, Raimondi SC, Hancock ML, et al. Immunologic, cytogenetic, and clinical characterization of childhood acute lymphoblastic leukemia with the t(1;19) (q23; p13) or its derivative. J Clin Oncol. 1994;12:2601–2606. doi: 10.1200/JCO.1994.12.12.2601. [DOI] [PubMed] [Google Scholar]
- 33.Felice MS, Gallego MS, Alonso CN, et al. Prognostic impact of t(1;19)/TCF3-PBX1 in childhood acute lymphoblastic leukemia in the context of Berlin-Frankfurt-Munster-based protocols. Leuk Lymphoma. 2011;52:1215–1221. doi: 10.3109/10428194.2011.565436. [DOI] [PubMed] [Google Scholar]
- 34.Jeha S, Pei D, Raimondi SC, et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia. 2009;23:1406–1409. doi: 10.1038/leu.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunger SP. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood. 1996;87:1211–1224. [PubMed] [Google Scholar]
- 36.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122:3407–3415. doi: 10.1172/JCI61203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collins EC, Rabbitts TH. The promiscuous MLL gene links chromosomal translocations to cellular differentiation and tumour tropism. Trends Mol Med. 2002;8:436–442. doi: 10.1016/s1471-4914(02)02397-3. [DOI] [PubMed] [Google Scholar]
- 38.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ford AM, Ridge SA, Cabrera ME, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363:358–360. doi: 10.1038/363358a0. [DOI] [PubMed] [Google Scholar]
- 40.Gale KB, Ford AM, Repp R, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A. 1997;94:13950–13954. doi: 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pieters R, Schrappe M, De Lorenzo P, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370:240–250. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 42.Dreyer ZE, Hilden JM, Jones TL, et al. Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3) Pediatric blood & cancer. 2015;62:419–426. doi: 10.1002/pbc.25322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pui CH, Chessells JM, Camitta B, et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia. 2003;17:700–706. doi: 10.1038/sj.leu.2402883. [DOI] [PubMed] [Google Scholar]
- 44.Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood. 2005;105:812–820. doi: 10.1182/blood-2004-06-2498. [DOI] [PubMed] [Google Scholar]
- 45.Stam RW, den Boer ML, Schneider P, et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood. 2005;106:2484–2490. doi: 10.1182/blood-2004-09-3667. [DOI] [PubMed] [Google Scholar]
- 46.Stam RW, Schneider P, de Lorenzo P, Valsecchi MG, den Boer ML, Pieters R. Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood. 2007;110:2774–2775. doi: 10.1182/blood-2007-05-091934. [DOI] [PubMed] [Google Scholar]
- 47.Annesley CE, Brown P. The Biology and Targeting of FLT3 in Pediatric Leukemia. Front Oncol. 2014;4:263. doi: 10.3389/fonc.2014.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernt KM, Armstrong SA. Targeting epigenetic programs in MLL-rearranged leukemias. Hematology Am Soc Hematol Educ Program. 2011;2011:354–360. doi: 10.1182/asheducation-2011.1.354. [DOI] [PubMed] [Google Scholar]
- 49.Dobbins SE, Sherborne AL, Ma YP, et al. The silent mutational landscape of infant MLL-AF4 pro-B acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2013;52:954–960. doi: 10.1002/gcc.22090. [DOI] [PubMed] [Google Scholar]
- 50.Andersson AK, Ma J, Wang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015 doi: 10.1038/ng.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arico M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med. 2000;342:998–1006. doi: 10.1056/NEJM200004063421402. [DOI] [PubMed] [Google Scholar]
- 52.Arico M, Schrappe M, Hunger SP, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:4755–4761. doi: 10.1200/JCO.2010.30.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biondi A, Schrappe M, De Lorenzo P, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol. 2012;13:936–945. doi: 10.1016/S1470-2045(12)70377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:5175–5181. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia. 2014;28:1467–1471. doi: 10.1038/leu.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang BH, Willis SG, Stork L, et al. Imatinib resistant BCR-ABL1 mutations at relapse in children with Ph+ ALL: a Children's Oncology Group (COG) study. Br J Haematol. 2012;157:507–510. doi: 10.1111/j.1365-2141.2012.09039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. 2014;123:1691–1698. doi: 10.1182/blood-2013-06-509794. [DOI] [PubMed] [Google Scholar]
- 58.Olsson L, Johansson B. Ikaros and leukaemia. Br J Haematol. 2015 doi: 10.1111/bjh.13342. [DOI] [PubMed] [Google Scholar]
- 59.Dorge P, Meissner B, Zimmermann M, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. 2013;98:428–432. doi: 10.3324/haematol.2011.056135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:5202–5207. doi: 10.1200/JCO.2008.21.6408. [DOI] [PubMed] [Google Scholar]
- 61.Moorman AV, Robinson H, Schwab C, et al. Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:3389–3396. doi: 10.1200/JCO.2013.48.9377. [DOI] [PubMed] [Google Scholar]
- 62.Heerema NA, Carroll AJ, Devidas M, et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:3397–3402. doi: 10.1200/JCO.2013.49.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harrison CJ, Moorman AV, Schwab C, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia. 2014;28:1015–1021. doi: 10.1038/leu.2013.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Schwab C, Ryan SL, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508:98–102. doi: 10.1038/nature13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Izraeli S, Vora A, Zwaan CM, Whitlock J. How I treat ALL in Down's syndrome: pathobiology and management. Blood. 2014;123:35–40. doi: 10.1182/blood-2013-07-453480. [DOI] [PubMed] [Google Scholar]
- 66.Maloney KW, Carroll WL, Carroll AJ, et al. Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood. 2010;116:1045–1050. doi: 10.1182/blood-2009-07-235291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lundin C, Forestier E, Klarskov Andersen M, et al. Clinical and genetic features of pediatric acute lymphoblastic leukemia in Down syndrome in the Nordic countries. J Hematol Oncol. 2014;7:32. doi: 10.1186/1756-8722-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buitenkamp TD, Izraeli S, Zimmermann M, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014;123:70–77. doi: 10.1182/blood-2013-06-509463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–2698. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- 70.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet. 2008;372:1484–1492. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 72.Kearney L, Gonzalez De Castro D, Yeung J, et al. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113:646–648. doi: 10.1182/blood-2008-08-170928. [DOI] [PubMed] [Google Scholar]
- 73.Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood. 2011;118:3080–3087. doi: 10.1182/blood-2011-03-341412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–4884. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 78.Loh ML, Zhang J, Harvey RC, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood. 2013;121:485–488. doi: 10.1182/blood-2012-04-422691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van der Veer A, Waanders E, Pieters R, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122:2622–2629. doi: 10.1182/blood-2012-10-462358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boer JM, Koenders JE, van der Holt B, et al. Expression profiling of adult acute lymphoblastic leukemia identifies a BCR-ABL1-like subgroup characterized by high non-response and relapse rates. Haematologica. 2015 doi: 10.3324/haematol.2014.117424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roberts KG, Pei D, Campana D, et al. Outcomes of children with BCR-ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:3012–3020. doi: 10.1200/JCO.2014.55.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–5321. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Attarbaschi A, Morak M, Cario G, et al. Treatment outcome of CRLF2-rearranged childhood acute lymphoblastic leukaemia: a comparative analysis of the AIEOP-BFM and UK NCRI-CCLG study groups. Br J Haematol. 2012;158:772–777. doi: 10.1111/j.1365-2141.2012.09221.x. [DOI] [PubMed] [Google Scholar]
- 86.Ensor HM, Schwab C, Russell LJ, et al. Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood. 2011;117:2129–2136. doi: 10.1182/blood-2010-07-297135. [DOI] [PubMed] [Google Scholar]
- 87.Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120:3510–3518. doi: 10.1182/blood-2012-03-415448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weston BW, Hayden MA, Roberts KG, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:e413–416. doi: 10.1200/JCO.2012.47.6770. [DOI] [PubMed] [Google Scholar]
- 89.Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. 2013;98:e146–148. doi: 10.3324/haematol.2013.095372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schwab C, Andrews R, Chilton L, et al. EBF1-PDGFR Fusion in Paediatric Acute Lymphoblastic Leukaemia (ALL): Genetic Profile and Clinical Implications. Blood. 2014;124 doi: 10.1182/blood-2015-09-670166. ASH Annual Meeting abstract #1068. [DOI] [PubMed] [Google Scholar]
- 91.Harvey RC, Kang H, Roberts KG, et al. Development and Validation of a Highly Sensitive and Specific Gene Expression Classifier to Prospectively Screen and Identify B-Precursor Acute Lymphoblastic Leukemia (ALL) Patients with a Phialdelphia Chromosome-Like Signature (“Ph-like” or “BCR-ABL1-Like”) for Therapeutic Targeting and Clinical Intervention. Blood. 2013;123 ASH annual meeting abstract #826. [Google Scholar]
- 92.Zaliova M, Zimmermannova O, Dorge P, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. 2014;28:182–185. doi: 10.1038/leu.2013.282. [DOI] [PubMed] [Google Scholar]
- 93.Clappier E, Auclerc MF, Rapion J, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28:70–77. doi: 10.1038/leu.2013.277. [DOI] [PubMed] [Google Scholar]
- 94.Pui C, Robison L, Look A. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 95.Wood BL, Winter SS, Dunsmore KP, et al. T-Lymphoblastic Leukemia (T-ALL) Shows Excellent Outcome, Lack of Significance of the Early Thymic Precursor (ETP) Immunophenotype, and Validation of the Prognostic Value of End-Induction Minimal Residual Disease (MRD) in Children's Oncology Group (COG) Study AALL0434. Blood. 2014;123 ASH Annual Meeting abstract #1. [Google Scholar]
- 96.Hastings C, Gaynon PS, Nachman JB, et al. Increased post-induction intensification improves outcome in children and adolescents with a markedly elevated white blood cell count (>/=200 × 10(9)/l) with T cell acute lymphoblastic leukaemia but not B cell disease: a report from the Children's Oncology Group. Br J Haematol. 2015;168:533–546. doi: 10.1111/bjh.13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berg SL, Blaney SM, Devidas M, et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:3376–3382. doi: 10.1200/JCO.2005.03.426. [DOI] [PubMed] [Google Scholar]
- 98.Gokbuget N, Basara N, Baurmann H, et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood. 2011;118:3504–3511. doi: 10.1182/blood-2011-01-329441. [DOI] [PubMed] [Google Scholar]
- 99.Dunsmore KP, Devidas M, Linda SB, et al. Pilot study of nelarabine in combination with intensive chemotherapy in high-risk T-cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2753–2759. doi: 10.1200/JCO.2011.40.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Winter SS, Dunsmore KP, Devidas M, et al. Safe integration of nelarabine into intensive chemotherapy in newly diagnosed T-cell acute lymphoblastic leukemia: Children's Oncology Group Study AALL0434. Pediatric blood & cancer. 2015 doi: 10.1002/pbc.25470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 102.Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. 2010;23:307–318. doi: 10.1016/j.beha.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 103.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Patrick K, Vora A. Update on biology and treatment of T-cell acute lymphoblastic leukaemia. Curr Opin Pediatr. 2015;27:44–49. doi: 10.1097/MOP.0000000000000171. [DOI] [PubMed] [Google Scholar]
- 105.De Keersmaecker K, Ferrando AA. TLX1-induced T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2011;17:6381–6386. doi: 10.1158/1078-0432.CCR-10-3037. [DOI] [PubMed] [Google Scholar]
- 106.Van Vlierberghe P, Palomero T, Khiabanian H, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42:338–342. doi: 10.1038/ng.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Van der Meulen J, Sanghvi V, Mavrakis K, et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood. 2015;125:13–21. doi: 10.1182/blood-2014-05-577270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 109.Ferrando AA. The role of NOTCH1 signaling in T-ALL. Hematology Am Soc Hematol Educ Program. 2009:353–361. doi: 10.1182/asheducation-2009.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Paganin M, Ferrando A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev. 2011;25:83–90. doi: 10.1016/j.blre.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Patrick K, Wade R, Goulden N, et al. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol. 2014;166:421–424. doi: 10.1111/bjh.12882. [DOI] [PubMed] [Google Scholar]
- 113.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Treanor LM, Zhou S, Janke L, et al. Interleukin-7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J Exp Med. 2014;211:701–713. doi: 10.1084/jem.20122727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125:1759–1767. doi: 10.1182/blood-2014-06-580480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43:932–939. doi: 10.1038/ng.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lo Nigro L, Mirabile E, Tumino M, et al. Detection of PICALM-MLLT10 (CALM-AF10) and outcome in children with T-lineage acute lymphoblastic leukemia. Leukemia. 2013;27:2419–2421. doi: 10.1038/leu.2013.149. [DOI] [PubMed] [Google Scholar]
- 118.Locatelli F, Schrappe M, Bernardo ME, Rutella S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood. 2012;120:2807–2816. doi: 10.1182/blood-2012-02-265884. [DOI] [PubMed] [Google Scholar]
- 119.Teachey DT, Hunger SP. Predicting relapse risk in childhood acute lymphoblastic leukaemia. Br J Haematol. 2013;162:606–620. doi: 10.1111/bjh.12442. [DOI] [PubMed] [Google Scholar]
- 120.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6 doi: 10.1038/ncomms7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bhojwani D, Kang H, Moskowitz NP, et al. Biologic pathways associated with relapse in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. Blood. 2006;108:711–717. doi: 10.1182/blood-2006-02-002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang JJ, Cheng C, Yang W, et al. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA. 2009;301:393–403. doi: 10.1001/jama.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Malinowska-Ozdowy K, Frech C, Schonegger A, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015 doi: 10.1038/leu.2015.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gang EJ, Hsieh YT, Pham J, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014;33:2169–2178. doi: 10.1038/onc.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–371. doi: 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Meyer JA, Wang J, Hogan LE, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45:290–294. doi: 10.1038/ng.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang JJ, Bhojwani D, Yang W, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112:4178–4183. doi: 10.1182/blood-2008-06-165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mar BG, Bullinger LB, McLean KM, et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014;5:3469. doi: 10.1038/ncomms4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bhatla T, Jones CL, Meyer JA, Vitanza NA, Raetz EA, Carroll WL. The biology of relapsed acute lymphoblastic leukemia: opportunities for therapeutic interventions. J Pediatr Hematol Oncol. 2014;36:413–418. doi: 10.1097/MPH.0000000000000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Irving J, Matheson E, Minto L, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124:3420–3430. doi: 10.1182/blood-2014-04-531871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Conter V, Valsecchi MG, Parasole R, et al. Childhood high-risk acute lymphoblastic leukemia in first remission: results after chemotherapy or transplant from the AIEOP ALL 2000 study. Blood. 2014;123:1470–1478. doi: 10.1182/blood-2013-10-532598. [DOI] [PubMed] [Google Scholar]
- 134.Paganin M, Fabbri G, Conter V, et al. Postinduction minimal residual disease monitoring by polymerase chain reaction in children with acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:3553–3558. doi: 10.1200/JCO.2014.56.0698. [DOI] [PubMed] [Google Scholar]
- 135.van Dongen JJ, Seriu T, Panzer-Grumayer ER, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352:1731–1738. doi: 10.1016/S0140-6736(98)04058-6. [DOI] [PubMed] [Google Scholar]
- 136.Peterson JK, Houghton PJ. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur J Cancer. 2004;40:837–844. doi: 10.1016/j.ejca.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 137.Norris RE, Adamson PC. Challenges and opportunities in childhood cancer drug development. Nat Rev Cancer. 2012;12:776–782. doi: 10.1038/nrc3370. [DOI] [PubMed] [Google Scholar]
- 138.Arico M, Conter V, Valsecchi MG, et al. Treatment reduction in highly selected standard-risk childhood acute lymphoblastic leukemia. The AIEOP ALL-9501 study. Haematologica. 2005;90:1186–1191. [PubMed] [Google Scholar]