

Abstract
The MYC oncogenes encode a family of transcription factors that feature prominently in cancer. MYC proteins are overexpressed or deregulated in a majority of malignancies, and drive tumorigenesis by inducing widespread transcriptional reprogramming that promotes cell proliferation, metabolism, and genomic instability. The ability of MYC to regulate transcription depends on its dimerization with MAX, which creates a DNA-binding domain that recognizes specific sequences in the regulatory elements of MYC target genes. Recently, we discovered that recognition of target genes by MYC also depends on its interaction with WDR5, a WD40-repeat protein that exists as part of several chromatin-regulatory complexes. Here, we discuss how interaction of MYC with WDR5 could create an avidity-based chromatin recognition mechanism that allows MYC to select its target genes in response to both genetic and epigenetic determinants. We rationalize how the MYC–WDR5 interaction provides plasticity in target gene selection by MYC, and speculate on the biochemical and genomic contexts in which this interaction occurs. Finally, we discuss how properties of the MYC–WDR5 interface make it an attractive point for discovery of small molecule inhibitors of MYC function in cancer cells.
Introduction
MYC oncogenes encode a family of related transcription factors that are overexpressed in a majority of malignancies (1). The ectopic appearance of MYC in a cell induces widespread transcriptional changes that drive cell cycle progression, enhance protein synthesis, reprogram cellular metabolism, and destabilize the genome (2). This near-perfect suite of pro-tumorigenic functions, together with their pervasive deregulation in cancer, has fueled the concept that blocking MYC function in cancer cells could have significant therapeutic impact. Indeed, in numerous mouse models, genetic inhibition of MYC promotes tumor regression (2), securing a place for MYC proteins as bonafide targets of anti-cancer therapies.
As transcription factors, the ability of MYC proteins to recognize regulatory elements in the promoters and enhancers of target genes is crucial for their function. Incapable of binding DNA alone, MYC heterodimerizes with MAX (3) to form a DNA-binding module that recognizes the “E-box” motif (CACGTG) common in MYC-responsive genes. Although interaction with MAX is required for MYC to bind DNA, precisely where MYC engages the genome is profoundly influenced by chromatin context. Indeed, MYC/MAX dimers associate exclusively with E-boxes found in regions of active chromatin, marked by specific sets of histone modifications—the most notable of which are H3 lysine 4 (H3K4) di- and tri-methylation (4). The molecular mechanisms through which chromatin context shapes target gene selection by MYC are largely unknown, but our recent work suggests that one way this occurs is via interaction of MYC with the prevalent chromatin-associated protein WDR5 (5). Here, we discuss how WDR5 influences target gene selection by MYC and speculate on the implications of our findings.
WDR5 is a co-factor for MYC
The major findings of our work (5) can be summarized as follows. MYC binds directly to WDR5, a highly-conserved protein found in multiple chromatin regulatory complexes (6), including the MLL histone methyltransferases that catalyze H3K4 methylation. MYC and WDR5 co-localize extensively on chromatin, with ∼80% of the genomic sites occupied by MYC also bound by WDR5. MYC binds WDR5 via a short sequence motif—EEIDVV—present in all MYC family members from all species. Structure-guided mutations in MYC that disrupt interaction with WDR5 do not impact the latter's recruitment to chromatin, nor do they disrupt the ability of MYC to bind E-boxes in naked DNA. These mutations do, however, prevent MYC from binding to ∼80% of its chromosomal locations and attenuate its tumorigenic potential in mice. Our findings demonstrate that the MYC–WDR5 interaction plays an important role in directing association of MYC with chromatin, and reveal that WDR5 is a critical co-factor for MYC-driven tumorigenesis.
We propose that stable association of MYC with target gene chromatin is governed by two sets of interactions: one between MYC/MAX dimers and DNA, and another between MYC and chromatin-bound WDR5. We refer to this mechanism of target gene recognition by MYC as “facilitated recruitment” (Fig. 1). Although key aspects of the facilitated recruitment model have yet to be challenged, this revised view of chromatin recognition by MYC proteins reconciles much of their behavior and raises a number of intriguing questions we discuss below.
Figure 1. Facilitated recruitment of MYC to chromatin by interaction with WDR5.
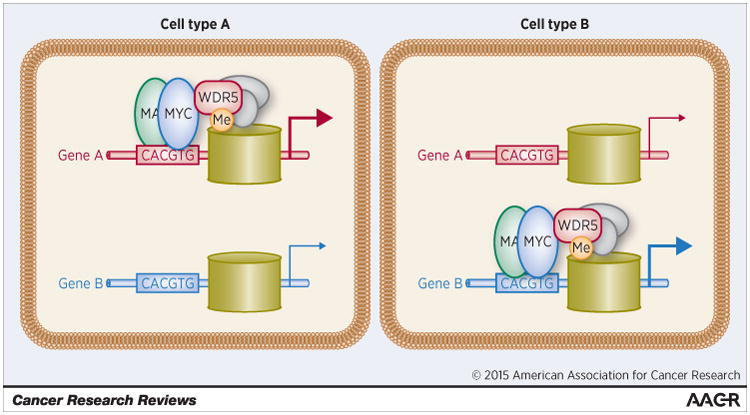
The cartoon represents two different genes in two different cell types. Both genes carry E-box consensus sites for binding MYC/MAX dimers. In Cell Type A, Gene A has recently been transcribed, and is marked by the presence of both H3K4 methylation (“Me”) and WDR5, which exists as part of a multi-protein complex (gray ovals). Because stable chromatin recognition by MYC depends on both DNA and WDR5 interactions, MYC associates with Gene A, and not Gene B, in this cell. In Cell Type B, the situation is reversed and only Gene B can recruit MYC.
Transcriptional plasticity and amplification
One notable feature of MYC is the breadth of transcriptional changes it induces. In any given cell type, it is not unusual to see changes in the expression of thousands of genes in response to MYC (2). What is equally notable, however, is that within this broad transcriptional response there is very little overlap between cell types, with only about 70 genes regulated by MYC in all contexts (7). The diversity of these transcriptional responses reveals that target gene selection by MYC is inherently plastic, and implies that additional mechanisms must exist to program which DNA elements in the genome capture MYC/MAX complexes.
The facilitated recruitment model can help explain the plasticity of target gene selection by MYC in several ways. As an avidity-based mechanism, facilitated recruitment integrates two distinct types of information—DNA sequence and WDR5 location—to determine where MYC will bind across the genome. In this view, if WDR5 is distributed differently in different cell types, different groups of E-boxes will be permissive for MYC binding (Fig. 1). Moreover, facilitated recruitment links to plasticity because WDR5 connects to both MYC and to H3K4 methylation. As mentioned, H3K4 di- and tri-methylation correlates tightly with MYC binding across the genome; so tightly that it has been called a “strict prerequisite” for target gene recognition by MYC (4). Importantly, these methylation events mark transcriptionally-active chromatin (8,9), and are plastic because their distribution is determined by which genes are active in a given cell type. Because WDR5 is both a component of MLL complexes, and capable of interacting specifically with H3K4 methylated histone tails (10), its distribution in cells closely mirrors H3K4 methylation patterns (11). It may be, therefore, that the correlation between MYC and H3K4 methylation has nothing to do with histone methylation per se, but with the distribution of WDR5 at these sites, which makes them available to bind MYC. By this logic, WDR5 may also play a key role in the recently-proposed ability of MYC to drive tumorigenesis by “amplifying” transcriptional programs already in place in a given cell type (12,13), which is based in large part on the concept that MYC can recognize which genes in the cell are marked as active.
Biochemical context of the MYC–WDR5 interaction
WDR5 is a versatile protein. In addition to its role in MLL complexes, WDR5 is also present in several other chromatin-modifying complexes, including the NSL histone acetyltransferase (14). One of the ways this small protein achieves such versatility is by reusing its interaction surfaces in different contexts. In MLL complexes, for example, WDR5 binds its essential partner protein RBBP5 via a shallow hydrophobic cleft that is draped in basic amino acid side chains (15). In NSL complexes, this same cleft of WDR5 binds a different protein, KANSL2 (14), that is critical for histone acetyltransferase activity. This shared cleft is poignant here because this is precisely the same surface that WDR5 uses to interact with MYC (5). The near-identical ways that RBBP5, KANSL2, and MYC associate with WDR5 reveals that no two proteins can bind WDR5 at the same time via this surface and implies a functional significance. But what can it be?
One possibility is that MYC engages WDR5 outside the context of an MLL or NSL complex. In this case, the mutually-exclusive binding would be a tidy way of keeping the complexes distinct, allowing WDR5 to serve critical, yet independent, roles in multiple processes. A more intriguing possibility, however, is that this shared interaction site plays a role in coordinating molecular events on chromosomes. If WDR5 is viewed as the central player in this process, it is easy to imagine how a chromatin-bound molecule of WDR5 could be a docking site for various proteins at a fixed genomic location. By depositing WDR5 at specific points on chromatin, and by employing mutually exclusive interactions, cells could tie specific molecular functions together in a way that allows coordination between them. For example, completion of a catalytic cycle by an MLL complex may trigger a transient dissociation of the complex (16), creating the opportunity for MYC to access WDR5 and be stably recruited to chromatin only after H3K4 methylation is safely deposited. Alternatively, MYC may actively disrupt chromatin-bound MLL complexes by displacing RBBP5 from WDR5, simultaneously binding its target genes and inhibiting further H3K4 methylation at these sites. It is interesting to note that MYC has been found to trigger transient and localized histone H3K4 demethylation upon binding target genes (17), and that this process leads to recruitment of additional proteins required for gene activation by MYC. By extension, we imagine that exchanges between resident WDR5 and its various interaction partners could be part of a cycle that not only brings MYC to chromatin but establishes an ordered series of events that prime the locus for enhanced transcription.
Key to understanding how WDR5 exerts its influence over MYC is to characterize the molecular context in which the two proteins associate and to learn how WDR5 itself is tethered to chromatin. Biochemical purification of the relevant MYC–WDR5 complex, and identification of proteins that associate with MYC via WDR5, will reveal whether MYC interacts with a novel WDR5-containing complex, or whether it is capable of disrupting MLL (or NSL) complexes.
Understanding how WDR5 is recruited or retained at MYC target genes, however, may be more challenging. Besides its role in the MLL and NSL complexes, WDR5 is an epigenetic “reader”, capable of binding histone H3 tails marked by K4 dimethylation (10), symmetric arginine 2 dimethylation (18), and threonine 11 phosphorylation (19). Like many WD40-repeat proteins, WDR5 can also associate with ubiquitin (20), a frequent modification of histones. And it can be recruited to specific sites on chromatin by long non-coding RNAs (21), which interestingly bind to the same cleft in WDR5 as MYC/RBBP5/KANSL2 (22). Compounded by the fact that WDR5 could be brought to different regions of chromatin by different mechanisms, distilling the mechanisms of target gene selectivity by WDR5, and how it relates to MYC, could take some time. But it should be possible to combine biochemical and genetic methods with approaches such as chromatin immunoprecipitation to learn how WDR5 is recruited to select genes, and use this information to build a global view of WDR5-targeting mechanisms.
Therapeutic implications
The utility of MYC proteins as targets for anti-cancer therapies is well-established (2), and the impact of bioavailable small molecule MYC inhibitors on cancer treatment options would likely be profound. A recent breakthrough was achieved with a class of compounds known as BET bromodomain inhibitors (2), which act against a family of proteins required for expression of MYC genes in certain cell types. While promising, these compounds are limited to situations where MYC expression is driven by BET bromodomain proteins. Targeting some property of the MYC protein itself may be expected to have broader utility, but this has proven difficult. MYC is largely unstructured in solution. Detailed understanding of how it associates with key partner proteins is scant, and restricted primarily to MAX. Although MAX is the most-highly validated and crucial MYC interaction partner, drug discovery efforts here are limited because of the extensive nature of the MYC–MAX interface, and because each protein is unstructured in the absence of the other; making structure-activity relationships difficult to pursue.
Identification of WDR5 as a co-factor for MYC presents a new target for drug discovery that is theoretically much more tractable. The MYC–WDR5 interface is small, relatively weak, and structurally well-defined. WDR5 is a compact protein whose structure is not significantly altered when bound to MYC. These unique properties of WDR5 and the way it associates with MYC make it possible to shift the focus of drug discovery efforts away from MYC and onto WDR5, via identification of small molecules that tightly associate with the MYC-binding cleft in WDR5. If the facilitated recruitment model is correct, such small molecules would be expected to chemically recapitulate the effects of point mutations in the EEIDVV motif of MYC (5); preventing its stable association with chromatin and disabling its tumorigenic properties.
The concept that WDR5 could be used as a platform to discover MYC inhibitors is exciting, but also raises a number of key questions that will need to be answered before their true potential can be realized. It is essential to know the extent to which MYC and WDR5 work together in different malignancies, to know which cancers may benefit from MYC–WDR5 inhibitors. It is important to determine whether association of WDR5 with MYC is required for tumor maintenance, as opposed to initiation, to reveal whether such inhibitors would have any utility for the treatment of pre-existing cancers. And careful consideration has to be given to the prospect of whether a therapeutic window for such inhibitors could be established, in which the activity of MYC can be reduced to a sufficient extent without sacrificing the normal, essential, functions of WDR5. This is a particularly relevant concern because it is difficult to envision a MYC–WDR5 inhibitor that would not also disrupt the WDR5–RBBP5 and WDR5–KANSL2 interactions. The only way to really know whether chemically inhibiting the MYC–WDR5 interaction can lead to a selective and useful therapeutic agent is by discovering and developing the inhibitors themselves. Fortunately, growing recognition of the pro-tumorigenic properties of WDR5 (23), and its value as anti-cancer drug target (24), provides impetus for discovering small molecules that bind key interaction surfaces on WDR5, such as the MYC/RBBP5/KANSL2-binding cleft, with the prospect that they will have anti-cancer activity in some setting. It seems likely, therefore, that chemical matter will be available soon to interrogate the true therapeutic potential of targeting the MYC–WDR5 nexus.
References
- 1.Spencer CA, Groudine M. Control of c-myc regulation in normal and neoplastic cells. Advances in cancer research. 1991;56:1–48. doi: 10.1016/s0065-230x(08)60476-5. [DOI] [PubMed] [Google Scholar]
- 2.Tansey WP. Mammalian MYC proteins and cancer. New Journal of Science. 2014;2013:1–27. [Google Scholar]
- 3.Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harbor perspectives in medicine. 2014;4(1):a014357. doi: 10.1101/cshperspect.a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall' Olio V, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8(7):764–70. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 5.Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, et al. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol Cell. 2015;58(3):440–52. doi: 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Migliori V, Mapelli M, Guccione E. On WD40 proteins: propelling our knowledge of transcriptional control? Epigenetics. 2012;7(8):815–22. doi: 10.4161/epi.21140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One. 2011;6(10):e26057. doi: 10.1371/journal.pone.0026057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A. 2002;99(13):8695–700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. Embo J. 2011;30(20):4198–210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121(6):859–72. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 11.Ang YS, Tsai SY, Lee DF, Monk J, Su J, Ratnakumar K, et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145(2):183–97. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151(1):68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dias J, Van Nguyen N, Georgiev P, Gaub A, Brettschneider J, Cusack S, et al. Structural analysis of the KANSL1/WDR5/KANSL2 complex reveals that WDR5 is required for efficient assembly and chromatin targeting of the NSL complex. Genes Dev. 2014;28(9):929–42. doi: 10.1101/gad.240200.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Odho Z, Southall SM, Wilson JR. Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J Biol Chem. 2010;285(43):32967–76. doi: 10.1074/jbc.M110.159921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song JJ, Kingston RE. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem. 2008;283(50):35258–64. doi: 10.1074/jbc.M806900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amente S, Bertoni A, Morano A, Lania L, Avvedimento EV, Majello B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene. 2010;29(25):3691–702. doi: 10.1038/onc.2010.120. [DOI] [PubMed] [Google Scholar]
- 18.Migliori V, Muller J, Phalke S, Low D, Bezzi M, Mok WC, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19(2):136–44. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 19.Kim JY, Banerjee T, Vinckevicius A, Luo Q, Parker JB, Baker MR, et al. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol Cell. 2014;54(4):613–25. doi: 10.1016/j.molcel.2014.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pashkova N, Gakhar L, Winistorfer SC, Yu L, Ramaswamy S, Piper RC. WD40 repeat propellers define a ubiquitin-binding domain that regulates turnover of F box proteins. Mol Cell. 2010;40(3):433–43. doi: 10.1016/j.molcel.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120–4. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang YW, Flynn RA, Chen Y, Qu K, Wan B, Wang KC, et al. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. eLife. 2014;3:e02046. doi: 10.7554/eLife.02046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Xie W, Gu P, Cai Q, Wang B, Xie Y, et al. Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci Rep. 2015;5:8293. doi: 10.1038/srep08293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53(2):247–61. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]