

Abstract
Patients with pancreatic and biliary carcinomas lack personalized treatment options, in part because biopsies are often inadequate for molecular characterization. Cell-free DNA (cfDNA) sequencing may enable a precision oncology approach in this setting. We attempted to prospectively analyze 54 genes in tumor and cfDNA for 26 patients. Tumor sequencing failed in nine patients (35%). In the remaining 17, 90.3% (95% CI: 73.1–97.5%) of mutations detected in tumor biopsies were also detected in cfDNA. The diagnostic accuracy of cfDNA sequencing was 97.7%, with 92.3% average sensitivity and 100% specificity across five informative genes. Changes in cfDNA correlated well with tumor marker dynamics in serial sampling (r=0.93). We demonstrate that cfDNA sequencing is feasible, accurate, and sensitive in identifying tumor-derived mutations without prior knowledge of tumor genotype or the abundance of circulating tumor DNA. cfDNA sequencing should be considered in pancreatobiliary cancer trials where tissue sampling is unsafe, infeasible, or otherwise unsuccessful.
Keywords: cell-free DNA, circulating tumor DNA, pancreatic adenocarcinoma, cholangiocarcinoma, next-generation sequencing
Introduction
Pancreatic ductal adenocarcinoma (PDA) has among the lowest survival rates of all cancers(1). Genomic profiling has not yet impacted treatment or diagnosis, leaving most PDA patients out of the molecular revolution reshaping the care of lung, melanoma, and breast cancers(2, 3). PDA remains largely refractory to genomic testing for at least two reasons. First, high stromal cell content often confounds analysis of biopsy specimens(4). Second, only 10% of PDA cases are resectable, and, of the resectable cases that have been enrolled in genome sequencing studies(5), most have tumor cellularity below 30%, with many much lower(6). As a result, most genomic research in PDA has centered around tumor-derived cell lines or xenografts in lieu of direct tumor sequencing(7), and expert bodies currently make no recommendations regarding molecular profiling for clinical decision-making in PDA(8), despite potentially sensitive subsets identified by sequencing(9).
Translational research and precision medicine efforts face similar tissue acquisition challenges in cancers of the biliary tree. These malignancies often harbor actionable mutations, and dramatic responses have been seen when such mutations are identified(10, 11), (12). However, biliary cancers are often diagnosed via ductal brushings obtained during endoscopic retrograde cholangiopancreatography (ERCP), which often yields insufficient material for molecular testing. Therefore, neither molecular profiling nor targeted therapy are standard practice(13), despite the presence of many molecular targets(14), (10).
The ability to detect cancer mutations in blood provides a possible solution to these and many other challenges. Advanced-stage tumors often shed cell-free DNA (cfDNA) into the bloodstream, which can be isolated from a non-invasive blood draw and then detected by polymerase chain reaction (PCR)-based assays(15) or next-generation sequencing (NGS)-based testing(16). Cell-free DNA sequencing could obviate the costs, complications, and delays associated with tissue biopsy in pancreatobiliary cancer patients. An added potential benefit of the cfDNA approach is the ability to monitor the quantities and identities of tumor-derived genetic lesions over time via routine and minimally invasive blood draws.
cfDNA is more abundant in pancreatic cancer patients than healthy controls, although the fraction that is tumor-derived is not known. More than 75% of metastatic PDAs have tumor-derived cfDNA detectable by PCR-based, single-gene methods(15). The concordance between mutations observed in cfDNA with those seen in primary or metastatic PDA tumor tissue also remains unknown. Additionally, it is not known how NGS-based multi-gene panels perform in biliary cancers, which is an especially relevant consideration in cholangiocarcinoma because numerous targetable driver mutations have been recently reported using tumor DNA sequencing(10, 14).
We designed a prospective clinical study to determine the diagnostic accuracy of cfDNA sequencing in pancreatobiliary patients (Figure 1A). This is the first study of its kind in these deadly diseases and one of the first prospective studies to measure concordance between commercially available NGS gene-panel tests of tumor tissue biopsies versus plasma-derived cfDNA.
Figure 1.
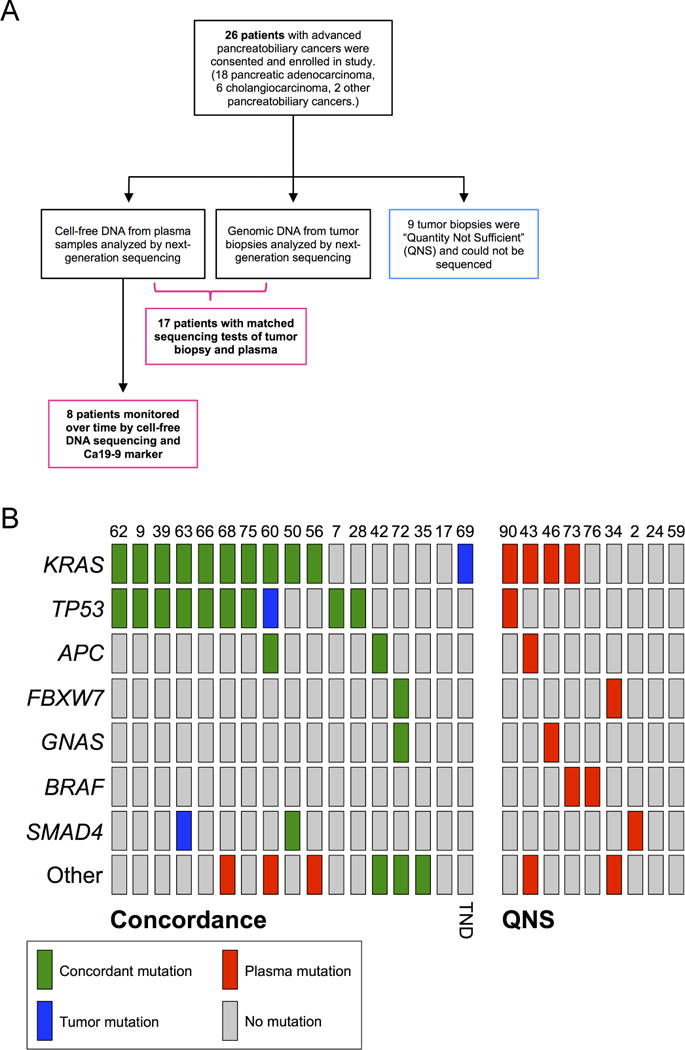
(A) Study design for measuring the feasibility, concordance, and accuracy of a plasma-based cell-free DNA sequencing test compared to biopsy-based sequencing tests for pancreatic and biliary cancer patients (in accordance with Standards for the Reporting of Diagnostic accuracy studies, STARD). Sixteen patients had mutations detected by either type of test, and one patient had no mutations detected in either plasma or in tumor tissue (n=17). (B) Oncoprint chart showing mutation occurrence for the top seven genes across all patients. The group at left (“Concordance”) shows mutations detected in the 17 samples used for the concordance analysis (see Figure 2B). The group at right (“QNS”) shows mutations detected in cfDNA from the eight QNS samples. TND, tumor not detected in cfDNA.
Results
To assess the feasibility and efficacy of cfDNA-sequencing-based testing, 26 patients with advanced pancreatic or biliary carcinomas that had tumor material available underwent blood draws for cfDNA testing. The cohort included 18 PDA cases and 8 biliary cancer cases, with 23 patients having metastatic disease (Supplementary Table 1). The blood samples were sent to a single commercial NGS cell-free DNA sequencing provider for gene panel sequencing (see Methods). In parallel, tumor biopsy specimens were sectioned from formalin-fixed, paraffin-embedded blocks and sent to two different commercial NGS test providers for tumor DNA gene-panel sequencing.
Seventy-three somatic mutations were reported from the union of all tumor and cell-free DNA tests across the entire cohort of 26 patients (mean = 2.8 mutations per patient; Supplementary Table 2). Mutation frequencies were determined per gene, and the mutation status of the seven most frequently mutated genes across the cohort were compared using an oncoprint(17) frequency chart (Figure 1B). KRAS and TP53 were the most commonly mutated genes, with APC, SMAD4, GNAS, FBXW7, and BRAF also being recurrently mutated in cfDNA (Figure 2A). Patients were grouped into four categories based on the two sets of reports: concordant, partially concordant, TND, or QNS. Reports from 13 patients were completely concordant (50%), three were partially concordant (12%), one was TND (4%), and eight were QNS (35%; Figure 2B). Three patients (the one TND and two partially concordant, 12% of cohort) each had one somatic mutation detected in tumor biopsy that should have been detected in cfDNA, were there sufficient quantities of tumor-derived DNA in circulation. Notably, nine patients (35% of cohort) had insufficient quantity or quality of biopsy material for genomic DNA sequencing, presumably due to the difficulties associated with tissue acquisition from these anatomically problematic and highly impure tumors. cfDNA sequencing identified mutations in 78% (7/9) of these QNS cases, with the other two patients having no mutations detected in plasma (Figure 1B).
Figure 2.
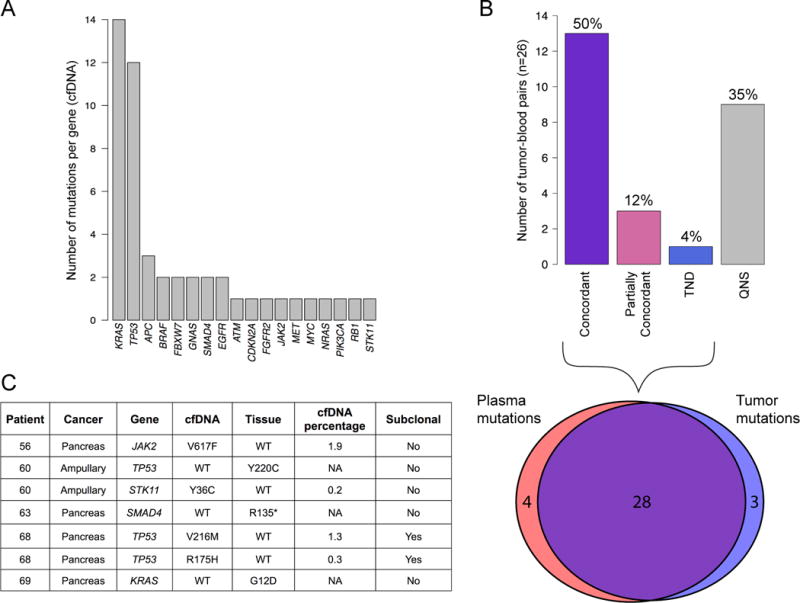
Summary of mutational concordance across 26 pancreatobiliary cancer patients. (A) Top: Numbers of matched biopsy DNA and plasma cfDNA samples in each patient-level concordance category. Concordant samples are those with all reported mutations found in both biopsy-based and plasma-based sequencing tests. Partial concordance occurred if at least one mutation, but not all mutations, were concordant between biopsy DNA and plasma cfDNA. TND: Tumor Not Detected. QNS: Quantity Not Sufficient. Bottom: Venn diagram showing overlap of reported mutations between cfDNA and tumor biopsy sequencing tests for 17 patients. (B) Numbers of mutations detected per gene by the cfDNA test across the cohort (49 mutations total). (C) Details of the seven non-concordant mutations from the 17 patients in (A).
We performed gene-level sensitivity and specificity analyses of the cfDNA test for five genes mutated in more than one patient’s tumor tissue biopsy. For the sake of comparisons herein, we considered the tissue biopsy gene-panel test as the gold standard. Across these five genes (KRAS, TP53, APC, FBXW7, and SMAD4) the average sensitivity was 92.3% (range 50–100%), specificity was 100%, and average diagnostic accuracy was 97.7% (range 93–100%) (Table 1). Only KRAS and TP53 were mutant in more than three patients’ cfDNA. Out of the 26 total mutations detected for both of these genes, only a single TP53 mutation was detected in tumor biopsy but not in cfDNA (Figure 2C, patient #60). Two of the three TP53 mutations observed in patient #68 were detected only in cfDNA and were not included in this analysis as they were likely subclonal (Figure 2C and Supplementary Table 2; see Methods for subclonal definition). When the cfDNA test was considered as the gold standard, the accuracy (97.7%), sensitivity (100%), and specificity (96.7%) were similar, reflecting the high level of concordance between the two platforms.
Table 1.
Contingency table to determine diagnostic sensitivity, specificity, and accuracy for the most frequently mutated genes among cohort. cfDNA-test-positive and cfDNA-test-negative mutation counts are shown for the top five genes in the first two columns. Only genes with at least two mutations in tumor tissue were included in this analysis. 95% confidence intervals are given below the totals in bottom row. PPV, positive predictive value; NPV, negative predictive value.
| Mutation present (tumor) | Mutation absent (tumor) | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | False Positive Rate (%) | False Negative Rate (%) | Diagnostic Accuracy (%) | |
|---|---|---|---|---|---|---|---|---|---|
| cfDNA positive (KRAS) | 10 | 0 | – | – | – | – | – | – | – |
| cfDNA negative (KRAS) | 0 | 7 | 100 | 100 | 100 | 100 | 0 | 0 | 100 |
| cfDNA positive (TP53) | 9 | 0 | – | – | – | – | – | – | – |
| cfDNA negative (TP53) | 1 | 7 | 90 | 100 | 100 | 88 | 0 | 10 | 94 |
| cfDNA positive (APC) | 2 | 0 | – | – | – | – | – | – | – |
| cfDNA negative (APC) | 0 | 15 | 100 | 100 | 100 | 100 | 0 | 0 | 100 |
| cfDNA positive (FBXW7) | 2 | 0 | – | – | – | – | – | – | – |
| cfDNA negative (FBXW7) | 0 | 15 | 100 | 100 | 100 | 100 | 0 | 0 | 100 |
| cfDNA positive (SMAD4) | 1 | 0 | – | – | – | – | – | – | – |
| cfDNA negative (SMAD4) | 1 | 15 | 50 | 100 | 100 | 94 | 0 | 50 | 94 |
| Total positive | 24 | 59 | – | – | – | – | – | – | – |
| Total negative | 2 | 0 | – | – | – | – | – | – | – |
| Total (positive + negative) | 26 | 59 |
92.3 (73.4–98.7) |
100 (92.4–100) |
100 (82.8–100) |
99.1 (87.6–99.4) |
0 (0–7.6) |
7.7 (1.3–26.6) |
97.7 (91–99.6) |
The cfDNA NGS test examined here interrogated 54 genes, allowing concordance assessments beyond binary present/absent metrics attendant to single-gene approaches. Seventeen patients were deemed evaluable for concordance analysis between plasma and tissue sequencing (i.e., they were not QNS). These 17 patients had a total of 35 mutations reported from plasma and from tumor biopsy. Of the 31 mutations detected by tumor-biopsy NGS, 28 were also detected by the cfDNA test (90.3% overlap, 95% CI: 73.1–97.5%). The seven mutations reported exclusively by the tumor-sequencing test or by the plasma sequencing test are shown in Figure 2C. As discussed below, one of these was a somatic mutation in a distinct tissue compartment (patient #56, JAK2V617F), and another was likely due to the level of circulating tumor DNA being below the limit of detection for cfDNA sequencing (patient #69, KRASG12D).
To test if mutation frequencies in cfDNA quantitatively correlated with disease progression or response to therapy, eight patients were monitored by serial cfDNA tests while on various therapies. Standard tumor markers were assayed concurrently and compared over time to mutation percentages in cfDNA (Supplementary Figure 1A–F, Supplementary Table 3). The cfDNA allele fraction of the most abundant mutation was compared to the level of tumor marker for each patient. For 19 blood-draw intervals (range of 41–166 days) across these eight patients, the direction of change in tumor marker and cfDNA-percentage agreed significantly more often than by chance (p=0.02, exact binomial test). Additionally, changes over time in the cfDNA mutation percentage correlated well with changes in tumor marker measurements (Pearson’s r = 0.69 for interval slopes, r = 0.93 for interval differences). These data suggested that cfDNA mutant allele fraction changes reflect changes in disease burden over time and treatment.
Clinically meaningful mutations were detected in patients in whom commercial tumor DNA sequencing was not possible due to tissue biopsy failure (QNS). Surprisingly, a canonical activating EGFR exon 19 deletion(18) was detected in the blood of PDA patient #34. Attempts at tumor NGS testing at the time of with the PDA diagnosis were made, but the tissue biopsy sample failed clinical tumor sequencing. The patient received FOLFIRINOX with stabilization of disease, but eventually developed hematologic toxicity requiring dose reduction followed by cessation of cytotoxic therapy. Upon progression, a repeat biopsy was obtained and again sent for commercial NGS, which this time confirmed the EGFR deletion seen at diagnosis in the blood, nearly seven months prior. The patient was then treated with capecitabine and the approved EGFR inhibitor erlotinib, resulting in radiographic response, functional improvement and CA 19–9 normalization (Figure 3A–C). This patient is alive and on erlotinib monotherapy (as of June 3, 2015). A second patient with cholangiocarcinoma had a BRAFD594G mutation, which is found in lung cancer and has also been reported in a cetuximab-responsive colorectal cancer patient(19), detected in cfDNA. This patient declined cytotoxic therapy (gemcitabine and cisplatin(20)) and was lost to follow up. An FGFR2S252L mutation was detected in the blood of patient #35 at diagnosis, which was confirmed by commercial tumor biopsy NGS. The patient first responded to and then progressed on gemcitabine and cisplatin, and enrolled in a FGFR inhibitor clinical trial using genotype selection at an outside institution (NCT02150967) upon progression.
Figure 3.
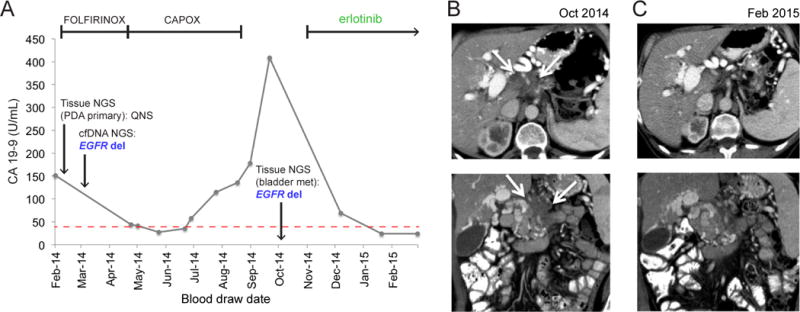
Treatment and diagnosis history, and response to targeted therapy, of a pancreatic adenocarcinoma patient with an EGFR exon19 deletion. (A) CA 19–9 levels (x axis) over time of treatment (y-axis) for patient #34. Dates of two biopsy-based NGS tests, and of EGFR indel detection by cfDNA test, are indicated along the CA 19–9 line. Time intervals for therapies administered to patient are indicated by bracketed lines at top. Erlotinib monotherapy is ongoing as of June 3, 2015. (B) Axial (top) and coronal (bottom) contrast-enhanced CT images from October 2014 demonstrate infiltrative tumor (arrows) arising from the pancreatic body and encasing the celiac axis. (D) Axial (top) and coronal (bottom) contrast-enhanced CT images from February 2015 demonstrate marked reduction in the size of the pancreatic mass.
Additional relevant mutations were observed in cfDNA that were not detected by tumor biopsy NGS. One patient (#56) showed a decrease in KRASG12V titer on gemcitabine and nab-paclitaxel, but a concurrent slight increase in JAK2V617F titer, an allele implicated in myeloproliferative diseases(21) (Supplementary Figure 1F). JAK2V617F was absent from the commercial tumor sequencing report, but was detected by polymerase chain reaction (PCR) in 0.47% of circulating white blood cells (data not shown). These findings suggest that the KRASG12V mutation was present in the PDA tumor cells, and which in turn responded to gemcitabine and nab-paclitaxel. On the other hand, the JAK2V617F mutation was present in hematopoietic cells and unaffected by this this treatment(22).
Discussion
Precision oncology requires DNA profiling of tumor-derived somatic mutations, which in turn can predict sensitivity or resistance to particular therapies. Despite a real unmet need for improved therapies for pancreatobiliary cancers, clinical efforts to develop genotype-specific treatments in these diseases have mostly failed at an early stage due to our inability to reliably, safely, and reproducibly obtain tissue for genomic analysis, especially in metastatic patients most in need of effective systemic therapies. As such, no molecular characterization is currently recommended for these diseases(8, 23), despite a desperate need for improved treatment approaches.
Our results are the first of their kind to demonstrate that cfDNA-based next generation sequencing of a gene panel is feasible and accurately detects tumor-derived mutations in advanced cancer without a priori knowledge of tumor genotype or cfDNA burden. Whereas tumor biopsy-based tests detected mutations in 62% of patients, cfDNA testing did so for 85% patients. Thus, 23% of our cohort (22 versus 16 patients) arguably stood to benefit from the cfDNA test over, or in addition to, tumor biopsy-based tests. The cfDNA test examined in this study detected over 90% of the mutations seen by tumor biopsy sequencing (within the 54 genes on the cfDNA test panel), and had high sensitivity and specificity across the five most frequently mutated genes. Because we were able to measure concordance on only 17 patients, future studies are needed to establish the robustness of this analysis. An additional 23 mutations were detected in 13 genes covered by the larger capture footprint of biopsy-based testing that were not covered by the cfDNA test panel (Supplementary Table 2), suggesting that expanded panel coverage could potentially capture additional useful information, possibly at increased cost, decreased sensitivity, or both. Furthermore, the sensitivity of cfDNA detection in early stage or resectable disease is also of potential interest, but was not addressed in this advanced stage cohort. Finally, we highlight biological scenarios that lead to discordance between tumor DNA and cfDNA sequencing including convergent, subclonal mutations in the TP53 gene (in patient #68), and a JAK2 mutation in a distinct tissue compartment from the primary pancreatic malignancy (in patient #56). Inaccurately labeling such results as “false negatives/positives” with respect to a given gold standard ignores these important biological nuances.
cfDNA mutation percentages correlated with clinically accepted tumor marker measurements, suggesting that quantitative cfDNA sequencing provides a useful measure of disease progression or response to therapy. Future studies should focus on the precise kinetics of cfDNA versus protein biomarker changes in response to therapy in pancreatobiliary cancers. In addition, three patients with cfDNA mutations had normal CA 19–9 levels, suggesting that cfDNA sequencing could provide both tumor-burden and mutational information in select patients who, for whatever reason, do not secrete antigenic tumor markers. Our analyses suggest that changes in tumor-derived cfDNA concentration and tumor marker concentration are closely correlated, and future studies may define unique and complementary uses for each.
The QNS rate for this advanced pancreatobiliary cancer cohort was 35%, which is considerably higher than the average QNS rate reported by commercial vendors across multiple tumor types(24). Due to difficulties in obtaining sufficient tumor genomic DNA from biopsy tissue for NGS, cfDNA testing may provide a substantial advantage for these cancers. In support of this idea, Lebofsky et al included two PDA patients in their comparison of matched tumor DNA and circulating, cell-free DNA on a NGS platform, with encouraging results(25).
The cfDNA sequencing approach has several advantages over biopsy-based NGS or targeted cfDNA profiling by PCR(26). Relative to targeted cfDNA profiling by PCR assays, cfDNA sequencing provides a much broader analytic footprint of the genome(16, 25). In addition to detecting more mutations per patient, copy number data and a broader scope of actionable mutations, cfDNA sequencing might also reveal a distinct mutational signature of tumor subclone emergence as drug resistance or disease progression occurs.
Of the 22 patients with cfDNA mutations, four had potentially actionable mutations in drug-able oncogenes (BRAF, EGFR, or FGFR2). PDA patient #34 harbored a tumor-encoded EGFR exon19 deletion, and was treated with erlotinib based on the results of a biopsy-based test obtained at progression because the primary tumor biopsy was QNS. This is the first report, to our knowledge, of an activating EGFR mutation in PDA responding to erlotinib, and would have been missed with single gene assays evaluating only KRAS, which is wild-type in this patient. Although the patient eventually experienced clinical benefit from EGFR blockade by erlotinib, this therapy was delayed by seven months due to failure of the initial tissue biopsy sequencing test ordered by the treating physician. This case illustrates that cfDNA testing can provide actionable findings earlier during patient management, and has the potential to reduce or eliminate invasive repeat biopsies when tissue sequencing is unsuccessful.
We achieved an analytic performance (clinical sensitivity and specificity) with this 54-gene NGS panel comparable(26, 27) or superior to(28) those achieved for a single gene (EGFR), PCR-based cfDNA methods in advanced lung cancer patients. Due to the lack of targeted therapies in pancreatobiliary cancers, acquired drug-resistance mutations were not observed, but they remain an important area of interest for cfDNA application in the clinic(29).
Our work highlights a critical discussion point for gastrointestinal oncologists, regarding the appropriate steps required to validate cfDNA analyses as surrogates for tissue analysis across clinical applications. Our results strongly support the clinical accuracy and feasibility of cfDNA testing, but prospective clinical trials with therapeutic decision-making based on cfDNA results are needed in the next generation of studies. These trials will offer unprecedented collaborative opportunities among commercial diagnostic and pharmaceutical entities as well as academic and community medical centers.
In conclusion, cfDNA sequencing has promise in advanced pancreatobiliary cancers. The clinical utility of the cfDNA diagnostic approach is evident in the increased detection of actionable mutations versus tissue-based NGS and the fact that many costs and potential complications associated with needle aspiration biopsies are obviated. The ability to reliably report accurate mutation findings over the course of cancer treatment could empower clinical trial execution in this challenging set of diseases. More generally, the approach presented here could augment tumor-tissue analysis for a variety of malignancies, and take its place for cases where tumor tissue testing fails.
Methods
Study design and patients
The institutional review board of the University of California, San Francisco Medical Center approved the study. All patients were recruited at a single center (UC San Francisco Medical Center) and provided informed consent under human subjects protocol CC# 13986. All patient studies were conducted in accordance with the Declaration of Helsinki. Diagnostic biopsies were sent for commercial NGS as a part of routine clinical care as described(24) (n=21) or were sequenced in-house (n=5). Baseline blood draws were performed as near to time of biopsy as possible (Supplementary Table 1). Results from cfDNA sequencing were not returned to patients, and treatment decisions were not made on cfDNA results. For patient #34, the initial diagnostic tissue biopsy sample failed processing by a commercial vendor. Seven months later, a second, transcystic biopsy was performed which confirmed progressive disease and was successfully sequenced.
Blood samples and cell-free DNA isolation
Venous blood was collected in Streck™ tubes during routine phlebotomy, and samples were shipped at room temperature overnight. 10mL of blood was processed upon receipt to isolate plasma by centrifugation at 1,600g for 10 minutes at 4°C. Plasma was immediately aliquoted and stored at −70°C. Cell-free DNA was extracted from 1mL aliquots of plasma using the QIAamp circulating nucleic acid kit (Qiagen), concentrated using Agencourt Ampure XP beads (Beckman Coulter), and quantified by Qubit fluorometer (Life Technologies, Carlsbad, CA, USA). All cell-free DNA sequencing and analysis was performed at Guardant Health (Redwood City, CA, USA).
Cell-free DNA sequencing
Barcoded sequencing libraries were generated from 5–30ng of cfDNA. The exons of 54 cancer genes, including all coding exons of 18 genes and the recurrently mutated exons in an additional 36 genes (Supplementary Table 4), were captured using biotinylated custom bait oligonucleotides (Agilent), resulting in a 78,000 base-pair (78 kb) capture footprint. Samples were paired-end sequenced on an Illumina Hi-Seq 2500, followed by algorithmic reconstruction of the digitized sequencing signals as described (Lanman et al., submitted). The coverage depth across all coding sequence in all samples averaged approximately 10,000x.
Illumina sequencing reads were mapped to the hg19/GRCh37 human reference sequence, and genomic alterations in cfDNA were identified from Illumina sequencing data by Guardant Health’s proprietary bioinformatics algorithms. These algorithms quantify the absolute number of unique DNA fragments at a given nucleotide position, thereby enabling circulating tumor DNA to be quantitatively measured as a fraction of total cfDNA. The mutant allele fraction for a given mutation was calculated as the fraction of cfDNA molecules harboring that mutation divided by the total number of unique cfDNA molecules mapping to the position of the mutation. The limit of detection for single-nucleotide variants in cfDNA by the Guardant360 assay is 0.1%. The EGFR deletion identified in patient #34 was confirmed by manual examination of sequencing reads in the Integrative Genomics Viewer (IGV).
Data analysis and accuracy assessments
Comparisons between tumor and plasma sequencing data were performed using clinical reports generated by Foundation Medicine (Cambridge, MA) or research test reports generated by Guardant Health (Redwood City, CA). Mutations called from cfDNA sequencing (plasma) were compared to mutations from tissue-biopsy DNA sequencing (tumor), and concordance was determined for all mutations across the cohort (mutation-level concordance) and for mutations detected in either cfDNA or tumor DNA per patient (patient-level concordance). The total set of mutations from the 17 patients with matched cfDNA and tumor sequencing was tabulated to calculate the total percentage agreement of cfDNA-sequencing versus tumor-sequencing tests. The numbers of cfDNA mutations versus tumor mutations per patient were tabulated, and patients were binned into four categories based on the relative concordance of each set of detected mutations: concordant, partially concordant, tumor DNA not detected in cfDNA (TND), or quantity not sufficient for tissue-based sequencing (QNS). The complete set of mutations for the 26 patients in this study is provided in Supplementary Table 2. Sensitivity, specificity, and diagnostic accuracy were calculated(30) by comparing cell-free DNA mutations to tumor DNA mutations for genes with at least two mutations detected in either type of sequencing test. Subclonal cfDNA mutations were defined as those exhibiting a mutant allele fraction less than 20% of the maximum mutant allele fraction in circulation in a given patient. Mutations that were present in minor subclones (TP53, patient #68) or in patients where the tumor was not detected in cfDNA (#69) were not included in the concordance or sensitivity/specificity analysis. An FBXW7 mutation from patient #34 was included in the sensitivity/specificity analysis (Figure 1B and Table 1). Patient #34 was evaluated for mutation concordance using the progression biopsy since the diagnostic tissue biopsy was QNS. Mutations detected for patient #34 were completely concordant (n=4) but were not counted in the overall concordance shown in Figure 2B.
Patient monitoring analysis
Eight patients’ tumor markers (e.g., CA 19–9) and cfDNA mutant allele fractions were followed over time by serial blood draws (separate draws for each type of test). The slope of the line connecting each pair of temporally separated CA 19–9 or CA 125 values, or mutant allele fractions (cfDNA percentage), was calculated across all intervals for all patients [(Value B – Value A)/Number of days between Draw A and Draw B)]. After excluding three intervals where the cfDNA-percentage slope was zero (mutation fell below limit of detection), there were 19 evaluable time intervals in total. Pearson correlations of CA 19–9 versus cfDNA percentage were determined on differences (Value B – Value A), and separately on slopes, across all 19 time intervals from the eight patients (Supplementary Figure 1A–F, Supplementary Table 3). The binomial test was used to test whether the directions of CA 19–9 change versus cfDNA percentage change coincided more often than chance (50/50) for the 19 intervals.
Supplementary Material
Statement of Significance.
Precision medicine efforts in biliary and pancreatic cancers have been frustrated by difficulties obtaining adequate tumor tissue for next-generation sequencing. Cell-free DNA sequencing reliably and accurately detects tumor-derived mutations, paving the way for precision oncology approaches in these deadly diseases.
Acknowledgments
We thank Dr. Richard Lanman for assistance with sensitivity/specificity calculations and helpful discussions. We thank members of the Collisson Lab and Guardant Health for helpful discussions.
Funding: American Cancer Society, SpinOdyssey Fellowship (O.A.Z.); Doris Duke Charitable Foundation (to E.A.C.); and Guardant Health (to E.A.C and P.N.M.).
Footnotes
Conflicts of interest: O.Z., D.S., L.S., and A.T. are employees of Guardant Health; A.T. acknowledges ownership of Guardant Health; E.A.C. received consulting fees and research funding from Guardant Health; P.M. received research funding from Guardant Health.
Author contributions: O.Z., A.T., and E.A.C designed the study. O.Z., C.G., J.L, L.S., D.S., A.T. and E.A.C performed research. O.Z., C.A., T.G.B., P.M., M.T., A.T. and E.A.C interpreted results. C.G., J.L, C.A., R.K.K., A.K., K.V.L., M.V., M.A.T., P.M. and E.A.C. enrolled patients. O.Z. and E.A.C. wrote the manuscript. All authors read and approved the manuscript.
References
- 1.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:2140–1. doi: 10.1056/NEJMc1412266. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 4.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–8. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 5.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song S, Nones K, Miller D, Harliwong I, Kassahn KS, Pinese M, et al. qpure: A Tool to Estimate Tumor Cellularity from Genome-Wide Single-Nucleotide Polymorphism Profiles. PLoS One. 2012;7:e45835. doi: 10.1371/journal.pone.0045835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Science. Vol. 321. New York, NY: 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses; pp. 1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tempero MA, Malafa MP, Behrman SW, Benson AB, 3rd, Casper ES, Chiorean EG, et al. Pancreatic adenocarcinoma, version 2.2014: featured updates to the NCCN guidelines. Journal of the National Comprehensive Cancer Network: JNCCN. 2014;12:1083–93. doi: 10.6004/jnccn.2014.0106. [DOI] [PubMed] [Google Scholar]
- 9.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Churi CR, Shroff R, Wang Y, Rashid A, Kang HC, Weatherly J, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9:e115383. doi: 10.1371/journal.pone.0115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loaiza-Bonilla A, Clayton E, Furth E, O’Hara M, Morrissette J. Dramatic response to dabrafenib and trametinib combination in a BRAF V600E-mutated cholangiocarcinoma: implementation of a molecular tumour board and next-generation sequencing for personalized medicine. Ecancermedicalscience. 2014;8:479. doi: 10.3332/ecancer.2014.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Javle M, Churi C, Kang HC, Shroff R, Janku F, Surapaneni R, et al. HER2/neudirected therapy for biliary tract cancer. Journal of hematology & oncology. 2015;8:58. doi: 10.1186/s13045-015-0155-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benson AB, 3rd, D’Angelica MI, Abrams TA, Are C, Bloomston PM, Chang DT, et al. Hepatobiliary cancers, version 2.2014. Journal of the National Comprehensive Cancer Network: JNCCN. 2014;12:1152–82. doi: 10.6004/jnccn.2014.0112. [DOI] [PubMed] [Google Scholar]
- 14.Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. The oncologist. 2014;19:235–42. doi: 10.1634/theoncologist.2013-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Science translational medicine. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nature medicine. 2014;20:548–54. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. Science. Vol. 304. New York, NY; 2004. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy; pp. 1497–500. [DOI] [PubMed] [Google Scholar]
- 19.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet Oncology. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 20.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 362:1273–81. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 21.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 22.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature medicine. 2014;20:1472–8. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benson AB, 3rd, Abrams TA, Ben-Josef E, Bloomston PM, Botha JF, Clary BM, et al. NCCN clinical practice guidelines in oncology: hepatobiliary cancers. Journal of the National Comprehensive Cancer Network: JNCCN. 2009;7:350–91. doi: 10.6004/jnccn.2009.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lebofsky R, Decraene C, Bernard V, Kamal M, Blin A, Leroy Q, et al. Circulating tumor DNA as a non-invasive subs to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Molecular oncology. 2015;9:783–90. doi: 10.1016/j.molonc.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Douillard JY, Ostoros G, Cobo M, Ciuleanu T, Cole R, McWalter G, et al. Gefitinib treatment in EGFR mutated caucasian NSCLC: circulating-free tumor DNA as a surrogate for determination of EGFR status. J Thorac Oncol. 2014;9:1345–53. doi: 10.1097/JTO.0000000000000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yung TK, Chan KC, Mok TS, Tong J, To KF, Lo YM. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res. 2009;15:2076–84. doi: 10.1158/1078-0432.CCR-08-2622. [DOI] [PubMed] [Google Scholar]
- 28.Goto K, Ichinose Y, Ohe Y, Yamamoto N, Negoro S, Nishio K, et al. Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol. 2012;7:115–21. doi: 10.1097/JTO.0b013e3182307f98. [DOI] [PubMed] [Google Scholar]
- 29.Luke JJ, Oxnard GR, Paweletz CP, Camidge DR, Heymach JV, Solit DB, et al. Realizing the potential of plasma genotyping in an age of genotype-directed therapies. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altman DG, Bland JM. Diagnostic tests. 1: Sensitivity and specificity. Bmj. 1994;308:1552. doi: 10.1136/bmj.308.6943.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.