

Abstract
Type 1 diabetes mellitus (T1DM) is the archetypal example of a T cell-mediated autoimmune disease characterized by selective destruction of pancreatic β cells. The pathogenic equation for T1DM presents a complex interrelation of genetic and environmental factors, most of which have yet to be identified. Based on the observed familial aggregation of T1DM, it is certain that there is a decided heritable genetic susceptibility for developing T1DM. The well-known association of T1DM with certain human histocompatibility leukocyte antigen (HLA) alleles of the major histocompatibility complex (MHC) was a major step toward understanding the role of inheritance in T1DM. Type 1 diabetes is a polygenic disease with a small number of genes having large effects, (e.g. HLA) and a large number of genes having small effects. Risk of T1DM progression is conferred by specific HLA DR/DQ alleles [e.g., DRB1*03-DQB1*0201 (DR3/DQ2) or DRB1*04-DQB1*0302 (DR4/DQ8)]. In addition, the HLA allele DQB1*0602 is associated with dominant protection from T1DM in multiple populations. A concordance rate lower than 100% between monozygotic twins indicates a potential involvement of environmental factors on disease development. The detection of at least two islet autoantibodies in the blood is virtually pre-diagnostic for T1DM. The majority of children who carry these biomarkers, regardless of whether they have an a priori family history of the disease, will develop insulin-requiring diabetes. Facilitating pre-diagnosis is the timing of seroconversion which is most pronounced in the first two years of life. Unfortunately the significant progress in improving prediction of T1DM has not yet been paralleled by safe and efficacious intervention strategies aimed at preventing the disease. Herein we summarize the chequered history of prediction and prevention of T1DM, describing successes and failures alike, and thereafter examine future trends in the exciting, partially explored field of T1DM prevention.
Introduction
Type 1 diabetes mellitus (T1DM) is a chronic autoimmune disease caused by an immune-mediated injury of pancreatic β cells (1). Genetic analyses of T1DM have linked the HLA complex, mainly class II alleles, to susceptibility to T1DM (2,3). Viral antigens may also play a role in the generation of β cell autoimmunity (4). The latter observations are supported by the increasing seasonal incidence of T1DM in many Western countries (5) and that enteroviruses may be involved in the autoimmune pathogenesis of T1DM (4,6,7,8) Type 1 diabetes was not always considered as the classical organ-specific disease it is now known to be. Insulin-dependent diabetes was known to occasionally occur in the Autoimmune Polyendocrine Syndrome I (APS I), a classic autoimmune syndrome with T-cell and B-cell antibody abnormalities directed at adrenal, parathyroid, gonadal, thyroid and other tissues. However, diabetes mellitus is not a constant, necessary or sufficient feature of APS I (9). This condition is now known to be caused by mutations in the autoimmune regulator gene (AIRE) (10,11). Similarly, the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome was later attributed to a mutation in FOXP3, which encodes a transcription factor that is involved in the function of regulatory T-cell responses (12,13). Furthermore, the recently described STAT3 (Signal Transducer and Activator of Transcription 3) polyautoimmunopathy (14) with early onset autoimmune diabetes and other autoimmune conditions, is due to a de novo germline activating STAT3 mutation. Bottazzo et al. reported that sections of human pancreas treated with sera of diabetic patients who also had Addison’s disease and myxedema, showed cytoplasmic fluorescence in the islets of Langerhans. This response was termed cytoplasmic islet cell antibodies (ICA) (15) and the existence of insulin autoantibodies as well as other autoantibodies against various islet proteins was not uncovered until years later. It was in 1983 that insulin autoantibodies were reported in sera of newly diagnosed patients with T1DM before any treatment with exogenous insulin (16). In this finding, improvements of the sensitivity of the insulin antibody assay were instrumental for the determination that about one-half of newly diagnosed patients had autoantibodies that bound radiolabeled insulin.
Following the early discoveries on humoral autoimmunity in T1DM, there has been a remarkable progress in the detection of T1DM-associated autoantibodies as well as in the characterization of the molecular basis of the antigenicity of their target proteins (17,18). This expansion has led to the uncovering of specific antigenic determinants for both antibodies and T cells involved in disease pathogenesis, the development and standardization of biochemically-defined immunoassays (19,20) and the improvement of T1DM prediction (20,21).
The genetic predisposition associated with prediction-high risk HLA haplotypes, and other genetic loci. The role of environmental factors
Evidence to suggest familial aggregation is provided by the knowledge that overall risk for developing T1DM varies from 1% to 15% in North American Caucasian siblings, parents and offspring of individuals with Type 1A diabetes (with documented autoimmune abnormalities, e.g. islet autoantibodies) as compared to 1.2/1,000 of the general population (22). A recent Finnish study showed that approximately 22% of children with newly diagnosed T1DM have an affected first- and/or second-degree relative (23). However, approximately 85% of T1DM cases occur in individuals with no apparent family history of the disease. In the remaining cases, this disease aggregates in families. The lifelong risk of T1DM is markedly increased in first degree relatives of patients, averaging approximately 6 percent in offspring, 5 percent in siblings, and between 23 and 50% percent in identical twins (24–27). A monozygotic twin of a patient with T1DM has a higher risk of diabetes than a dizygotic twin; in addition, the expression of β cell autoimmunity does not differ between potential dizygotic twins and siblings of patients with T1DM (28). Alleles at each locus on a single chromosome are usually inherited in combination as a unit. This combination is termed haplotype. Since each individual inherits one set of chromosomes from each parent, each individual has two haplotypes. HLA genes are codominant and follow a simple Mendelian form of transmission in families. Therefore, both alleles are expressed at a given HLA locus. There is a 25% chance that two siblings share the same haplotype, a 50% chance that they will share one haplotype, and a 25% chance that they share no haplotype. Siblings with the highest risk HLA DR and DQ alleles (e.g., DR3/DR4 heterozygotes), who inherit both HLA regions identical by descent to their diabetic sibling, may have a risk of developing islet autoimmunity as high as 70 percent and a similar long-term risk of diabetes (29,30).
The current opinion is that both genetic and environmental factors contribute to the risk of progression to clinical disease. This opinion is supported by the fact that T1DM has the tendency to cluster in families and that environmental agents, including exposure to viral agents, may play a role in the generation of β-cell autoimmunity (22–26). Genes within the major histocompatibility complex (MHC) and elsewhere in the genome can influence risk. HLA alleles have a large effect, followed by insulin gene polymorphisms and PTPN22.
The environmental factors leading to T1DM development is a subject of intense investigation. A number of candidates have been identified, including dietary factors (breast feeding vs. infant formula, highly hydrolysed infant formula vs. conventional infant formula, early/late exposure to gluten, vitamin D deficiency etc.), exposure to certain viral elements or helminths anthropometric and psychosocial factors (31). Some viruses manipulate the immune system, compromising lymphopoiesis and causing B and T cell lymphopenia in mice (32) and humans (33). Lymphopenia was observed in human Norovirus acute infection (34), suggesting that this virus may compromise immune homeostasis. Whether noroviruses have pancreatic β-cells tropism as shown by other enteral viruses remains to be elucidated (35,36). Observations on macaques suggested that a virus within the same family as noroviruses, an enteric calicivirus, replicates in B cells from mesenteric lymph nodes (MLN) (37), and therefore spreading of the virus to other lymphoid organs as well as the pancreas is conceivable. The ongoing prospective study high intensive infection follow-up Study, INDIS, reported norovirus among the four most prevalent viruses detected in T1DM genetically predisposed toddlers (38,39). The follow-up of this study will be very informative.
The involvement of the gastrointestinal system in T1DM etiology is suggested by differences in intestinal microbiota composition observed in individuals diagnosed with T1DM or with evidence for islet autoimmunity (40,41). The gut microbiota or microbial compounds such as lipopolysaccaride (LPS), could be the sine qua non for enteric viruses to gain access to the body, as exemplified by the abrogation of virus-induced disease in germ-free or antibiotic treated mice (42). Interestingly, abrogation of bacterial sensing (MyD88−/−) significantly protected NOD mice from T1DM development (43). The possibility that viruses and bacteria synergize to cause autoimmunity is also suggested by the protective role shown by antibiotics in virus-induced T1DM in Kilham rats (44). Several studies have reported examples of dysbiosis in patients with T1DM (45,46), but the cause-effect relationship between dysbiosis, metabolic disorder and/or infection is unclear.
The time has come to carefully design adequately powered longitudinal studies in humans to determine to what extent changes in the gut microbiota or in the metabolome, affect islet autoimmunity and T1DM progression.
HLA genes
The short arm of human chromosome 6 (6p21) accommodates a ~3.5 megabase genetic segment containing a group of immune response genes termed the major histocompatibility complex (MHC). The main genes localized within the MHC encode human leukocyte antigens, or HLA, two molecular classes of cell surface glycoproteins differing in structure, function, and tissue distribution. While class I MHC molecules are expressed in virtually all nucleated cells, class II molecule expression is restricted to B lymphocytes, dendritic cells, macrophages and activated T lymphocytes. Both class I and class II antigens are involved in the presentation of antigen to T cells. Cytotoxic T cells (CD8+) mainly recognize antigen in the context of class I, whereas helper/inducer cells (CD4+) usually recognize antigen in the context of the class II molecules.
Many immunologically mediated diseases, including certain endocrine syndromes are genetically associated with specific HLA molecules and several hypotheses have been suggested to explain HLA-disease associations (47). Some HLA-associated diseases have been linked with polymorphisms of the genes encoding the class II molecule. One typical example is T1DM.
The ability of these class II molecules to present antigens is dependent in part upon the amino acid composition of their α and β chains. Substitutions at one or two critical positions can markedly increase or decrease binding of relevant autoantigens and yet the susceptibility to T1DM (48,3). In particular, more than 90 percent of patients with T1DM carry either HLA-DR3, DQB1*0201 (also referred to as DR3-DQ2) or -DR4, DQB1*0302 (also referred to as DR4-DQ8), versus 40 percent of controls with either haplotype; furthermore, about 30 percent of patients have both haplotypes (DR3, DQB1*0201/DR4, DQB1*0302 heterozygotes), which confers the greatest susceptibility (49). The prevalence of this high-risk genotype is remarkably high in some populations. As an example, 8.9 percent of healthy white teenagers in Washington state have the DR3, DQB1*0201/DR4, DQB1*0302 genotype and 2.4 percent of the general population of Denver, Colorado. Approximately 5 percent of children with this genotype develop T1DM versus approximately 0.3 percent of children overall (50,51). A subset of DR4 alleles, such as DRB1*0403 and DRB1*0402, decrease the risk of development of diabetes, even with the high risk DQB1*0302 allele (52,53). Moreover, we provided evidence to suggest that HLA-DQ typing can identify a seronegative phenotype associated with two HLA-DQ high-risk haplotypes and insulin-requiring diabetes progression (54).
Surprisingly, the HLA allele DQB1*0602 confers dominant protection against the development of T1DM. This allele is present in approximately 20 percent of the general US population, but less than 1 percent of children developing T1DM (55). A prospective study evaluated 72 relatives with islet-cell antibodies (ICA), 75 percent of whom carried the high-risk alleles DQB1*0302 and/or *0201 (56). Diabetes developed in 28 of the 64 subjects who did not have the DQB1*0602 allele versus none of the eight with it. No other DQ allele provides such a dramatic protection. At present, carrying a “protective” DQB*0602 allele is considered as an exclusion criteria for enrolling first degree relatives of diabetic patients in clinical prevention trials, such as the National Institutes of Health-sponsored, international TrialNet Pathways to Prevention Program, evolved from the Diabetes Prevention Trial (DPT-1) (55).
Non-HLA genes
Although important, the MHC susceptibility genes are not sufficient to induce T1DM, suggesting polygenic inheritance in most cases (47,57–59). Polymorphisms of non-MHC genes are reported to influence the risk of T1DM diabetes including, the insulin gene, the protein tyrosine phosphatase, non-receptor type 22 (PTPN22) gene, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), IL-2 receptor (CD25), a lectin-like gene (KIA0035), ERBB3e, and an undefined gene at chromosome 12q (47,60,61). Genome-wide association studies confirmed the above associations and identified four additional risk loci associated with an increased risk of T1DM (62). Most loci have small effects, and the variants studied are common.
The Type 1 Diabetes Genetics Consortium (T1DGC) has captured the majority of the genetic risk for T1DM (62,59). Genome-wide association studies of T1DM have identified 55 non-MHC susceptibility loci, which required more detailed mapping in order to identify their candidate genes and causal variants.
Insulin Gene
The insulin gene (INS) region on chromosome 11p15 became a premier candidate for genetic association with T1DM in the early 1980s (63–65). Insulin’s central role in metabolism and blood glucose homeostasis and its unique distinction as the only known β cell-specific antigen made it a likely frontrunner to account for an inherited susceptibility to diabetes. Julier et al. provided evidence for genetic linkage for the insulin gene (IDDM2) with T1DM in a collection of multiplex families from France, USA and North Africa (66). Subsequently, the investigations of Bain et al. have confirmed the evidence of linkage between IDDM2 and T1DM (67). They demonstrated linkage for IDDM2 independent from the influence of HLA alleles (i.e., IDDM1) and the parental source of the IDDM2 susceptibility allele.
Detailed sequence analysis of the insulin gene region identified a polymorphic locus which consists of a variable number of tandem repeats, or VNTR, present within the 5′ regulatory region (promoter) adjacent to the coding sequence of the insulin gene. Each repeat element consists of a 14- to 15-bp DNA segment having the consensus nucleotide sequence A(C/T)AGGGGT(G/C)C(T/C/G)(G/A/T) (G/T/A)G(G/C/T). The number of repeats within sequenced alleles ranges from 26 to >200, with three classes of alleles identified on the basis of overall size: class I, class II and class III. Class I INS VNTR alleles consist of 26–63 repeats, averaging 570 bp in length, and are associated with T1DM susceptibility. Class III alleles consist of 140–200 or more repeats and are considered to be protective from diabetes. In size, class III alleles are the largest variants, averaging over 2.2 kb in length. Finally, class II alleles (1.2 kb average length) are too rare in the populations studied to draw any conclusion about their association with T1DM susceptibility.
A number of studies have suggested that the INS VNTR may have a biological role in the genetic regulation of insulin expression in the thymus (68,69). The proximity of this polymorphism to the INS transcriptional start site (<400 bp upstream) makes this an attractive hypothesis.
It has previously been reported that INS mRNA levels in the thymus were correlated with VNTR alleles in opposite fashion to that observed in the pancreas (70). INS transcripts in cis with class III VNTR alleles are transcribed at much higher levels (on average 2–3 fold) than those in cis with class I VNTR alleles (71). The higher transcription levels detected in thymus could underlie the protective effect associated with class III VNTR alleles, as higher insulin levels in the thymus may more efficiently induce negative selection of insulin-specific T-lymphocytes (or improved selection of regulatory T cells). In contrast, homozygosity for diabetes-associated class I VNTR alleles determines lower insulin levels that may be associated with a less efficient deletion of insulin-specific autoreactive T-cells (or impaired selection of regulatory T cells). Proinsulin appears to be the main product of the insulin gene in the thymus (71). Direct support for the hypothesis that levels of INS expression in thymus and lymphoid organs could influence T1DM susceptibility was provided by studies in insulin gene knockout mice and transgenic mice (72,73) and more recently in humans (74).
Protein tyrosine phosphatase, non-receptor type 22 (PTPN22)
Bottini and coworkers evaluated a functional polymorphism in the lyp gene in two series of T1DM patients, one from Denver and one from Sardinia (61). The Lyp molecule, coded by the PTPN22 locus, is a lymphoid tyrosine phosphatase located on chromosome 1p13. The relevant diabetes associated polymorphism appears to be a missense mutation that changes an arginine at position 620 to a tryptophan and thereby abrogates the ability of the molecule to bind to the signaling molecule Csk (61,75,76). The lyp-Csk complex downregulates T cell receptor signaling and thus loss of this interaction is believed to enhance T cell receptor signaling, though a study by Bottini et al. indicated a gain of function with the missense mutation and inhibition of T-cell receptor signaling (76). Of note, the minor tryptophan encoded allele is associated with a series of autoimmune disorders including T1DM, rheumatoid arthritis and lupus erythematosus (76). A number of studies have confirmed the association of this missense mutation with T1DM including a large study from Great Britain. A gain of function with a missense polymorphism probably also explains why the R620W change is the most widely studied polymorphism within the PTPN22 locus clearly associated with T1DM.
Cytotoxic T-lymphocyte-associated protein-4 (CTLA-4)
A polymorphism in the CTLA-4 gene was shown to be associated with the risk of T1DM in a meta-analysis of 33 studies involving over 5000 patients (77). This chromosomal region 2q33 contains the CTLA-4 and CD28 genes, which encode for two molecules that are intimately involved in the regulation of T-cell activation and proliferation. Differential regulation of these molecules could easily affect T-cell function and hence the regulation of immune responses. The CTLA-4 gene is a strong candidate gene for autoimmune diseases since it encodes a protein that functions as a key negative regulator of T-cell activation, and the linked markers encompass a region containing an (AT)n microsatellite located in the 3′ UTR of the CTLA-4 gene. Moreover, the analysis of an A-G transition in the first exon of the CTLA-4 gene, coding for a Thr/Ala substitution in the leader peptide, also showed preferential transmission to affected siblings. Although linkage was not observed in families from Sardinia, U.K., and U.S.A., preferential transmission was observed considering all of the above families together (n= 818). Further confirmation of association with the IDDM12-CTLA-4 locus came through linkage disequilibrium analysis using a multi-ethnic collection of families with one or more affected children, which included families from Spain, France, China, Korea, and Mexican-Americans. In this study, the transmission disequilibrium test (TDT) revealed a highly significant deviation for transmission of alleles at the (AT)n microsatellite marker in the 3′ untranslated region as well as the A/G polymorphism in the first exon of the CTLA-4 gene (78).
Predictors of risk for T1DM Progression
Over the past two decades major efforts have been devoted to identify islet autoantigens which are targets of T1DM-specific autoimmune responses. An ongoing search has identified several islet autoantigens which are considered targets of immune-mediated responses. Seminal studies have suggested that using a combination of autoantibodies directed to islet autoantigens (insulin, GAD65, IA-2 and ZnT8) gives a high predictive value for T1DM progression (79).
As autoimmunity in T1DM progresses from initial activation to a chronic state, there is often a higher number of islet autoantigens reacting with T cells and autoantibodies. This condition is termed “epitope spreading”. Compelling evidence indicates that islet autoantibody responses directed to two or more of the islet autoantigens: insulin, glutamic acid decarboxylase (GAD), islet antigen 2 (IA-2), and zinc transporter 8 (ZnT8) are associated with progression to overt disease (79–81). A number of additional T1DM-related autoantigens have been identified, which include islet cell autoantigen 69 kDa (ICA69) (82,83), the islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) (84), chromogranin A (ChgA)(85), the insulin receptor, heat shock proteins, the antigens jun-B,16 (86), CD38 (81), peripherin (87) and glial fibrillary acidic protein (GFAP) (88).
During normal immune system development, lymphocytes that react to self-antigens in the thymus and bone marrow are deleted. Host molecules, in particular proteins and nucleic acids, are constantly being modified in the course of normal physiological events. A key post-translational modification in autoimmunity appears to be the citrullination of arginine amino acid residues, by the enzymatic deimination of arginine to citrulline (89,90). This reaction is catalyzed by the enzyme peptidyl arginine deiminase (PAD) (91). In multiple sclerosis and RA, citrullinated isoforms of myelin basic protein (92) and fibrin (93) have been found in the brain and synovia respectively. It must be pointed out that the detection of anti-citrullinated protein antibodies (ACPA) has proven extremely useful in the early diagnosis and assessment of prognosis in rheumatoid arthritis (RA), and has also led to insights into gene/environment effects in autoimmune diseases (94).
a. Insulin Autoantibodies
Insulin is a peptide hormone produced by the pancreatic β-cells, which has a pathological role as T1DM autoantigen. Proinsulin/Insulin is the predominant secretory product of pancreatic β cells whose autoimmune destruction leads to insulin deficiency and consequent metabolic de-compensation of glucose homeostasis (72). A high titer of insulin autoantibodies (IAA) at younger ages is consistent with the concept that these patients develop a rapid disease course. In particular, insulin autoantibodies levels greater than 2000 nU/ml are almost exclusively found in patients who progress to T1DM prior to age 5 years, and less than half of individuals developing T1DM after age 15 years carry detectable levels of IAA (95).
It is believed that T cell responses to proinsulin/insulin drive autoreactive B cell responses and insulin autoantibody formation. Investigations on the immunologically relevant regions of the insulin molecule have focused on T cell epitopes. Studies in a spontaneous model of murine autoimmune diabetes, revealed that amino acids 9–23 in the insulin B chain (termed B:9–23) and the effect of intracellular processing of molecules, such as insulin, within the β-cell can lead to formation of immunogenic epitopes (96). In human T1DM, many T cell responses have been reported to proinsulin/insulin, including insulin B:9–23), from the peripheral blood and pancreatic lymph nodes (97–99). A recent study just reported CD4+ T cells cloned from the pancreas of a T1DM patient in which 53 distinct T cell clones were identified. Approximately one in four clones responded to proinsulin epitopes restricted to HLA DQ8 and DQ8 transdimers that form in DQ8/DQ2 heterozygous individuals, again implicating proinsulin/insulin specific T cell responses in human T1DM pathogenesis (100).
b. Glutamic Acid Decarboxylase (GAD) Autoantibodies
In an early study, incubation of rat islets with radioactively labeled [35S]-methionine and subsequent immunoprecipitation of solubilized membranes with serum from newly diagnosed patients with T1DM or controls showed that an antigen with a molecular weight of 64 kDa was precipitated by sera from T1DM patients (101). Autoantibodies against the 64 kDa antigen were present in approximately 80% of newly diagnosed T1DM patients and in pre-diabetics prior to clinical disease. The nature of the 64 kDa antigen remained unknown until the report by Solimena et al showing autoantibodies to GABA-ergic neurons and pancreatic β-cells in an unusual condition termed stiff person syndrome (102). Glutamic acid decarboxylase is the enzyme which catalyzes the conversion of glutamic acid to gamma amino butyric acid (GABA), a potent inhibitory neurotransmitter. This led Baekkeskov et al. to identify GAD as the 64 kDa autoantigen in T1DM (103,104,105). Other molecular-related forms of GAD, such as the 67 kDa isoform, have subsequently been identified (106). Autoantibodies against GAD are a predictor of progression to overt diabetes. When coupled with insulin autoantibodies and islet cell antibodies (ICA), they also contribute to our ability to predict the likelihood of developing T1DM in asymptomatic first-degree relatives of T1DM patients.
c. IA-2 (ICA512) Autoantibodies
The neuroendocrine antigen IA-2 (ICA512) is another major autoantigen in T1DM (107). It is an enzymatically inactive member of the tyrosine phosphatase family, involved in regulating insulin secretion. Assessment of the presence of IA-2 autoantibodies contributes to the predictability of the likelihood of developing T1DM. As shown by Verge et al. and others, ICA512 (IA-2) and its homologue IA-2β (phogrin) are both neuroendocrine molecules (54,79,108). IA-2 is 979-amino acid protein with a single transmembrane region and with significant homology to the receptor-type PTP(RT-PTPase). An insulin granule membrane protein-tyrosine phosphatase (PTP) homologue, termed phogrin, was subsequently identified. Subcellular fractionation of insulinoma tissue showed that both IA-2 and phogrin had a very similar cellular distribution to that of insulin and carboxypeptidase H, and these two molecules are predominantly localized in the secretory granules of neuroendocrine cells (109,110).
While the main immune reactive region of the IA-2 molecule was thought to reside within its intracellular domain (amino acids 601–979), the inclusion of this region together with other reactive epitope regions of the molecule, (encompassing aa residues 256–979), has permitted the development of highly accurate constructs to assess the presence of antibodies against IA-2 (111). However, we have demonstrated that humoral autoimmunity is also directed to epitopes residing in the extracellular domain of IA-2 and antibody responses to these epitopes are associated with a more rapid T1DM development (20) (Figure 1).
Figure 1.
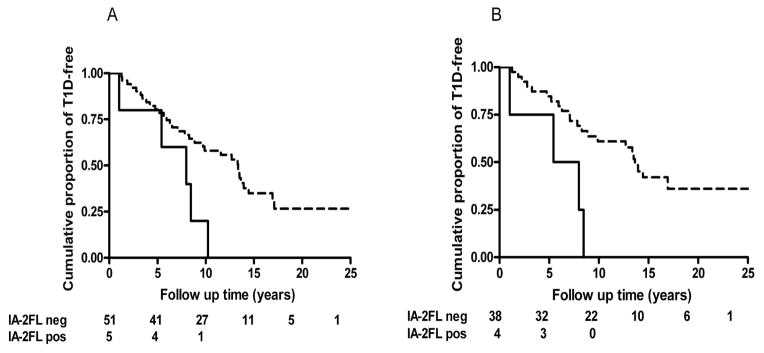
A: The rate of progression to T1DM development in relatives negative for ICA512bdc carrying GAD65 in the absence (dashed line) or presence (solid line) of autoantibodies reacting with IA-2 full length (IA-2FL) (amino acids 1–979) (log rank P = 0.016). In this subgroup of relatives, the cumulative risk of developing insulin requiring diabetes in IA-2 full length antibody positive FDR was strikingly high, being 100% by 11 years of follow-up in GAD65 antibody positives (log rank P = 0.026).
B: This effect was also observed in FDR selected for being negative for ICA512bdc having at least 2 islet autoantibodies in the absence (dashed line) or presence (solid line) of autoantibodies reacting with IA-2 full length (IA-2FL) (amino acids 1–979) (log rank, P = 0.003). In these relatives the cumulative risk of progressing to overt diabetes was 100% by 10 years of follow-up (log rank P = 0.022) (20). (With permission from Endocrinology)
d. Zinc transporter family member 8 (ZnT8) and Other Autoantibodies
Zinc transporter family member 8 (ZnT8) is a member of the cation diffusion facilitator family, with abundant expression in pancreatic β-cells, although it is also expressed in extra-pancreatic tissues (112,113). In the β-cell it plays an important physiological role since zinc, which is highly concentrated in β-cells, is needed for normal insulin storage. β-cell-specific deletion of ZnT8 in mice results in glucose-intolerance, reduced β-cell zinc accumulation and anomalous insulin granules, as well as blunted first-phase glucose-stimulated insulin secretion, reduced insulin processing enzyme transcripts and increased proinsulin levels (114). Its relevance as an important T1DM autoantigen was first described by Wenzlau and coworkers (80), following an evaluation of 68 candidate islet autoantigens compiled from multidimensional analyses of microarray mRNA expression profiling. The assessment of ZnT8 (the gene Slc30A8 encodes ZnT8), indicated that it was targeted by autoantibodies in 60–80% of new-onset T1DM compared with <2% of healthy controls, <3% type 2 diabetic patients and in up to 30% of patients with other T1DM-associated autoimmune pathologies. Interestingly, ZnT8 antibodies were found in 26% of T1DM subjects who had not exhibited antibody positivity to other commonly measured autoantigens such as GAD, (IA-2), insulin, or in the assay for cytoplasmic islet cell antibodies (ICA). Further research has revealed polymorphisms in ZnT8 that are relevant to its role as a major T1DM autoantigen. There are three polymorphic variants located in the intracellular (C-terminus) domain of the transporter protein, namely Arg 325, Trp325 and Gln 325. Of these variants, Trp325 (W) and Arg325 (R) have been shown to be the major autoantigenic polymorphisms in T1DM and use of a construct containing the W and R variants (ZnT8WR) (115) has proven its efficacy as a screen for T1DM associated humoral autoimmunity. More recently, a chimeric construct containing amino acid residues 609–979 of the intracellular domain of IA-2, linked to peptides containing both ZnT8 W and R polymorphisms has been successfully developed and tested as a broader and more economical screen to detect patients exhibiting humoral autoimmunity against IA-2 and/or ZnT8 (116).
Processing of molecules such as insulin within the β-cell generates peptides that are then taken up by APCs either as whole dead β-cells or specifically granules of β-cells for eventual further processing/presentation of islet peptides to self-reactive T cells (96). Furthermore, Stadinski et al. have shown that chromogranin A (ChgA) is an autoantigen in T1DM, and that the peptide WE14 from ChgA stimulates diabetogenic CD4+ T cell clones (85). More recently, the same group provided evidence to suggest that WE14 is recognized by T cells from diabetic subjects vs. controls in a dose dependent manner (117). The natural form of the antigen in β cell extracts is far more potent than an unmodified synthetic WE14 peptide, suggesting that this peptide may be post-translationally modified with a carbonyl group in pancreatic islets.
Prediction based on autoantibody characteristics and their combination with metabolic markers
There is a wealth of studies demonstrating that a combination of islet autoantibody biomarkers gives a high predictive value for T1DM progression, and great sensitivity without significant loss of specificity (21,54,79,108). Moreover, combining both immunologic and metabolic strategies (e.g. oral glucose tolerance test or the first phase (1 + 3 min.) insulin response of an intravenous glucose tolerance), the current opinion is that type 1 diabetes progression can be predicted with approximately 80% accuracy within 10-years (118).
A study on the Diabetes Autoimmunity Study in the Young (DAISY) cohort showed that 89% of children who progressed to T1DM had two or more islet-related autoantibodies (119). Age of diagnosis of diabetes was strongly correlated with age of appearance of first autoantibody and IAA levels. By life-table analysis, children exhibiting two or more autoantibodies showed a nearly linear progression to diabetes (P < 0.0001). Children with persistently positive IAA levels had a higher progression rate to overt T1DM (nearly 100% by 5.6 years) as compared to children with fluctuating IAA levels (63% by the 10-year follow-up) (P < 0.0001). In children enrolled in the DAISY study followed to the development of diabetes onset, only high IAA titer correlated with rapid progression to T1DM onset (P < 0.0001). As a matter of fact, this effect was not evident with respect to the presence of high GAD65 or IA-2/ICA512 autoantibody titer. Therefore, insulin autoantibody levels at the time of diagnosis are inversely related to the age of the patient being highest in those less than 5 years of age and hence, IAA appear to be an early marker of β-cell destruction. The titer of IAA along with the insulin secretory response judged by the first phase insulin levels at 1 and 3 minutes after an intravenous glucose challenge, has also been successfully employed to construct mathematical models to predict likelihood of clinical diabetes in asymptomatic first-degree relatives of patients (120). Investigators from the DAISY study reported that 5 children were found to have persistent IAA before 1 year of age, and 4 of them went on to develop the clinical onset of T1DM (all before 3.5 years of age). In contrast, children not exhibiting persistent IAA before the age of 1 year, rarely rapidly developed T1DM. When analyzing only children followed from birth, that progressed to clinical diabetes, the two major predictors of diabetes diagnosis were the age at which autoantibodies first appeared and the mean level of insulin autoantibodies. These observations emphasize the utility of IAA, particularly in younger populations and justify the need to design trials focused on such a young group. However, the success of these strategies depends on the safety and effectiveness of therapeutic regimens. Indeed, this was the strategy in the trial to prevent development of T1DM (DPT-1), which successfully predicted the development of diabetes in first degree relatives of T1DM patients (121).
A seminal study showed that progression to T1DM in children with multiple islet autoantibodies was faster for those children who had islet autoantibody seroconversion younger than age 3 years (21). We have demonstrated that autoantibodies against epitopes localized within the extracellular domain of IA-2 are associated with accelerated progression to T1DM (Figure 1) (20).
We provided evidence to suggest that a subset of cytoplasmic islet cell antibodies (ICA) is related to a more rapid progression to insulin-requiring diabetes in GAD65 and IA-2 antibody positive relatives of T1DM patients, compared to relatives with GAD65 and IA-2 antibodies without ICA (122). The precise nature of the antigen(s) detected a subset of ICA remains to be elucidated; Both our observation and an additional study indicate that other yet unidentified autoantigen(s) may be reactive to the pool of immunoglobulins of ICA (122,123). In conclusion, measurement of serum antibodies directed against proteins in β cells (insulin, GAD, IA-2 and ZnT8) can effectively assess T1DM risk, and further research is warranted to correlate autoantibody profiles as well as mechanistic biomarkers with progression to clinical T1DM in at risk individuals.
There is evidence indicating that antigen specificity–related heterogeneity during the initiation of β-cell autoimmunity. For example, IAA are considered the first autoantibodies that occur during the natural history of the disease. They appear at a very young age and are associated with the DR4-DQ8 haplotype, whereas GAD autoantibodies appear at a later age and are associated with the DR3/DQ2 haplotype. This notion might be useful in the search for the environmental triggers and strategies to prevent the disease (124).
Challenges and Hope for the Prevention of Type 1 diabetes
Several immunomodulatory, immunosuppressive agents and other drugs have been tested alone or in combination in an effort to halt the immune-mediated destruction of β cells that occurs in T1DM and are reviewed below. The majority of the studies have been performed in newly diagnosed T1DM and, in some studies, in patients with established T1DM (125–129). In both situations most of the β cell function has already been lost, and the anticipated outcome is preservation of remaining β cell function, usually measured as area under the insulin secretion curve in response to stimulation (125). Furthermore, large scale prevention trials have been conducted using several preparations of insulin in an attempt to delay and prevent the onset of T1DM in an at risk setting (130). Several ongoing prevention trials include the use of oral insulin, an anti-CD3 monoclonal antibody or abatacept (CTLA4-Ig) in secondary prevention trials (NCT 00419562, 01122446, 01773707). While intervention in first degree relatives of T1DM patients, is entirely justified, the time for implementation of a large-scale screening in the general population is approaching. Increasing evidence suggests that T1DM risk should be evaluated in the general population which includes: a dramatically increasing incidence (131–134), approximately 85% of T1DM cases without family history, life-threatening diabetic ketoacidosis at onset (134) and improved islet autoantibody assays (135–137). Although there are still a number of barriers to overcome for general population screening (cost, repeated assessments, and integration into clinical practice), an effective method for delaying or preventing clinical T1DM onset would greatly aid in screening the general population for islet autoantibodies.
A number of critical issues should be considered for the prevention of T1DM. It must be emphasized that T1DM can be managed with the use of exogenous insulin therapy and monitoring of blood glucose levels, which is in contrast to life threatening conditions such as cancer or severe systemic autoimmune diseases. Multiple daily insulin injections and self-monitoring blood glucose, although imperfect and tedious, can delay diabetic complications, but this is not a cure for T1DM. The hope is that one day a new safe therapeutic strategy prevents or at least significantly delays overt T1DM. Major challenges include what prevention strategies could be used to alter the disease process, when to intervene, the dosing, the duration of treatment, and what end points should be considered. Given that clinical prevention trials require a very large number of subjects, are costly and time consuming, it is imperative to that the selected agents are safe, ethically acceptable, and offer the promise of efficacy.
Prevention strategies are defined as primary, secondary or tertiary. Primary prevention is conducted in asymptomatic individuals prior to T1DM onset (i.e. those with high genetic risk). Typical examples of primary prevention measures are regular physical activity to reduce the risk of developing cardiovascular disease and vaccination against infectious diseases. If obesity or insulin resistance are risk factors for T1DM (138), population strategies could also be applicable to the prevention of T1DM. Ideally the burden of T1DM and other autoimmune diseases would be reduced by primary prevention aimed at the environmental factors or other risk factors thought to precipitate disease in genetically at risk individuals.
Secondary prevention attempts to identify a disease at its earliest stage so that prompt and appropriate management can be initiated. Successful secondary prevention reduces the impact of the disease. For T1DM, secondary prevention occurs in individuals with multiple islet autoantibodies. Appropriate pharmacologic treatment in the earliest stage of T1DM could help prevent later stages of the disease. This is the case for rheumatoid arthritis in which at early diagnosis (within 6 months of symptom onset), followed by the use of disease-modifying antirheumatic drugs (DMARDs) has been shown to provide the most effective disease control and joint preservation (139).
Tertiary prevention focuses on reducing or minimizing the consequences of a disease once it has developed. The goal of tertiary prevention is to eliminate, or at least delay, the onset of complications and disability due to the disease. In medicine most medical interventions fall into this category. A classic example refers to subjects with diabetes keeping their blood glucose under tight control in an effort to prevent diabetic complications.
Examples of attempts to primary prevention in T1DM include the Trial to Reduce Insulin-Dependent Diabetes Mellitus in the Genetically at Risk (TRIGR), which is a prevention study in infants carrying high-risk HLA alleles and who have a first-degree relative with T1DM (140). The pilot study included 230 children and showed that avoidance of foods containing bovine protein in the first 6–8 months of life and supplementation of breast milk with highly hydrolyzed milk formula (Nutramigen) as opposed to a cow’s milk-based formula decreased the risk of developing diabetes related autoantibodies by approximately 50%. The mechanism(s) whereby elemental formula may reduce T1DM risk remains to be elucidated. Unfortunately, after seven years of follow-up, this trial with 2159 randomized children showed that hydrolyzed formula failed to reduce the incidence of islet autoantibodies in the participants compared to those that received conventional formula. The final outcome of TRIGR is clinical T1DM and that endpoint will be reached in 2017 (141).
Similar to the TRIGR study, the BABYDIET study examined the impact of timing of nutritional exposure to islet cell autoimmunity. The hypothesis that delaying exposure to dietary gluten reduces the risk of islet cell autoimmunity was supported by mouse models and epidemiologic human data. In this pilot study of 150 infants with a first-degree relative with T1D, delaying gluten exposure until 12 months of age resulted in no reduction in islet autoimmunity (142). However, the study was not powered for efficacy, and compliance was problematic with this open label study design.
Vitamin D as an immuno-modulator has received attention as a primary intervention strategy in T1DM. Based on epidemiological studies, vitamin D deficiency seems to be associated with TIDM as well as other autoimmune diseases. In an analysis from a Norwegian study, vitamin D deficiency during pregnancy increased the offspring’s risk of developing TIDM, whereas supplementing with vitamin D during pregnancy was associated with a 50% decrease in the rate of autoantibody development in the offspring (143). A Finnish study showed that supplementing vitamin D in the first year of life was associated with a decreased risk of developing T1DM (144). Although intriguing, these studies hinge upon associations and, to the best of our knowledge, are not substantiated by solid mechanistic data, at least in humans.
For those at risk individuals with 2 or more islet autoantibodies, with or without evidence of β-cell dysfunction, a number of intervention strategies have been utilized in an attempt to prevent progression to T1DM (secondary prevention). The Diabetes Prevention Trial-1 (DPT-1) was a randomized, controlled, non-blinded North American study to determine if the administration of insulin prior to the onset of T1DM would prevent or delay progression to overt disease in subjects with islet cell antibodies (ICA). Neither parenteral nor oral insulin had effect on preventing or delaying T1DM onset (145,146). However, in a posthoc analysis, subjects receiving oral insulin who had higher baseline insulin autoantibody titers appeared to have a statistically significant delay in the onset of T1DM by 4.5 year follow up (147). Furthermore, the rate of progression seemed to increase when oral insulin therapy was stopped, suggesting that the therapy was probably effective but required on-going administration (148). This analysis has prompted a larger and justified follow-up study with oral insulin to confirm these preliminary studies (NCT00419562).
Anti-inflammatory agents have also been explored as a secondary prevention therapy for T1DM. As a result of animal and small human pilot studies, the European Nicotinamide Diabetes Intervention Trial (ENDIT) evaluated the effects of nicotinamide, a free radical scavenger, as a secondary prevention strategy for T1DM development. There was no difference in the percentage of subjects who progressed to T1DM between the placebo and treatment groups (149). One interpretation of these results was the dose being too low or administered too late in the disease process.
It is noteworthy to emphasize that earlier intervention may result in greater efficacy in preventing T1DM. This has prompted the use of immunomdulatory interventions in as T1DM preventing agents. A monoclonal antibody directed against CD3 on T cells that affects T cell signaling is now being studied in the highest-risk members of the prevention group (multiple autoantibodies and impaired glucose tolerance) (125,150). The abatacept prevention trial has been launched for similar reasons. Abatacept (also called CTLA4-Ig) is a soluble fusion protein comprising cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and the Fc portion of IgG1. This drug prevents CD28 from binding to its counter-receptor, CD80/CD86, due to its higher affinity for CD80/CD86. Administration of CTLA4-Ig or successful transfer of its gene prevents full T cell activation (151). It is noteworthy acknowledging that costimulation modulation with abatacept slowed decline of β-cell function and improved HbA1c in recent-onset T1D. The beneficial effect was sustained for at least 1 year after cessation of abatacept infusions or 3 years from T1DM diagnosis (152).
One of the increasingly recognized challenges in T1DM prevention is the heterogeneity of the disease. Variations in clinical characteristics at diagnosis and afterwards may reflect important pathogenic differences. For example, age at diagnosis affects the risk of concordance for T1DM among monozygotic twins of patients (153) and, as noted above, the interpretation of insulin autoantibodies on disease risk. Children who are obese or overweight at the time of T1DM diagnosis have greater β-cell function (154). Residual β-cell function at onset define phenotypically distinct types of T1DM that likely have etiological and pathogenic differences (31,155). It is possible that non-immunological genetic or environmental factors may also play a role in progression to T1DM in defined subgroups of patients; for example, we recently reported that children who express a single islet autoantibody at the onset of T1DM are more likely to carry a diabetogenic polymorphism of the transcription factor 7 like 2 (TCF7L2) gene, which is the strongest risk factor for type 2 diabetes but has not been associated with T1DM (156). Understanding the heterogeneous factors that contribute to T1DM and designing strategies that address those differences is critical to our success in T1DM prevention.
New Treatments for Type 1 Diabetes: What is on the Horizon?
Although great strides have been made in the development of agents to treat T1DM, with many therapies currently under investigation, the time is ripe for the beginning of a new phase of treatment strategies. A critical question is whether or not animal models of autoimmune diabetes tell us to intervene in humans based on the results obtained in these models. There are examples in both biobreeding (BB) rats and nonobese diabetic (NOD) mice that have provided good insight into the natural history of the disease, although an effect in delaying or preventing the disease in these models can be achieved by a large number of interventions (157). In these animal models, immunologic therapies usually succeed if administered prior to the appearance of islet autoimmunity and less frequently after disease onset.
A commonly cited concern is the tendency to rapidly transition from the NOD model into human disease. It is prudent to test the efficacy and the safety of interventions in several animal models and human ex vivo cells prior to humans, especially as some of these therapies could theoretically accelerate the disease. Numerous therapeutic strategies have been applied in both NOD mice and BB rat and listing these interventions is beyond the scope of this article. Any intervention that is promising and reproducible in animal models of diabetes, regardless of the disadvantages of these models, should be provided serious consideration in terms of understanding the mechanisms involved in the disease process, safety, cost and feasibility for a preventative therapy in humans. To date NOD mouse model of spontaneous autoimmune diabetes provides the most homology to human disease in that both are T cell mediated diseases resulting in insulitis, insulin autoantibodies appear in both, and MHC class II alleles confer significant disease risk (murine IA is homologous to human DQ). Furthermore, it provides a model of spontaneous autoimmune diabetes to test specific scientific hypotheses and perform mechanistic studies from target tissues (thymus, pancreatic lymph nodes and islets). These studies must continue to flourish, including careful evaluation of safety, before moving to human investigation.
At the current time, T1DM is predictable with the measurement of islet autoantibodies but not yet preventable (158). The only Food and Drug Administration approved treatments for T1DM are insulin preparations and pramlintide, which is infrequently used in treatment regimens. It is our belief that safe and specific therapies are needed to delay, prevent and ultimately cure T1DM.
A growing number of intervention strategies have targeted key components of the autoimmune response associated with T1DM. Regulatory T cells (Tregs) play a major role in controlling effector T cells (Teffs) responding to self-antigens associated to autoimmune diseases (159). An altered Treg/Teff balance contributes to most autoimmune diseases, including T1DM. To restore a proper balance, attempts have been made blocking Teffs with immunosuppressants, but some of these agents were partially effective and had adverse effects. It appears that expanding/activating Tregs with low-dose interleukin-2 (IL-2) could provide immunoregulation rather than immunosuppression and yet prevent the progression to diabetes in the NOD mouse model (160). A clinical study of low dose IL-2 showed seems to have a good safety profile and Treg expansion/activation in T1DM patients (161). This initial study paves the way for new clinical trials to test low-dose IL-2 in the prevention of T1DM, such as a multicenter European phase II-b clinical trial using ultra-low dosing of IL-2, the DIABil-2 trial.
Sotagliflozin (LX4211) is an orally-delivered small molecule compound that inhibits both sodium-glucose cotransporter type 2, or SGLT2, a transporter responsible for most of the glucose reabsorption performed by the kidney, and sodium-glucose cotransporter type 1, or SGLT1, a transporter responsible for glucose and galactose absorption in the gastrointestinal tract, and to a lesser extent than SGLT2, glucose reabsorption in the kidney (162). Thus far, the clinical efficacy of sotagliflozin has been encouraging, suggesting robust HbA1c reduction, potential synergy with DPP4 inhibitors, possible efficacy in type 2 diabetes with renal impairment and the opportunity to improve the overall benefit/risk of treating T1DM. Larger and longer term studies are warranted in T1DM.
Gene and cell therapy strategies as well as differentiated derivatives of stem cells are being used as reagents to test for drugs that may have an impact in delaying clinical T1DM, or correct disease phenotypes found in several degenerative disorders (163–167).
Additional investigations and therapies have targeted the critical components in T1DM pathogenesis depicted in Figure 2 and 3, self-reactive CD4 T cell receptor– self antigen – HLA class II molecule, termed the trimolecular complex (168–170). Specifically, targeting autoreactive T cells directed against insulin presented by HLA DQ8 was championed by the late George Eisenbarth in his 2009 Banting Lecture (171). As previously mentioned, specific HLA molecules confer significant disease risk with 50–60% of all T1DM patients having the HLA-DQ8 (DQB*03:02) allele (172). Proinsulin/insulin peptides are self-antigens that lead to T cell responses and promote autoantibody production. Targeting the highest risk genetic components (HLA molecules and insulin) for T1DM is a rational approach. There are a number of therapeutic modalities in preclinical testing that target one or more of the components of the anti-insulin trimolecular complex.
Figure 2.

Schematic representation of initiation of the immunologic response to an autoantigen. The antigen binds to a groove in MHC class II molecules on antigen-presenting cells (APCs). This binding allows the antigen to be presented to antigen receptors on autoreactive CD4 inducer or helper T cells which, in T1DM, mediate immune-mediated injury to the pancreatic β cells. Furthermore, the respective binding of B7 proteins and lymphocyte functional antigen-3 (LFA-3) on APCs to CD28 and CD2 on T cells are important costimulatory pathways that further enhance T cell activation. Other molecules can also participate in the immune response, such as the binding of interleukin-2 to its receptor (IL-2R). Both regulatory (Treg) and effector T lymphocytes are produced and their balance is crucial for maintaining tolerance (183).
Figure 3.
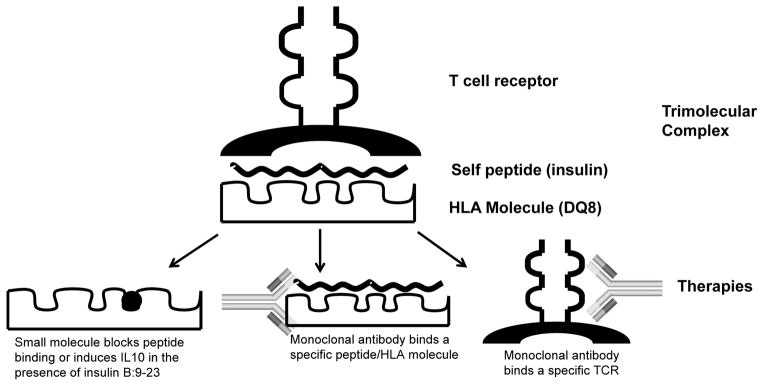
The trimolecular complex and therapies targeting each component. The trimolecular complex consists of a T cell receptor, self-peptide, and HLA class II molecule. Potential therapies include: (a) a small molecule (black circle) occupies a pocket in the HLA peptide binding groove thereby blocking peptide presentation and subsequent T cell activation or a small molecule induces a protective immune response (IL10 production), (b) a monoclonal antibody can bind a specific peptide/MHC complex to block T cell activation, or (c) a monoclonal antibody targets a specific T cell receptor α or β chain to deplete pathogenic T cells.
One such approach targets HLA DQ8 in T1DM with small ‘drug-like’ molecules to block peptide presentation to T cells. Using a supercomputer to perform in silico molecular modeling and docking, drug candidates were scored on their ability to bind to one of four pockets in the peptide binding groove of the HLA molecule (173). In vitro and subsequent preclinical studies in the NOD mouse have shown this to be a potential pathway for immune intervention in diabetes. This pathway has now been translated from bench to bedside utilizing a clinically well-established medication, methyldopa (Aldomet) to block DQ8 antigen presentation. A current phase 1b dose escalation trial is underway as methyldopa is being administered to recent onset adult T1D patients with residual endogenous insulin production and the presence of the DQ8 gene (NCT01883804). This approach and trial uses personalized medicine, as the inclusion criteria requires the presence of the high risk diabetes gene, DQ8. Methyldopa is administered orally, safe as it has been used clinically for the last 50 years, and currently indicated for the treatment of pregnancy induced hypertension. With all therapies, safety is a concern; however, all individuals have three class II molecules (termed DQ, DR, and DP), and by blocking a single class II molecule, there are two others that can function to permit normal immune system processes.
Another approach that stemmed from this line of investigation is the use of a small drug molecule in the presence of an autoantigen peptide to induce protective immune responses. Glyphosine, an immune stimulatory molecule, induces the anti-inflammatory and regulatory T cell cytokine, IL10, in the presence of the insulin B:9–23 peptide (173). The compound was able to completely prevent diabetes while administered to NOD mice and induced IL10 production from DQ8 positive T1DM peripheral blood mononuclear cells at nanomolar concentration. This approach represents an intriguing possibility to improve antigen specific therapy and is currently undergoing preclinical drug development.
A second therapeutic strategy stemming from the anti-insulin trimolecular complex is using insulin as an antigen specific therapy. As previously mentioned in the prevention of T1DM section, there have been many large scale trials evaluating insulin preparation for disease prevention, unfortunately with limited success. Recent understanding of how insulin peptides are presented by diabetogenic MHC class II molecules to stimulate T cells offers new hope to insulin antigen specific therapies. Insulin B chain amino acids 9–23 (B:9–23) is critical for the development of NOD diabetes (72) and insulin B:9–23 is presented by both murine and human diabetogenic MHC class II molecules in an unfavorable position or “register” of binding (72,174,175). A modified insulin B:9–23 peptide (mutating B22 arginine to glutamic acid) is capable of detecting robust inflammatory T cell responses from the peripheral blood of new-onset T1D patients at a much greater frequency than the native insulin sequence (175). Strikingly, healthy control subjects also responded to the mutated insulin peptide but predominantly made an IL10 response dependent upon their DQ genotype indicating that T1DM risk may be related to how DQ molecules influence the balance of T cell inflammatory and regulatory responses to insulin (175). Furthermore the insulin mimotope stimulates T cells about 100 fold better than the wild type peptide and has been successfully used to prevent diabetes onset in the NOD mouse. Using low dose continuous administration of the mimotope, but not wild type peptide, naïve T cells were converted into regulatory T cells leading to diabetes prevention (176). Using this strong T cell agonist holds promise as a tolerogenic vaccine in those at risk for T1DM.
Another potential antigen specific therapy is a monoclonal antibody that specifically blocks the insulin/MHC complex where insulin is presented in a functional register to activate T cells. Specific monoclonal antibodies were developed by immunizing NOD mice with recombinant protein, insulin B:9–23 complexed to the murine MHC class II molecule, IAg7, where the peptide was in a functional register needed to activate autoreactive T cells (177,178). A specific monoclonal antibody, named mAb287, blocked insulin specific responses in vitro but not those of other specificities presented by IAg7. Administration of mAb287 to pre-diabetic NOD significantly reduced the severity of insulitis and delayed diabetes onset (179). Interestingly, treatment with mAb287 also blocked islet infiltrating cells with other specificities such as chromogranin A reactive CD4 T cells and islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) responding CD8 T cells (Figure 4). These results suggest that targeting insulin/MHC class II complexes may be sufficient to alter the disease course. Translation of these findings to human disease with a monoclonal antibody targeting insulin B:9–23/DQ8 is an area of active research.
Figure 4.

Monoclonal antibody mAb287 delays diabetes and islet lymphocyte infiltration in NOD mice. (A) Groups of 4-wk-old female NOD mice were treated weekly with PBS (n = 18; black squares), 0.1 mg of mouse IgG1 (n = 18; blue diamonds), 0.1 mg of mAb287 (n = 15; green circles), or 0.5 mg of mAb287 (n = 18; red triangles), and followed up to 30 wk. The percentages of diabetes free mice are shown. (B–D) Groups of eight mice were treated weekly from age 4–11 wk with 0.5 mg of control IgG1 (red bars) or mAb287 (blue bars) at which time pancreatic islets were pooled in each group. (B) The average number of live CD4 T cells, CD8 T cells, and 220+ B cells per pancreas. (C) The percentages of CD4 T cells specifically binding the IAg7–R3:RE, IAg7-R3:RGE or IAg7-chromogranin A (ChgA) tetramers, or CD8 T cells specifically binding the Kd-IGRP tetramer. (D) Average number of islet-infiltrating tetramer-positive cells per pancreas were calculated from the data in B and C (179) (With permission from the Proceedings of the National Academy of Sciences).
The final component of the trimolecular complex and target for therapies is the CD4 T cell and its T cell receptor (TCR). Most TCRs interact with peptide/MHC through six complementarity determining regions (CDR) with the α and β chains having three each. In the NOD mouse, it is appreciated that a specific V α gene family (TRAV-5) is utilized to recognize insulin with approximately 70% of TCRs responding to the insulin B:9–23 peptide using a single α chain, TRAV5D-4 (180). These TCRs are able to induce insulin autoantibodies, insulitis, and diabetes when the specific α chain is introduced into an α-chain only retrogenic animal model (181). In addition to the NOD mouse, rat models of diabetes also use predominant T cell receptors with the BB rat having a germline encoded TCR, Vβ 13, predisposing diabetes risk. An anti-rat V β 13 depleting monoclonal antibody is able to prevent diabetes in induced and spontaneous autoimmune diabetes rat models, while a specific anti-Vβ 16 antibody did not change disease frequency (182) (Figure 3c). These studies indicate that is possible to prevent diabetes by targeting a limited segment of the T cell repertoire. Active research is underway to understand human autoreactive TCR usage, especially for insulin specific T cells.
In conclusion, advancements in the understanding of T1DM pathogenesis and prediction have been profound over the last two decades. Type 1 diabetes is now predictable with the measurement of islet autoantibodies and prevention should naturally follow. Our view over the horizon is targeting the disease specific trimolecular complex, especially those focused on proinsulin/insulin. The individual components of the trimolecular complex provide defined targets for diabetes intervention. Tailoring therapies to specific HLA alleles, autoantigen, and autoreactive T cell receptor is personalized medicine which we believe will delay disease onset and ultimately prevent T1DM. We envisage other modalities evolving towards, such as expanding Tregs, orally-delivered small molecule compounds, differentiated derivatives of stem cells, that in combination with new drugs enhancing β cell function will aid in finding a cure for Type 1 diabetes.
Acknowledgments
This work was supported by the National Institutes of Health (Grant Number: R01 DK53456 and R01 DK56200 to MP, K08DK095995 to AM) and by the Juvenile Diabetes Research Foundation to AM and MP. We greatly acknowledge the McNair Medical Institute for its support.
Abbreviations
- AIRE
Autoimmune Regulator
- APS I
Autoimmune Polyendocrine Syndrome I
- CTLA4
Cytotoxic T-lymphocyte-associated Protein 4
- IPEX
Immunodysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome
- IDDM1
Major type 1 diabetes gene within HLA class II
- IDDM2
Major type 1 diabetes gene, the insulin gene
- IGRP
Islet-specific Glucose-6-phosphatase Catalytic Subunit-related Protein
- MYD88
Myeloid differentiation primary response gene 88
- PTPN22
Protein tyrosine phosphatase, non-receptor type 22
- STAT3
Signal Transducer and Activator of Transcription 3
- ZnT8
Zinc transporter family member 8
References
- 1.Eisenbarth GS. Type I diabetes mellitus: a chronic autoimmune disease. N Engl J Med. 1986:1360–1368. doi: 10.1056/NEJM198605223142106. [DOI] [PubMed] [Google Scholar]
- 2.Todd JA, Bell JI, McDevitt HO. HLA-DQβ gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- 3.Morel PA, Dorman JS, Todd JA, McDevitt HO, Trucco M. Aspartic acid at position 57 of the HLA-DQ beta chain protects against Type I diabetes: a family study. Proc Natl Acad Sci (USA) 1988:8111–8115. doi: 10.1073/pnas.85.21.8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lonnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, Savola K, Muona P, Simell T, Koskela P, Hyoty H. Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes. 2000;8:1314–1318. doi: 10.2337/diabetes.49.8.1314. [DOI] [PubMed] [Google Scholar]
- 5.Orchard TJ, Forman JS, LaPorte RE, Ferrell RE, Drash AL. Host and environmental interactions in diabetes mellitus. J Chron Dis. 1986:979–999. doi: 10.1016/0021-9681(86)90135-9. [DOI] [PubMed] [Google Scholar]
- 6.Zipris D, Lien E, Nair A, Xie JX, Greiner DL, Mordes JP, Rossini AA. TLR9-signaling pathways are involved in Kilham rat virus-induced autoimmune diabetes in the biobreeding diabetes-resistant rat. J Immunol. 2007;2:693–701. doi: 10.4049/jimmunol.178.2.693. [DOI] [PubMed] [Google Scholar]
- 7.Hyoty H. Enterovirus infections and type 1 diabetes. Ann Med. 2002;3:138–147. [PubMed] [Google Scholar]
- 8.Jaberi-Douraki M, Schnell S, Pietropaolo M, Khadra A. Unraveling the contribution of pancreatic beta-cell suicide in autoimmune type 1 diabetes. J Theor Biol. 2014 doi: 10.1016/j.jtbi.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. N Engl J Med. 2004;20:2068–2079. doi: 10.1056/NEJMra030158. [DOI] [PubMed] [Google Scholar]
- 10.Pitkänen J, Peterson P. Autoimmune regulator: from loss of function to autoimmunity. Genes Immun. 2003;1:12–21. doi: 10.1038/sj.gene.6363929. [DOI] [PubMed] [Google Scholar]
- 11.Husebye ES, Anderson MS. Autoimmune polyendocrine syndromes: clues to type 1 diabetes pathogenesis. Immunity. 2010;4:479–487. doi: 10.1016/j.immuni.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;5609:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 13.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;5899:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 14.Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Lango AH, De FE, McDonald TJ, Rajala H, Ramelius A, Barton J, Heiskanen K, Heiskanen-Kosma T, Kajosaari M, Murphy NP, Milenkovic T, Seppanen M, Lernmark Å, Mustjoki S, Otonkoski T, Kere J, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;8:812–814. doi: 10.1038/ng.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet. 1974:1279–1282. doi: 10.1016/s0140-6736(74)90140-8. [DOI] [PubMed] [Google Scholar]
- 16.Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science. 1983:1337–1339. doi: 10.1126/science.6362005. [DOI] [PubMed] [Google Scholar]
- 17.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;9277:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 18.Pietropaolo M, Eisenbarth GS. Autoantibodies in human diabetes. Curr Dir Autoimmun. 2001:252–282. doi: 10.1159/000060541. [DOI] [PubMed] [Google Scholar]
- 19.Bonifacio E, Yu L, Williams AK, Eisenbarth GS, Bingley PJ, Marcovina SM, Adler K, Ziegler AG, Mueller PW, Schatz DA, Krischer JP, Steffes MW, Akolkar B. Harmonization of glutamic acid decarboxylase and islet antigen-2 autoantibody assays for national institute of diabetes and digestive and kidney diseases consortia. J Clin Endocrinol Metab. 2010;7:3360–3367. doi: 10.1210/jc.2010-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morran MP, Casu A, Arena VC, Pietropaolo S, Zhang YJ, Satin LS, Nelson P, Omenn GS, Trucco M, Becker DJ, Pietropaolo M. Humoral autoimmunity against the extracellular domain of the neuroendocrine autoantigen IA-2 heightens the risk of type 1 diabetes. Endocrinology. 2010;6:2528–2537. doi: 10.1210/en.2009-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, Bonifacio E, Eisenbarth GS. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA. 2013;23:2473–2479. doi: 10.1001/jama.2013.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LaPorte RE, Matsushima M, Chang Y-F. 199519953746.4646. [Google Scholar]
- 23.Parkkola A, Härkönen T, Ryhänen S, Ilonen J, Knip M the Finnish Pediatric Diabetes Register. Extended family history of type 1 diabetes and phenotype and genotype of newly diagnosed children. Diabetes Care. 2013;36:348–354. doi: 10.2337/dc12-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redondo MJ, Eisenbarth GS. Genetic control of autoimmunity in Type I diabetes and associated disorders. Diabetologia. 2002:605–622. doi: 10.1007/s00125-002-0781-1. [DOI] [PubMed] [Google Scholar]
- 25.Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, Stengard J, Kesaniemi YA. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia. 1992:1060–1067. doi: 10.1007/BF02221682. [DOI] [PubMed] [Google Scholar]
- 26.Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med. 2008;26:2849–2850. doi: 10.1056/NEJMc0805398. [DOI] [PubMed] [Google Scholar]
- 27.Gale EA, Bingley PJ, Eisenbarth GS, Redondo MJ, Kyvik KO, Petersen JS. Reanalysis of twin studies suggests that diabetes is mainly genetic. BMJ. 2001;7319:997–998. [PMC free article] [PubMed] [Google Scholar]
- 28.Redondo MJ, Fain PR, Krischer JP, Yu L, Cuthbertson D, Winter WE, Eisenbarth GS. Expression of beta-cell autoimmunity does not differ between potential dizygotic twins and siblings of patients with type 1 diabetes. J Autoimmun. 2004;3:275–279. doi: 10.1016/j.jaut.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Aly TA, Ide A, Jahromi MM, Barker JM, Fernando MS, Babu SR, Yu L, Miao D, Erlich HA, Fain PR, Barriga KJ, Norris JM, Rewers MJ, Eisenbarth GS. Extreme genetic risk for type 1A diabetes. Proc Natl Acad Sci U S A. 2006;38:14074–14079. doi: 10.1073/pnas.0606349103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner A. Up Hill and Down Dale in the Genomic Landscape: The Odd Distribution of Matching Segments. J Genet Geneal. 2010;1:4. [Google Scholar]
- 31.Elding Larsson H, Larsson C, Lernmark Å. Baseline heterogeneity in glucose metabolism marks the risk for type 1 diabetes and complicates secondary prevention. Acta Diabetol. 2014 Nov 8; doi: 10.1007/s00592-014-0680-1. [DOI] [PubMed] [Google Scholar]
- 32.Althof N, Whitton JL. Coxsackievirus B3 infects the bone marrow and diminishes the restorative capacity of erythroid and lymphoid progenitors. J Virol. 2013;5:2823–2834. doi: 10.1128/JVI.03004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binder D, Fehr J, Hengartner H, Zinkernagel RM. Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J Exp Med. 1997;3:517–530. doi: 10.1084/jem.185.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexander WJ, Holmes JR, Shaw JF, Riley WE, Roper WL. Norwalk virus outbreak at a college campus. South Med J. 1986;1:33–6. 40. doi: 10.1097/00007611-198601000-00011. [DOI] [PubMed] [Google Scholar]
- 35.Trucco M, LaPorte R. Exposure to superantigens as an immunogenetic explanation of type I diabetes mini-epidemics. J Pediatric Endocrinol. 1995:3–10. doi: 10.1515/jpem.1995.8.1.3. [DOI] [PubMed] [Google Scholar]
- 36.Roivainen M, Rasilainen S, Ylipaasto P, Nissinen R, Ustinov J, Bouwens L, Eizirik DL, Hovi T, Otonkoski T. Mechanisms of coxsackievirus-induced damage to human pancreatic beta-cells. J Clin Endocrinol Metab. 2000;1:432–440. doi: 10.1210/jcem.85.1.6306. [DOI] [PubMed] [Google Scholar]
- 37.Sestak K, Feely S, Fey B, Dufour J, Hargitt E, Alvarez X, Pahar B, Gregoricus N, Vinje J, Farkas T. Experimental inoculation of juvenile rhesus macaques with primate enteric caliciviruses. PLoS One. 2012;5:e37973. doi: 10.1371/journal.pone.0037973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simonen-Tikka ML, Klemola P, Suomenrinne S, Kaijalainen S, Soderstrom D, Savolainen-Kopra C, Nanto-Salonen K, Ilonen J, Simell T, Simell O, Roivainen M. Virus infections among young children--the first year of the INDIS study. J Med Virol. 2013;9:1678–1684. doi: 10.1002/jmv.23625. [DOI] [PubMed] [Google Scholar]
- 39.Savolainen-Kopra C, Simonen-Tikka ML, Klemola P, Blomqvist S, Suomenrinne S, Nanto-Salonen K, Simell O, Roivainen M. Human rhinoviruses in INDIS-study material-evidence for recovery of viable rhinovirus from fecal specimens. J Med Virol. 2013;8:1466–1472. doi: 10.1002/jmv.23593. [DOI] [PubMed] [Google Scholar]
- 40.Reyes A, Wu M, McNulty NP, Rohwer FL, Gordon JI. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci U S A. 2013;50:20236–20241. doi: 10.1073/pnas.1319470110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nielsen DS, Krych L, Buschard K, Hansen CH, Hansen AK. Beyond genetics. Influence of dietary factors and gut microbiota on type 1 diabetes. FEBS Lett. 2014;22:4234–4243. doi: 10.1016/j.febslet.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 42.Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;6053:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;7216:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hara N, Alkanani AK, Ir D, Robertson CE, Wagner BD, Frank DN, Zipris D. Prevention of virus-induced type 1 diabetes with antibiotic therapy. J Immunol. 2012;8:3805–3814. doi: 10.4049/jimmunol.1201257. [DOI] [PubMed] [Google Scholar]
- 45.Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, Casella G, Drew JC, Ilonen J, Knip M, Hyoty H, Veijola R, Simell T, Simell O, Neu J, Wasserfall CH, Schatz D, Atkinson MA, Triplett EW. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;10:e25792. doi: 10.1371/journal.pone.0025792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, Queipo-Ortuno MI. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;46 doi: 10.1186/1741-7015-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morran MP, Vonberg A, Khadra A, Pietropaolo M. Immunogenetics of type 1 diabetes mellitus. Mol Aspects Med. 2015 Apr;42:42–60. doi: 10.1016/j.mam.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, McDermott M, Sinha AA, Timmerman L, Steinman L, McDevitt HO. A molecular basis for MHC class II associated autoimmunity. Science. 1988:1003–1009. doi: 10.1126/science.3368786. [DOI] [PubMed] [Google Scholar]
- 49.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 50.Rowe RE, Leech NJ, Nepom GT, McCulloch DK. High genetic risk for IDDM in the Pacific Northwest: first report from the Washington State diabetes prediction study. Diabetes. 1994:87–94. doi: 10.2337/diab.43.1.87. [DOI] [PubMed] [Google Scholar]
- 51.Barker JM, Barriga KJ, Yu L, Miao D, Erlich HA, Norris JM, Eisenbarth GS, Rewers M. Prediction of autoantibody positivity and progression to type 1 diabetes: Diabetes Autoimmunity Study in the Young (DAISY) J Clin Endocrinol Metab. 2004;8:3896–3902. doi: 10.1210/jc.2003-031887. [DOI] [PubMed] [Google Scholar]
- 52.Undlien DE, Friede T, Rammensee HG, Joner G, Dahl-Jorgensen K, Sovik O, Akselsen HE, Knutsen I, Ronningen KS, Thorsby E. HLA-encoded genetic predisposition in IDDM: DR4 subtypes may be associated with different degrees of protection. Diabetes. 1997;1:143–149. doi: 10.2337/diab.46.1.143. [DOI] [PubMed] [Google Scholar]
- 53.Baschal EE, Aly TA, Babu SR, Fernando MS, Yu L, Miao D, Barriga KJ, Norris JM, Noble JA, Erlich HA, Rewers MJ, Eisenbarth GS. HLA-DPB1*0402 protects against type 1A diabetes autoimmunity in the highest risk DR3-DQB1*0201/DR4-DQB1*0302 DAISY population. Diabetes. 2007;9:2405–2409. doi: 10.2337/db07-0029. [DOI] [PubMed] [Google Scholar]
- 54.Pietropaolo M, Becker DJ, LaPorte RE, Dorman JS, Riboni S, Rudert WA, Mazumdar S, Trucco M. Progression to insulin-requiring diabetes in seronegative prediabetic subjects: the role of two HLA-DQ high risk haplotypes. Diabetologia. 2002:66–76. doi: 10.1007/s125-002-8246-5. [DOI] [PubMed] [Google Scholar]
- 55.Greenbaum CJ, Schatz DA, Cuthbertson D, Zeidler A, Eisenbarth GS, Krischer JP. Islet cell antibody-positive relatives with human leukocyte antigen DQA1*0102, DQB1*0602: identification by the Diabetes Prevention Trial-type 1. J Clin Endocrinol Metab. 2000;3:1255–1260. doi: 10.1210/jcem.85.3.6459. [DOI] [PubMed] [Google Scholar]
- 56.Pugliese A, Gianani R, Moromisato R, Awdeh ZL, Alper CA, Erlich HA, Jackson RA, Eisenbarth GS. HLA-DQB1*0602 is associated with dominant protection from diabetes even among islet cell antibody positive first degree relatives of patients with insulin-dependent diabetes. Diabetes. 1995:608–613. doi: 10.2337/diab.44.6.608. [DOI] [PubMed] [Google Scholar]
- 57.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;7:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rich SS, Akolkar B, Concannon P, Erlich H, Hilner JE, Julier C, Morahan G, Nerup J, Nierras C, Pociot F, Todd JA. Current status and the future for the genetics of type I diabetes. Genes Immun. 2009:S128–S131. doi: 10.1038/gene.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Howson JM, Cooper JD, Smyth DJ, Walker NM, Stevens H, She JX, Eisenbarth GS, Rewers M, Todd JA, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Pociot F, Rich SS. Evidence of gene-gene interaction and age-at-diagnosis effects in type 1 diabetes. Diabetes. 2012;11:3012–3017. doi: 10.2337/db11-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nistico L, Buzzetti R, Pritchard LE, Van der AB, Giovannini C, Bosi E, Larrad MT, Rios MS, Chow CC, Cockram CS, Jacobs K, Mijovic C, Bain SC, Barnett AH, Vandewalle CL, Schuit F, Gorus FK, Tosi R, Pozzilli P, Todd JA. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet. 1996;7:1075–1080. doi: 10.1093/hmg/5.7.1075. [DOI] [PubMed] [Google Scholar]
- 61.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;4:337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 62.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;6:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bell GI, Karam JH, Rutter WJ. Polymorphic DNA region adjacent to the 5′ end of the human insulin gene. Proc Natl Acad Sci USA. 1981:5759–5763. doi: 10.1073/pnas.78.9.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rotwein P, Chyn R, Chirgwin J, Cordell B, Goodman HM, Permutt MA. Polymorphism in the 5′-flanking region of the human insulin gene and its possible relation to Type II diabetes. Science. 1981:1117–1120. doi: 10.1126/science.6267694. [DOI] [PubMed] [Google Scholar]
- 65.Owerbach D, Nerup J. Restriction fragment length polymorphism of the insulin gene in diabetes mellitus. Diabetes. 1982:275–277. doi: 10.2337/diab.31.3.275. [DOI] [PubMed] [Google Scholar]
- 66.Julier C, Hyer RN, Davies J, Merlin F, Soularue P, Briant L, Cathelineau G, Deschamps I, Rotter JI, Froguel P, Boitard C, Bell JI, Lathrop GM. Insulin-IGF2 region encodes a gene implicated in HLA-DR4-dependent diabetes susceptibility. Nature. 1991:155–159. doi: 10.1038/354155a0. [DOI] [PubMed] [Google Scholar]
- 67.Bain SC, Prins JB, Hearne CM, Rodrigues NR, Rowe BR, Pritchard LE, Ritchie RJ, Hall JRS, Undlien DE, Ronningen KS, Dunger DB, Barnett AH, Todd JA. Insulin gene region-encoded susceptibility to type I diabetes is not restricted to HLA-DR4-positive individuals. Nature Genet. 1992:212–215. doi: 10.1038/ng1192-212. [DOI] [PubMed] [Google Scholar]
- 68.Lucassen AM, Screaton GR, Julier C, Elliot TJ, Lathrop M, Bell JI. Regulation of insulin gene expression by the IDDM associated, insulin locus haplotype. Hum Mol Genet. 1994;4:501–506. doi: 10.1093/hmg/4.4.501. [DOI] [PubMed] [Google Scholar]
- 69.Kennedy GC, German MS, Rutter WJ. The minisatellite in the diabetes susceptibility locus IDDM2 regulates insulin transcription. Nature Genet. 1995:293–298. doi: 10.1038/ng0395-293. [DOI] [PubMed] [Google Scholar]
- 70.Bennett ST, Lucassen AM, Gough SCL, Powell EE, Undlien DE, Pritchard LE, Merriman ME, Kawaguchi Y, Dronsfield MJ, Pociot F, Nerup J, Bouzekri N, Cambon-Thomsen A, Ronningen KS, Barnett AH, Bain SC, Todd JA. Susceptibility to human type I diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nature Genet. 1995:284–292. doi: 10.1038/ng0395-284. [DOI] [PubMed] [Google Scholar]
- 71.Pugliese A, Zeller M, Fernandez JrA, Zalcberg LJ, Barlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlate with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nature Genet. 1997:293–297. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- 72.Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;7039:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J. 2009;18:2812–2824. doi: 10.1038/emboj.2009.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Durinovic-Bello I, Gersuk VH, Ni C, Wu R, Thorpe J, Jospe N, Sanda S, Greenbaum CJ, Nepom GT. Avidity-dependent programming of autoreactive T cells in T1D. PLoS One. 2014;5:e98074. doi: 10.1371/journal.pone.0098074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol. 2006;4:207–213. doi: 10.1016/j.smim.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 76.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;12:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 77.Kavvoura FK, Ioannidis JP. CTLA-4 gene polymorphisms and susceptibility to type 1 diabetes mellitus: a HuGE Review and meta-analysis. Am J Epidemiol. 2005;1:3–16. doi: 10.1093/aje/kwi165. [DOI] [PubMed] [Google Scholar]
- 78.Marron MP, Raffel LJ, Garchon HJ, Jacob CO, Serrano-Rios M, Martinez Larrad MT, Teng WP, Park Y, Zhang ZX, Goldstein DR, Tao YW, Beaurain G, Bach JF, Huang HS, Luo DF, Zeidler A, Rotter JI, Yang MC, Modilevsky T, Maclaren NK, et al. Insulin-dependent diabetes mellitus (IDDM) is associated with CTLA4 polymorphisms in multiple ethnic groups. Hum Mol Genet. 1997;8:1275–1282. doi: 10.1093/hmg/6.8.1275. [DOI] [PubMed] [Google Scholar]
- 79.Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase PH, Eisenbarth GS. Prediction of type I diabetes mellitus in first degree relatives using a combination of insulin, glutamic acid decarboxylase and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996:926–933. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- 80.Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, Rewers M, Eisenbarth GS, Jensen J, Davidson HW, Hutton JC. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A. 2007;43:17040–17045. doi: 10.1073/pnas.0705894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pietropaolo M, Towns R, Eisenbarth GS. Humoral autoimmunity in type 1 diabetes: prediction, significance, and detection of distinct disease subtypes. Cold Spring Harb Perspect Med. 2012;10 doi: 10.1101/cshperspect.a012831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pietropaolo M, Castaño L, Babu S, Buelow R, Kuo Y-LS, Martin S, Martin A, Powers AC, Prochazka M, Naggert J, Leiter EH, Eisenbarth GS. Islet cell autoantigen 69 kDa (ICA69): Molecular cloning and characterization of a novel diabetes associated autoantigen. J Clin Invest. 1993:359–371. doi: 10.1172/JCI116574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fan Y, Gualtierotti G, Tajima A, Grupillo M, Coppola A, He J, Bertera S, Owens G, Pietropaolo M, Rudert WA, Trucco M. Compromised central tolerance of ICA69 induces multiple organ autoimmunity. J Autoimmun. 2014:10–25. doi: 10.1016/j.jaut.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clin Immunol. 2008;3:359–365. doi: 10.1016/j.clim.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, Marrack P, Mahata SK, Kappler JW, Haskins K. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;3:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bach J-F, Chatenaud L. Tolerance to islet autoantigens in type 1 diabetes. Annu Rev Immunol. 2001:131–161. doi: 10.1146/annurev.immunol.19.1.131. [DOI] [PubMed] [Google Scholar]
- 87.Boitard C, Villa MC, Becourt C, Gia HP, Huc C, Sempe P, Portier MM, Bach JF. Peripherin: An islet antigen that is cross-reactive with nonobese diabetic mouse class II gene products. Proc Natl Acad Sci (USA) 1992:172–176. doi: 10.1073/pnas.89.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, Sampson A, Afifiyan F, Elford A, Jackowski G, Becker DJ, Santamaria P, Ohashi P, Dosch HM. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nature Medicine. 2003:198–205. doi: 10.1038/nm818. [DOI] [PubMed] [Google Scholar]
- 89.Eggleton P, Haigh R, Winyard PG. Consequence of neo-antigenicity of the ‘altered self’. Rheumatology (Oxford) 2008;5:567–571. doi: 10.1093/rheumatology/ken014. [DOI] [PubMed] [Google Scholar]
- 90.Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol. 2012;1:112–118. doi: 10.1016/j.coi.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soderlin MK, Kastbom A, Kautiainen H, Leirisalo-Repo M, Strandberg G, Skogh T. Antibodies against cyclic citrullinated peptide (CCP) and levels of cartilage oligomeric matrix protein (COMP) in very early arthritis: relation to diagnosis and disease activity. Scand J Rheumatol. 2004;3:185–188. doi: 10.1080/03009740310004856. [DOI] [PubMed] [Google Scholar]
- 92.Moscarello MA, Mastronardi FG, Wood DD. The role of citrullinated proteins suggests a novel mechanism in the pathogenesis of multiple sclerosis. Neurochem Res. 2007;2:251–256. doi: 10.1007/s11064-006-9144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Masson-Bessiere C, Sebbag M, Girbal-Neuhauser E, Nogueira L, Vincent C, Senshu T, Serre G. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J Immunol. 2001;6:4177–4184. doi: 10.4049/jimmunol.166.6.4177. [DOI] [PubMed] [Google Scholar]
- 94.Kastbom A, Strandberg G, Lindroos A, Skogh T. Anti-CCP antibody test predicts the disease course during 3 years in early rheumatoid arthritis (the Swedish TIRA project) Ann Rheum Dis. 2004;9:1085–1089. doi: 10.1136/ard.2003.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu L, Robles DT, Abiru N, Rewers M, Kelemen K, Eisenbarth GS. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci USA. 2000;4:1701–1706. doi: 10.1073/pnas.040556697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, Marrack P, Eisenbarth G, Kappler JW. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A. 2011;40:16729–16734. doi: 10.1073/pnas.1113954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alleva DG, Crowe PD, Jin L, Kwok WW, Ling N, Gottschalk M, Conlon PJ, Gottlieb PA, Putnam AL, Gaur A. A disease-associated cellular immune response in type 1 diabetics to an immunodominant epitope of insulin. J Clin Invest. 2001;2:173–180. doi: 10.1172/JCI8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, Roep BO, Peakman M. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. 2004;3:451–463. doi: 10.1172/JCI19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kent SC, Chen Y, Bregoli L, Clemmings SM, Kenyon NS, Ricordi C, Hering BJ, Hafler DA. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;7039:224–228. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 100.Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PT, Brodnicki TC, O’Connell PJ, Kedzierska K, Rodda C, Bergman P, Hill E, Purcell AW, Dudek NL, Thomas HE, Kay TW, Mannering SI. Proinsulin-Specific, HLA-DQ8, and HLA-DQ8-Transdimer-Restricted CD4+ T Cells Infiltrate Islets in Type 1 Diabetes. Diabetes. 2015;1:172–182. doi: 10.2337/db14-0858. [DOI] [PubMed] [Google Scholar]
- 101.Baekkeskov S, Warnock G, Christie M, Rajotte RV, Larsen PM, Fey S. Revelation of specificity of 64K autoantibodies in IDDM serums by high-resolution 2-D gel electrophoresis: unambiguous identification of 64K target antigen. Diabetes. 1989:1133–1141. doi: 10.2337/diab.38.9.1133. [DOI] [PubMed] [Google Scholar]
- 102.Solimena M, Folli F, Denis-Donini S, Comi GC, Pozza G, De Camilli P, Vicari AM. Autoantibodies to glutamic acid decarboxylase in a patient with stiffman syndrome, epilepsy, and Type I diabetes mellitus. N Engl J Med. 1988:1012–1020. doi: 10.1056/NEJM198804213181602. [DOI] [PubMed] [Google Scholar]
- 103.Baekkeskov S, Aanstoot H, Christgau S, Reetz A, Solimena MS, Cascalho M, Folli F, Richter-Olsen H, DeCamilli P. Identification of the 64K autoantigen in insulin dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990:151–156. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 104.Atkinson MA, Kaufman DL, Newman D, Tobin AJ, Maclaren NK. Islet cell cytoplasmic autoantibody reactivity to glutamate decarboxylase in insulin-dependent diabetes. J Clin Invest. 1993:350–356. doi: 10.1172/JCI116192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karlsen AE, Hagopian WA, Grubin CE, Dube S, Disteche CM, Adler DA, Barmeier H, Mathewes S, Grant FJ, Foster D, Lernmark Å. Cloning and primary structure of a human isoform of glutamic acid decarboxylase from chromosome 10. Proc Natl Acad Sci (USA) 1991:8337–8341. doi: 10.1073/pnas.88.19.8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hagopian WA, Michelsen B, Karlsen AE, Larsen F, Moody A, Grubin CE, Rowe R, Petersen J, McEvoy R, Lernmark Å. Autoantibodies in IDDM primarily recognize the 65,000-Mr rather than the 67,000-Mr isoform of glutamic acid decarboxylase. Diabetes. 1993:631–636. doi: 10.2337/diab.42.4.631. [DOI] [PubMed] [Google Scholar]
- 107.Lan MS, Lu J, Goto Y, Notkins AL. Molecular cloning and identification of a receptor-type protein tyrosine phosphatase, IA-2, from human insulinoma. DNA Cell Biol. 1994:505–514. doi: 10.1089/dna.1994.13.505. [DOI] [PubMed] [Google Scholar]
- 108.Achenbach P, Warncke K, Reiter J, Naserke HE, Williams AJ, Bingley PJ, Bonifacio E, Ziegler AG. Stratification of type 1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes. 2004;2:384–392. doi: 10.2337/diabetes.53.2.384. [DOI] [PubMed] [Google Scholar]
- 109.Wasmeier C, Hutton JC. Molecular cloning of phogrin, a protein tyrosyne phosphatase homologue localized to insulin secretory granule membranes. J Biol Chem. 1996;30:18161–18170. doi: 10.1074/jbc.271.30.18161. [DOI] [PubMed] [Google Scholar]
- 110.Mziaut H, Trajkovski M, Kersting S, Ehninger A, Altkruger A, Lemaitre RP, Schmidt D, Saeger HD, Lee MS, Drechsel DN, Muller S, Solimena M. Synergy of glucose and growth hormone signalling in islet cells through ICA512 and STAT5. Nat Cell Biol. 2006;5:435–445. doi: 10.1038/ncb1395. [DOI] [PubMed] [Google Scholar]
- 111.Kawasaki E, Yu L, Gianani R, Verge CF, Babu S, Bonifacio E, Eisenbarth GS. Evaluation of islet cella antigen (ICA) 512/IA-2 autoantibody radioassay using overlapping ICA512/IA-2 constructs. J Clin Endocrinol Metab. 1997:375–380. doi: 10.1210/jcem.82.2.3723. [DOI] [PubMed] [Google Scholar]
- 112.Chimienti F, Devergnas S, Favier A, Seve M. Identification and cloning of a beta-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes. 2004;9:2330–2337. doi: 10.2337/diabetes.53.9.2330. [DOI] [PubMed] [Google Scholar]
- 113.Wijesekara N, Chimienti F, Wheeler MB. Zinc, a regulator of islet function and glucose homeostasis. Diabetes Obes Metab. 2009:202–214. doi: 10.1111/j.1463-1326.2009.01110.x. [DOI] [PubMed] [Google Scholar]
- 114.Wijesekara N, Dai FF, Hardy AB, Giglou PR, Bhattacharjee A, Koshkin V, Chimienti F, Gaisano HY, Rutter GA, Wheeler MB. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia. 2010;8:1656–1668. doi: 10.1007/s00125-010-1733-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wenzlau JM, Moua O, Liu Y, Eisenbarth GS, Hutton JC, Davidson HW. Identification of a major humoral epitope in Slc30A8 (ZnT8) Ann N Y Acad Sci. 2008:252–255. doi: 10.1196/annals.1447.028. [DOI] [PubMed] [Google Scholar]
- 116.Yu L, Liu Y, Miao D, Wenzlau J, Davidson H, Hutton J, Eisenbarth GS. Triple chimeric islet autoantigen IA2-ZnT8WR to facilitate islet autoantibody determination. J Immunol Methods. 2010;1–2:20–23. doi: 10.1016/j.jim.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 117.Gottlieb PA, Delong T, Baker RL, Fitzgerald-Miller L, Wagner R, Cook G, Rewers MR, Michels A, Haskins K. Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun. 2014:38–41. doi: 10.1016/j.jaut.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xu P, Wu Y, Zhu Y, Dagne G, Johnson G, Cuthbertson D, Krischer JP, Sosenko JM, Skyler JS. Prognostic performance of metabolic indexes in predicting onset of type 1 diabetes. Diabetes Care. 2010;12:2508–2513. doi: 10.2337/dc10-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Steck AK, Johnson K, Barriga KJ, Miao D, Yu L, Hutton JC, Eisenbarth GS, Rewers MJ. Age of islet autoantibody appearance and mean levels of insulin, but not GAD or IA-2 autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes autoimmunity study in the young. Diabetes Care. 2011;6:1397–1399. doi: 10.2337/dc10-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Eisenbarth GS, Gianani R, Yu L, Pietropaolo M, Verge CF, Chase HP, Redondo MJ, Colman P, Harrison LC, Jackson R. Dual-parameter model for prediction of type I diabetes mellitus. Proc Ass Am Phys. 1998;2:126–135. [PubMed] [Google Scholar]
- 121.Orban T, Sosenko JM, Cuthbertson D, Krischer JP, Skyler JS, Jackson R, Yu L, Palmer JP, Schatz D, Eisenbarth G. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;12:2269–2274. doi: 10.2337/dc09-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pietropaolo M, Yu S, Libman IM, Pietropaolo SL, Riley K, LaPorte RE, Drash AL, Mazumdar S, Trucco M, Becker DJ. Cytoplasmic islet cell antibodies remain valuable in defining risk of progression to type 1 diabetes in subjects with other islet autoantibodies. Pediatr Diabetes. 2005;4:184–192. doi: 10.1111/j.1399-543X.2005.00127.x. [DOI] [PubMed] [Google Scholar]
- 123.Andersson C, Kolmodin M, Ivarsson SA, Carlsson A, Forsander G, Lindblad B, Ludvigsson J, Kockum I, Marcus C, Samuelsson U, Ortqvist E, Lernmark Å, Elding LH, Torn C. Islet cell antibodies (ICA) identify autoimmunity in children with new onset diabetes mellitus negative for other islet cell antibodies. Pediatr Diabetes. 2014;5:336–344. doi: 10.1111/pedi.12093. [DOI] [PubMed] [Google Scholar]
- 124.Ilonen J, Hammais A, Laine AP, Lempainen J, Vaarala O, Veijola R, Simell O, Knip M. Patterns of beta-cell autoantibody appearance and genetic associations during the first years of life. Diabetes. 2013;10:3636–3640. doi: 10.2337/db13-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;22:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 126.Hagopian W, Ferry RJ, Jr, Sherry N, Carlin D, Bonvini E, Johnson S, Stein KE, Koenig S, Daifotis AG, Herold KC, Ludvigsson J. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protege trial. Diabetes. 2013;11:3901–3908. doi: 10.2337/db13-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gitelman SE, Gottlieb PA, Rigby MR, Felner EI, Willi SM, Fisher LK, Moran A, Gottschalk M, Moore WV, Pinckney A, Keyes-Elstein L, Aggarwal S, Phippard D, Sayre PH, Ding L, Bluestone JA, Ehlers MR. Antithymocyte globulin treatment for patients with recent-onset type 1 diabetes: 12-month results of a randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2013;4:306–316. doi: 10.1016/S2213-8587(13)70065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rigby MR, DiMeglio LA, Rendell MS, Felner EI, Dostou JM, Gitelman SE, Patel CM, Griffin KJ, Tsalikian E, Gottlieb PA, Greenbaum CJ, Sherry NA, Moore WV, Monzavi R, Willi SM, Raskin P, Moran A, Russell WE, Pinckney A, Keyes-Elstein L, et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013;4:284–294. doi: 10.1016/S2213-8587(13)70111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Rosenthal SM, Shuster JJ, Zou B, Brusko TM, Hulme MA, Wasserfall CH, Mathews CE, Atkinson MA, Schatz DA. Anti-thymocyte globulin/G-CSF treatment preserves beta cell function in patients with established type 1 diabetes. J Clin Invest. 2015;1:448–455. doi: 10.1172/JCI78492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Simmons K, Michels AW. Lessons from type 1 diabetes for understanding natural history and prevention of autoimmune disease. Rheum Dis Clin North Am. 2014;4:797–811. doi: 10.1016/j.rdc.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.The DIAMOND Project Group. Incidence and trends of childhood Type 1 diabetes worldwide 1990–1999. Diabet Med. 2006:857–866. doi: 10.1111/j.1464-5491.2006.01925.x. [DOI] [PubMed] [Google Scholar]
- 132.Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet. 2009;9680:2027–2033. doi: 10.1016/S0140-6736(09)60568-7. [DOI] [PubMed] [Google Scholar]
- 133.Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet. 2008;9626:1777–1782. doi: 10.1016/S0140-6736(08)60765-5. [DOI] [PubMed] [Google Scholar]
- 134.Barker JM, Goehrig SH, Barriga K, Hoffman M, Slover R, Eisenbarth GS, Norris JM, Klingensmith GJ, Rewers M. Clinical characteristics of children diagnosed with type 1 diabetes through intensive screening and follow-up. Diabetes Care. 2004;6:1399–1404. doi: 10.2337/diacare.27.6.1399. [DOI] [PubMed] [Google Scholar]
- 135.Yu L, Miao D, Scrimgeour L, Johnson K, Rewers M, Eisenbarth GS. Distinguishing persistent insulin autoantibodies with differential risk: nonradioactive bivalent proinsulin/insulin autoantibody assay. Diabetes. 2012;1:179–186. doi: 10.2337/db11-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Miao D, Guyer KM, Dong F, Jiang L, Steck AK, Rewers M, Eisenbarth GS, Yu L. GAD65 autoantibodies detected by electrochemiluminescence assay identify high risk for type 1 diabetes. Diabetes. 2013;12:4174–4178. doi: 10.2337/db13-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yu L, Dong F, Miao D, Fouts AR, Wenzlau JM, Steck AK. Proinsulin/Insulin autoantibodies measured with electrochemiluminescent assay are the earliest indicator of prediabetic islet autoimmunity. Diabetes Care. 2013;8:2266–2270. doi: 10.2337/dc12-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia. 2004;10:1661–1667. doi: 10.1007/s00125-004-1507-3. [DOI] [PubMed] [Google Scholar]
- 139.Hirata S, Tanaka Y. Combination therapy for early rheumatoid arthritis: a treatment holiday perspective. Expert Rev Clin Pharmacol. 2015;1:115–122. doi: 10.1586/17512433.2015.984689. [DOI] [PubMed] [Google Scholar]
- 140.Knip M, Virtanen SM, Seppa K, Ilonen J, Savilahti E, Vaarala O, Reunanen A, Teramo K, Hamalainen AM, Paronen J, Dosch HM, Hakulinen T, Akerblom HK. Dietary intervention in infancy and later signs of beta-cell autoimmunity. N Engl J Med. 2010;20:1900–1908. doi: 10.1056/NEJMoa1004809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Knip M, Akerblom HK, Becker D, Dosch HM, Dupre J, Fraser W, Howard N, Ilonen J, Krischer JP, Kordonouri O, Lawson ML, Palmer JP, Savilahti E, Vaarala O, Virtanen SM. Hydrolyzed infant formula and early beta-cell autoimmunity: a randomized clinical trial. JAMA. 2014;22:2279–2287. doi: 10.1001/jama.2014.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hummel S, Pfluger M, Hummel M, Bonifacio E, Ziegler AG. Primary dietary intervention study to reduce the risk of islet autoimmunity in children at increased risk for type 1 diabetes: the BABYDIET study. Diabetes Care. 2011;6:1301–1305. doi: 10.2337/dc10-2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sorensen IM, Joner G, Jenum PA, Eskild A, Torjesen PA, Stene LC. Maternal serum levels of 25-hydroxy-vitamin D during pregnancy and risk of type 1 diabetes in the offspring. Diabetes. 2012;1:175–178. doi: 10.2337/db11-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001;9292:1500–1503. doi: 10.1016/S0140-6736(01)06580-1. [DOI] [PubMed] [Google Scholar]
- 145.Pugliese A. The multiple origins of Type 1 diabetes. Diabet Med. 2013;2:135–146. doi: 10.1111/dme.12081. [DOI] [PubMed] [Google Scholar]
- 146.Diabetes Prevention Trial--Type 1 Diabetes Study G. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med. 2002;22:1685–1691. doi: 10.1056/NEJMoa012350. [DOI] [PubMed] [Google Scholar]
- 147.Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, Cuthbertson D, Rafkin-Mervis LE, Chase HP, Leschek E. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diabetes Care. 2005;5:1068–1076. doi: 10.2337/diacare.28.5.1068. [DOI] [PubMed] [Google Scholar]
- 148.Vehik K, Cuthbertson D, Ruhlig H, Schatz DA, Peakman M, Krischer JP. Long-term outcome of individuals treated with oral insulin: diabetes prevention trial-type 1 (DPT-1) oral insulin trial. Diabetes Care. 2011;7:1585–1590. doi: 10.2337/dc11-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gale EA, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;9413:925–931. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- 150.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin JM, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;25:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 151.Stumpf M, Zhou X, Bluestone JA. The B7-independent isoform of CTLA-4 functions to regulate autoimmune diabetes. J Immunol. 2013;3:961–969. doi: 10.4049/jimmunol.1201362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, Moran A, Peakman M, Raskin P, Russell WE, Schatz D, Wherrett DK, Wilson DM, Krischer JP, Skyler JS. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care. 2014;4:1069–1075. doi: 10.2337/dc13-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, Leslie RD. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia. 2001;3:354–362. doi: 10.1007/s001250051626. [DOI] [PubMed] [Google Scholar]
- 154.Redondo MJ, Rodriguez LM, Escalante M, O’Brian SE, Balasubramanyam A, Haymond MW. Beta cell function and BMI in ethnically diverse children with newly diagnosed autoimmune type 1 diabetes. Pediatr Diabetes. 2012;7:564–571. doi: 10.1111/j.1399-5448.2012.00875.x. [DOI] [PubMed] [Google Scholar]
- 155.Redondo MJ, Rodriguez LM, Escalante M, Smith EO, Balasubramanyam A, Haymond MW. Types of pediatric diabetes mellitus defined by anti-islet autoimmunity and random C-peptide at diagnosis. Pediatr Diabetes. 2013;5:333–340. doi: 10.1111/pedi.12022. [DOI] [PubMed] [Google Scholar]
- 156.Redondo MJ, Muniz J, Rodriguez LM, Iyer D, Vaziri-Sani F, Haymond MW, Hampe CS, Metzker ML, Grant SF, Balasubramanyam A. Association of TCF7L2 variation with single islet autoantibody expression in children with type 1 diabetes. BMJ Open Diabetes Res Care. 2014;1:e000008. doi: 10.1136/bmjdrc-2013-000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang Y, Santamaria P. Lessons on autoimmune diabetes from animal models. Clin Sci (Lond) 2006;6:627–639. doi: 10.1042/CS20050330. [DOI] [PubMed] [Google Scholar]
- 158.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;9911:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tang Q, Bluestone JA. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med. 2013;11 doi: 10.1101/cshperspect.a015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, Cagnard N, Carpentier W, Tang Q, Bluestone J, Chatenoud L, Klatzmann D, Salomon BL, Piaggio E. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med. 2010;9:1871–1878. doi: 10.1084/jem.20100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, Lorenzon R, Long SA, Buckner JH, Afonso G, Pham HP, Hartemann A, Yu A, Pugliese A, Malek TR, Klatzmann D. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun. 2015:48–58. doi: 10.1016/j.jaut.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lapuerta P, Zambrowicz B, Strumph P, Sands A. Development of sotagliflozin, a dual sodium-dependent glucose transporter 1/2 inhibitor. Diab Vasc Dis Res. 2015;2:101–110. doi: 10.1177/1479164114563304. [DOI] [PubMed] [Google Scholar]
- 163.D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;11:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 164.Pagliuca FW, Millman JR, Gurtler M, Segel M, Van DA, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;2:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fox IJ, Daley GQ, Goldman SA, Huard J, Kamp TJ, Trucco M. Stem cell therapy. Use of differentiated pluripotent stem cells as replacement therapy for treating disease. Science. 2014;6199:1247391. doi: 10.1126/science.1247391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, Yang YH, Johnson JD, Kieffer TJ. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;11:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- 167.Di CV, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N. Involvement of suppressive B-lymphocytes in the mechanism of tolerogenic dendritic cell reversal of type 1 diabetes in NOD mice. PLoS One. 2014;1:e83575. doi: 10.1371/journal.pone.0083575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Michels AW, Nakayama M. The anti-insulin trimolecular complex in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;4:329–334. doi: 10.1097/MED.0b013e32833aba41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Michels AW. Targeting the trimolecular complex. Clin Immunol. 2013;3:339–344. doi: 10.1016/j.clim.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Michels AW. Targeting the trimolecular complex: the pathway towards type 1 diabetes prevention. Diabetes Technol Ther. 2013:S2–S2. doi: 10.1089/dia.2013.0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Eisenbarth GS. Banting Lecture 2009: An unfinished journey: molecular pathogenesis to prevention of type 1A diabetes. Diabetes. 2010;4:759–774. doi: 10.2337/db09-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P. HLA DR-DQ Haplotypes and Genotypes and Type 1 Diabetes Risk: Analysis of the Type 1 Diabetes Genetics Consortium Families. Diabetes. 2008 doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Michels AW, Ostrov DA, Zhang L, Nakayama M, Fuse M, McDaniel K, Roep BO, Gottlieb PA, Atkinson MA, Eisenbarth GS. Structure-based selection of small molecules to alter allele-specific MHC class II antigen presentation. J Immunol. 2011;11:5921–5930. doi: 10.4049/jimmunol.1100746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yang J, Chow IT, Sosinowski T, Torres-Chinn N, Greenbaum CJ, James EA, Kappler JW, Davidson HW, Kwok WW. Autoreactive T cells specific for insulin B:11–23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci U S A. 2014;41:14840–14845. doi: 10.1073/pnas.1416864111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nakayama M, McDaniel K, Fitzgerald-Miller L, Kiekhaefer C, Snell-Bergeon JK, Davidson HW, Rewers M, Yu L, Gottlieb P, Kappler JW, Michels A. Regulatory vs. inflammatory cytokine T-cell responses to mutated insulin peptides in healthy and type 1 diabetic subjects. Proc Natl Acad Sci U S A. 2015;14:4429–4434. doi: 10.1073/pnas.1502967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Daniel C, Weigmann B, Bronson R, von BH. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med. 2011;7:1501–1510. doi: 10.1084/jem.20110574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang L, Stadinski BD, Michels A, Kappler JW, Eisenbarth GS. Immunization with an insulin peptide-MHC complex to prevent type 1 diabetes of NOD mice. Diabetes Metab Res Rev. 2011;8:784–789. doi: 10.1002/dmrr.1252. [DOI] [PubMed] [Google Scholar]
- 178.Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A. 2010;24:10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, Kappler JW, Eisenbarth GS. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc Natl Acad Sci U S A. 2014;7:2656–2661. doi: 10.1073/pnas.1323436111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang L, Jasinski JM, Kobayashi M, Davenport B, Johnson K, Davidson H, Nakayama M, Haskins K, Eisenbarth GS. Analysis of T cell receptor beta chains that combine with dominant conserved TRAV5D-4*04 anti-insulin B:9–23 alpha chains. J Autoimmun. 2009;1:42–49. doi: 10.1016/j.jaut.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nakayama M, Castoe T, Sosinowski T, He X, Johnson K, Haskins K, Vignali DA, Gapin L, Pollock D, Eisenbarth GS. Germline TRAV5D-4 T-cell receptor sequence targets a primary insulin peptide of NOD mice. Diabetes. 2012;4:857–865. doi: 10.2337/db11-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Liu Z, Cort L, Eberwine R, Herrmann T, Leif JH, Greiner DL, Yahalom B, Blankenhorn EP, Mordes JP. Prevention of type 1 diabetes in the rat with an allele-specific anti-T-cell receptor antibody: Vbeta13 as a therapeutic target and biomarker. Diabetes. 2012;5:1160–1168. doi: 10.2337/db11-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008:371–390. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]