

Currently, a substantial amount of effort is focused on developing actively targeted, receptor-specific nanoparticles for the delivery of anticancer drugs and diagnostic imaging contrast agents.[1–7] Receptor-targeted nanoparticles hold much promise and are often shown to provide nanoparticle delivery beyond that seen with the enhanced permeability and retention (EPR) effect alone. However, these nanoparticles still face considerable challenges as a result of the significant heterogeneity in receptor expression, not only between patients and tumor types, but also within individual tumors.[8] In many cases, the overexpressed receptor may also be present on normal tissues leading to detrimental off-target effects. Perhaps even more troubling is that several recent studies have shown that cancer stem cells may not even possess any known up-regulated receptor.[9] Therefore, targeting strategies that are more generalizable across a broad range of tumors than receptor-specific targeting are highly desirable.
The microenvironment of solid tumors has several unique characteristics, including high interstitial fluid pressure (IFP), low oxygen tension or hypoxia, and low extracellular pH (pHe) that differentiate it from normal tissues.[10,11] For example, many studies have shown that nearly all human and animal tumors exhibit an extracellular pH (pHe) lower than 7.0, even reaching as low as 6.3.[12,13] This subphysiologic pHe is thought to arise from increased glucose uptake and metabolism, a phenomena known as the Warburg effect.[14,15] This altered acidic tumor microenvironment is a promising target for delivery of nanoparticles carrying drugs and imaging agents to a wide variety of malignancies, however a major hurdle is the lack of practical ways to target this subphysiologic pH. Ideally, such nanocarriers should maintain an inactive state at physiologic pH and transition to an active state within the acidic tumor microenvironment.
To date, several approaches have been developed to target the acidic tumor microenvironment. One strategy involves the use of a pH low insertion peptide (pHLIP) that is soluble at physiologic pH, but forms a rigid transmembrane complex at pH < 7.0.[16] This hydrophilic-to-hydrophobic transition leads to improved cellular uptake within the acidic tumor microenvironment. In another approach, an outer shielding layer on the nanoparticle surface is removed upon exposure to the acidic tumor microenvironment, revealing an active agent, such as an underlying polycationic surface[17,18] or cell-penetrating peptide.[19] While these approaches have shown promise for targeting the acidic tumor microenvironment, several significant obstacles to clinical applications remain, including immunogenicity, toxicity, instability in vivo, and cost. Therefore, we sought to develop a platform that can overcome these barriers allowing for the efficient delivery of therapeutic agents into acidic tumor sites.
In an effort to identify pH-responsive materials for targeting low pHe tumor microenvironment, we recently discovered that native glycol chitosan (GC), a water-soluble and low-cost biopolymer with a pH-titratable charge, can achieve this purpose.[20,21] Compared with existing pH-sensitive materials, glycol chitosan shows many promising advantages including its abundant natural precursor, ease of synthesis, biodegradability, biocompatibility, and low cost. In this study, we investigated whether GC-coated liposomes could form a highly efficient drug delivery system that targets low pHe microenvironments (Figure 1). Liposomes were chosen as the model system since they have been extensively studied as nanocarriers for the delivery of anticancer drugs. Liposomes were prepared using hydrogenated soy phosphatidylcholine (HSPC) and cholesterol (CHOL). For GC-conjugation, a small percentage (5 mol%) of COOH-termi nated phospholipid, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG2K-COOH), was also included during film preparation. Nanometer-scale liposomes were then formed by subjecting the sample to multiple freeze–thaw cycles followed by extrusion through a 100 nm polycarbonate filter. The anti-cancer drug doxorubicin (DOX) was encapsulated into pre-formed liposomes with the well-established transmembrane pH gradient loading method.[22] The conjugation of GC onto the surface of DOX-loaded liposomes was achieved using carbodiimide chemistry, i.e., 1-ethyl-3-(3-dimethylaminoisopropyl) carbodiimide (EDC)/N-hydroxysulfosuccinimide (Sulfo-NHS) coupling. It was hypothesized that GC-DOX-liposomes would transition from a negative surface charge at physiologic pH to a positive surface charge in the slightly acidic tumor extracellular environment, thus leading to enhanced cellular uptake and improved cytotoxicity.
Figure 1.
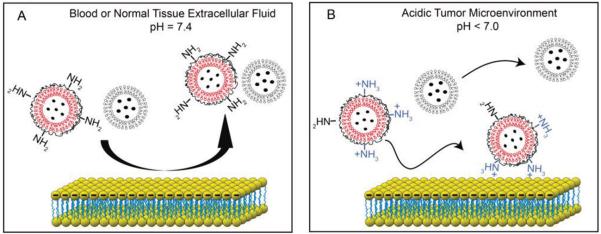
pH-titratable surface charge of GC-coated liposomes enables targeting of cells in the acidic tumor microenvironment. A) Both GC-liposomes and control liposomes (i.e., no GC coating) exhibit negative surface charge at physiologic pH, which limits their association with blood components and normal tissue. B) In the acidic extracellular environment, the GC-liposomes loaded with anticancer drugs become positively charged, leading to an increase in cell labeling and improved anticancer efficacy.
Dynamic light scattering (DLS) revealed that the unmodified liposomes (i.e., no GC) had a mean diameter of 123.0 ± 2.0 nm (PDI = 0.039) (Figure 2). After GC conjugation, an increase in liposome diameter (135.4 ± 0.8 nm, PDI = 0.063) was observed, suggesting the successful conjugation of glycol chitosan to the surface of liposomes. To ensure that the GC chemically bound to the liposome surface, the liposomes were also prepared in the absence of DSPE-PEG2K-COOH, and then incubated with GC and EDC/Sulfo-NHS. In contrast to the results from the DSPE-PEG2K-COOH-incorporated liposomes, these control vesicles did not exhibit an increased diameter, suggesting that the bound GC were conjugated through a surface chemical reaction and not from nonspecific absorption. The GC conjugation was further confirmed by fourier transform infrared spectroscopy (FTIR). In addition to a weak absorption at 1732 cm−1, due to the carboxyl group (Figure S1, Supporting Information), signals related to GC segments also appeared, indicating that GC was successfully conjugated to the liposomal surface.
Figure 2.
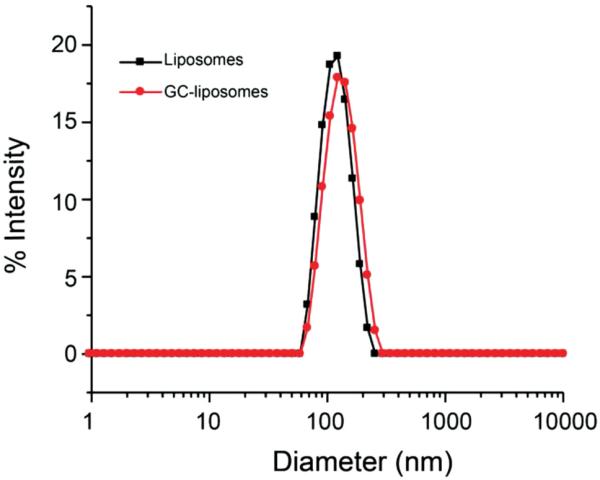
Intensity-weighted size distribution of liposomes with and without GC conjugation as measured by dynamic light scattering (DLS).
To investigate the titratability of the liposome surface charge, the zeta potential of unmodified liposomes and GC-liposomes was measured at different pH values from 6.0 to 7.75. As shown in Figure 3, the surface charge of the unmodified liposomes was independent of pH, with zeta potential values clustering around −25 mV for all pH levels. The negative surface charge of the unmodified liposomes was due to their surface COOH groups. The GC-liposomes, however, show a distinctly pH-dependent surface charge, with zeta potential values increasing from −14.3 mV at pH 7.4 to +9.08 mV upon exposure to pH 6.0, further confirming the successful incorporation of the GC polymer. These results suggest that GC-liposomes may be capable of enhanced retention in tumors through electrostatic interactions (between positively charged liposomes and negatively charged cell membranes and extracellular matrix components) that occur preferentially in the acidic microenvironment.
Figure 3.
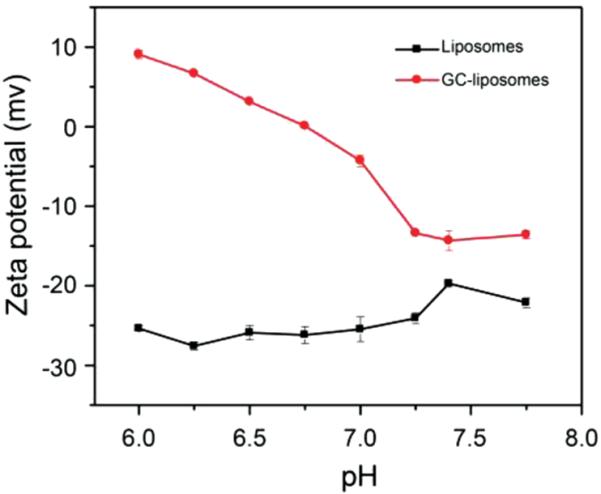
Zeta potential of liposomes with and without surface-bound glycol chitosan. Liposomes were incubated in buffers with pH values ranging from 6.0 to 7.75.
To prepare pH-responsive and drug-loaded liposomes, DOX-loaded liposomes were prepared and then conjugated with GC. The encapsulation efficiency of DOX in this lipo-some formulation was above 90%. The average diameter of GC-DOX-liposomes was found to be 142.7 ± 2.7 nm (PDI = 0.068). As a comparison, the DOX-liposomes had a diameter of 123.7 ± 1.4 nm (PDI = 0.040) (Figure S2, Supporting Information). These measurements indicated that loading DOX into preformed liposomes did not lead to any significant change in hydrodynamic diameter. Cryogenic transmission electron microscopy (Cryo-TEM) indicated that the mean diameter of GC-DOX-liposomes was very close to that of DOX-liposomes (Figure S3, Supporting Information). Moreover, all liposomes were uniformly dispersed with little to no cohesion resulting from GC conjugation, implying that no liposomal aggregation was caused by the EDC/Sulfo-NHS reaction. The zeta potential of DOX-loaded liposomes with or without GC was also tested. It was concluded that loading DOX into liposomes did not result in a significant change in surface charge and GC-DOX-liposomes remained equally responsive to a change in pH.
To demonstrate the feasibility of utilizing GC-coated liposomes to improve cellular targeting in acidic microenvironments, the labeling of HT1080 cells with GC-DOX-liposomes was evaluated at two different pH values, pH 7.4 and pH 6.5 (Figure 4). After a 2 h incubation, the cells were washed three times with PBS and fluorescence images (using DOX as the fluorescent marker) were then acquired to assess the extent of cell association. When GC-DOX-liposomes were incubated with cells in culture medium at pH 6.5, a bright fluorescence signal was observed, indicating significant cellular association. When GC-DOX-liposomes were incubated with cells in culture medium at pH 7.4, however, fluorescence was significantly less, confirming that cellular binding of the GC-liposomes at physiological pH was much lower than the binding at a more acidic pH. For comparison, DOX-loaded liposomes without GC coating were also tested under similar conditions, revealing a low level of fluorescence without any appreciable pH dependence. These results confirm that GC-liposomes exhibit greater cancer cell association than unmodified liposomes, and this association is largely mediated by a decrease in the extracellular pH.
Figure 4.
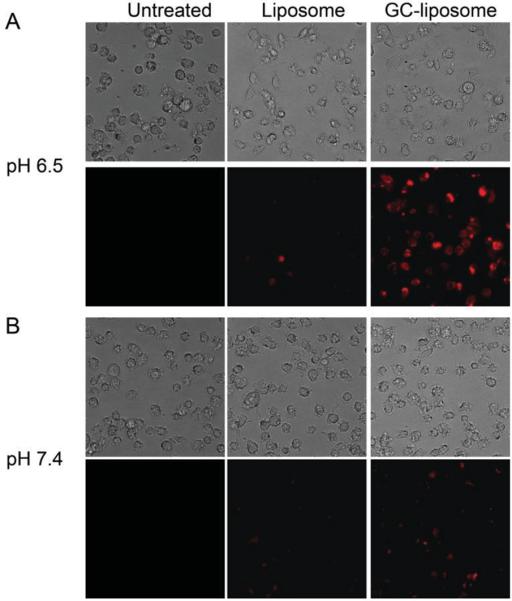
Microscopy images of HT1080 cells incubated with GC-DOX-liposomes and DOX-loaded liposomes at two different pH. A) HT1080 cells were incubated with liposomes at the pH 6.5. The top row shows phase contrast images and the bottom row shows fluorescence images. B) HT1080 cells were incubated with liposomes at the pH 7.4. In all cases, cells were incubated with DOX-liposomes or GC-DOX-liposomes at an equivalent DOX concentration of 10 μg mL−1. All liposomes were incubated with HT1080 cells for 2 h before images were acquired.
To provide further quantitative evidence that GC-liposomes could improve cellular uptake in a subphysiologic pH environment, flow cytometric analysis was carried out for determination of DOX uptake by HT1080 cells (Figure 5). The fluorescence intensity was directly proportional to the amount of DOX internalized. The results revealed that the GC-liposomes had a significantly greater uptake at pH 6.5 compared with pH 7.4. However, no difference was observed between pH 7.4 and 6.5 when the unmodified liposomes (i.e., no GC) were incubated with the cells. This result was consistent with the fluorescence imaging studies, confirming that transition from a negative to positive surface charge indeed facilitated the cellular uptake of the GC-liposomes.
Figure 5.
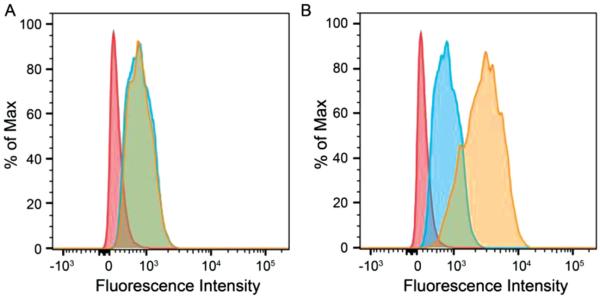
Flow cytometry of HT1080 cells labeled with A) DOX-liposomes and B) GC-DOX-liposomes. HT1080 cells were incubated with liposomes at pH 7.4 and 6.5 at 37 °C for 2 h. Red: control cells; blue: pH 7.4; and brown: pH 6.5.
Next, cytotoxicity assays were used to confirm that GC-liposomes lacking DOX payload are inherently nontoxic. Specifically, HT1080 cells were incubated with the various liposome formulations and their viability was then assessed via an MTT cell proliferation assay (where MTT is 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide). HT1080 cells were incubated at 37 °C, both at pH 7.4 and 6.5, with various concentrations of either unmodified liposomes or GC-liposomes. After a 4 hour incubation with the liposomes, cells were incubated for another 20 h in fresh medium without liposomes, and then the cell viability was determined. As shown in Figure 6A, both empty liposome formulations showed very low cytotoxicity (>90% viability compared to untreated cells) at HSPC concentrations up to 200 μg mL−1, both at pH 7.4 and pH 6.5, indicating that these two liposome formulations are nontoxic and biocompatible.
Figure 6.
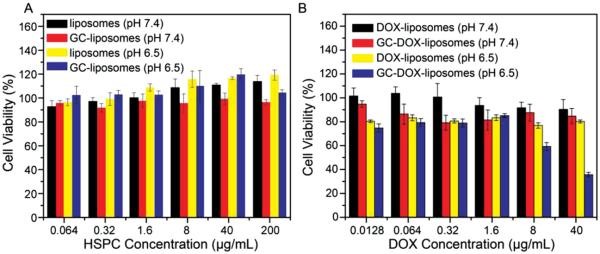
A) Cell viability of HT1080 cells after incubation with empty liposomes (i.e., no DOX) at pH 7.4 and 6.5 for 4 h. B) Cell viability of HT1080 cells after incubation with DOX-liposomes or GC-DOX-liposomes at various DOX concentrations for 4 h followed by 20 h in fresh medium at 37 °C. Data are presented as the average ± standard deviation (n = 4).
Next, the pH-dependent cytotoxic efficacy of DOX-loaded GC-liposomes was demonstrated. HT1080 cells were again incubated at 37 °C, at both pH 7.4 and 6.5, with either DOX-liposomes or GC-DOX-liposomes at various DOX concentrations. Again, after a 4 h incubation with liposomes, cells were incubated for another 20 h in fresh medium without liposomes, and then the cell viability was determined. As shown in Figure 6B, GC-DOX-liposomes resulted in a 64% reduction in cell viability, compared with untreated cells, at a DOX concentration of 40 μg mL−1, in cell medium at pH 6.5. In contrast, at pH 7.4 GC-DOX-liposomes resulted in <15% reduction in viability. The DOX-liposomes without the GC coating led to <20% reduction in viability regardless of pH. Similarly, no difference in cytotoxicity was observed for free DOX at pH 7.4 and 6.5, indicating that slightly acidic conditions did not affect the toxicity of DOX (Figure S4, Supporting Information). These findings further confirm that the improved anticancer efficacy of GC-DOX-liposomes was predominantly mediated through the pH-responsive GC coating on the surface of the DOX-loaded liposomes.
To explore the potential of targeting the acidic tumor microenvironment in vivo, a biodistribution and antitumor study were performed in a murine xenograft tumor model. For biodistribution studies, various organs were excised 24 h following the administration of DOX-liposomes or GC-DOX-liposomes and fluorescent images of DOX were acquired (Figure 7A). It is seen that the biodistribution of the GC-DOX- and DOX-liposomes in the visceral organs is not homogenous. Most liposomes accumulated in the liver, kidneys, and tumor tissue for both liposome formulations and their contents in the heart, spleen, and lung were comparatively low. As expected, the fluorescent intensity of GC-DOX-liposomes in the tumor was significantly higher than that of DOX-liposomes (p < 0.05), as seen in Figure 7B. This convincingly demonstrates the targeting effect of the GC moieties in GC-DOX-liposomes.
Figure 7.

A) Fluorescent images of excised organs 24 h postadministration of DOX-liposomes or GC-DOX-liposomes at a DOX concentration of 5 mg kg−1 body weight (n = 3). B) Semiquantitative analysis of organ fluorescence from (A) (mean ± S.D., * P < 0.05). C) In vivo antitumor activity after i.v. injection of PBS, free DOX, DOX-liposomes, or GC-DOX-liposomes at a DOX concentration of 5 mg kg−1 body weight (n = 6) (mean ± S.D., * P < 0.05). D) H&E-stained tumor sections excised from T6-17 tumor-bearing mice following 16 d treatment with (i) PBS, (ii) free DOX, (iii) DOX-liposomes, or (iv) GC-DOX-liposomes. The images of tumor were obtained by a Zeiss microscope at low magnification (20×).
To study the in vivo antitumor effect of GC-DOX- and DOX-liposomes, T6-17 xenografts were established by injecting T6-17 cells in the back left flank of nu/nu mice. When the tumor grew to a size of 50–150 mm3, 7 d after inoculation of the cancer cells, the mice were randomly divided into four groups with six mice in each group. Mice were then treated with either free DOX, DOX-liposomes, GC-DOX-liposomes, or PBS (i.e., untreated control). The equivalent dose of DOX (5 mg kg−1) was given three times (on day 0, 2, and 4) via an i.v. tail injection and the sizes of the tumor were measured every other day (Figure 7C). In the untreated group (i.e., PBS), the tumor volume increased most rapidly. The group treated with free DOX showed a slight antitumor response. In contrast, a significant antitumor response was observed for mice treated with DOX-liposomes or GC-DOX-liposomes. The GC-DOX-liposomes showed the strongest antitumor effect. Statistical analysis between DOX-liposomes and GC-DOX-liposomes group indicted a P value of 0.024. Furthermore, hematoxylin and eosin (H&E) staining of the tumor sections for each group of treated animals is shown in Figure 7D. The antitumor effects were most notable in the animals treated with GC-DOX-liposomes compared to controls. These data suggest that a pH-responsive drug delivery platform based on glycol chitosan–coated liposomes can enhance tumor accumulation and in turn provide an improved therapeutic effect.
In summary, we have developed a pH-responsive liposomal platform for enhanced DOX delivery to cells residing in a subphysiologic pH environment. The glycol chitosan–modified liposomal system successfully mediated increased cellular uptake and increased therapeutic efficacy of DOX both in vitro and in vivo due to its pH-dependent transition in surface charge. We believe that this approach could be further employed to enhance the intracellular delivery of other anticancer agents, proteins, genes, imaging agents, etc., and offers a promising approach for intelligent drug delivery.
Supplementary Material
Acknowledgements
This work was supported in part by the National Institutes of Health NCI 1R21CA140695 (A.T.), NCI R01CA157766 (A.T.), NCI R01CA175480 (Z.C.), and pilot grants of TBIC (A.T. and Z.C.) and CT3N (Z.C.) of ITMAT of the University of Pennsylvania. The authors gratefully acknowledge Prof. Shu Yang and Dr. Yu Xia for technical support with the FTIR, and Dewight Williams for assistance with the cryo-TEM.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Cheng ZL, Thorek DLJ, Tsourkas A. Angew. Chem. Int. Ed. 2010;49:346. doi: 10.1002/anie.200905133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yang LL, Peng XH, Wang YA, Wang XX, Cao ZH, Ni CC, Karna P, Zhang XJ, Wood WC, Gao XH, Nie SM, Mao H. Clin. Cancer Res. 2009;15:4722. doi: 10.1158/1078-0432.CCR-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Davis ME, Chen Z, Shin DM. Nat. Rev. Drug Discovery. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- [4].Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS, Langer R, Farokhzad OC. Sci. Transl. Med. 2013 doi: 10.1126/scitranslmed.3007049. 10.1126/scitranslmed.3007049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tu CQ, Ng TSC, Sohi HK, Palko HA, House A, Jacobs RE, Louie AY. Biomaterials. 2011;32:7209. doi: 10.1016/j.biomaterials.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Du X, Shi BY, Liang J, Bi JX, Dai S, Qiao SZ. Adv. Mater. 2013;25:5981. doi: 10.1002/adma.201302189. [DOI] [PubMed] [Google Scholar]
- [7].Du X, Shi BY, Tang YH, Dai S, Qiao SZ. Biomaterials. 2014;35:5580. doi: 10.1016/j.biomaterials.2014.03.051. [DOI] [PubMed] [Google Scholar]
- [8].Cheng ZL, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Science. 2012;338:903. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cho RW, Clarke MF. Curr. Opin. Genet. Dev. 2008;18:48. doi: 10.1016/j.gde.2008.01.017. [DOI] [PubMed] [Google Scholar]
- [10].Cairns R, Papandreou I, Denko N. Mol. Cancer Res. 2006;4:61. doi: 10.1158/1541-7786.MCR-06-0002. [DOI] [PubMed] [Google Scholar]
- [11].Du X, Xiong L, Dai S, Kleitz F, Qiao SZ. Adv. Funct. Mater. 2014;24:7627. [Google Scholar]
- [12].Gillies RJ, Raghunand N, Garcia-Martin ML, Gatenby RA. IEEE Eng. Med. Biol. Mag. 2004;23:57. doi: 10.1109/memb.2004.1360409. [DOI] [PubMed] [Google Scholar]
- [13].Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. J. Magn. Reson. Imaging. 2002;16:430. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]
- [14].Ferreira LMR. Exp. Mol. Pathol. 2010;89:372. doi: 10.1016/j.yexmp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- [15].Schornack PA, Gillies RJ. Neoplasia. 2003;5:135. doi: 10.1016/s1476-5586(03)80005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Proc. Natl. Acad. Sci. USA. 2008;105:15340. doi: 10.1073/pnas.0804746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Poon Z, Chang D, Zhao X, Hammond PT. ACS Nano. 2011;5:4284. doi: 10.1021/nn200876f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Popat A, Liu J, Lu GQ, Qiao SZ. J. Mater. Chem. 2012;22:11173. [Google Scholar]
- [19].Kale AA, Torchilin VP. Methods Mol. Biol. 2010;605:213. doi: 10.1007/978-1-60327-360-2_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Crayton SH, Tsourkas A. ACS Nano. 2011;5:9592. doi: 10.1021/nn202863x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nwe K, Huang CH, Tsourkas A. J. Med. Chem. 2013;56:7862. doi: 10.1021/jm4012565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Haran G, Cohen R, Bar LK, Barenholz Y. Biochim. Biophys. Acta. 1993;1151:201. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.