

Abstract
Damaged mitochondria are detrimental to cellular homeostasis. One mechanism for removal of damaged mitochondria involves the PINK1-PARKIN pathway, which poly-ubiquitylates damaged mitochondria to promote mitophagy. We report that assembly of ubiquitin chains on mitochondria triggers the recruitment of autophagy adaptors concomitantly with activation of the TBK1 protein kinase, which physically associates with OPTN, NDP52, and SQSTM1. Full TBK1 activation in HeLa cells requires OPTN and NDP52, and OPTN ubiquitin chain binding. In addition to the known role of S177 phosphorylation in OPTN on ATG8 recruitment, TBK1-dependent phosphorylation on S473 and S513 promotes ubiquitin chain binding in vitro as well as TBK1 activation, OPTN mitochondrial retention, and efficient mitophagy in vivo. These data reveal a self-reinforcing positive feedback mechanism that coordinates TBK1-dependent autophagy adaptor phosphorylation with the assembly of ubiquitin chains on mitochondria to facilitate efficient mitophagy. Analogous mechanisms may facilitate adaptor-protein mediated delivery of other types of cargo to autophagosomes.
Graphical Abstract
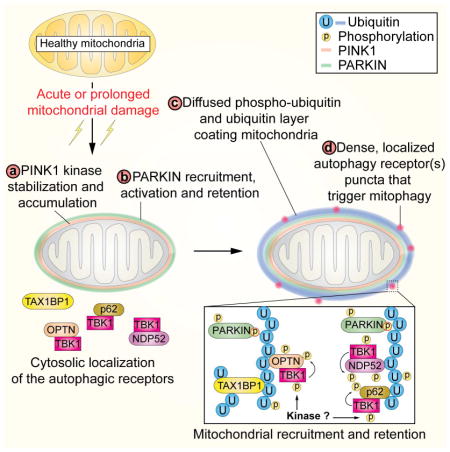
INTRODUCTION
Mitochondrial quality control via mitophagy is central to the health of the cell and is linked to several neurodegenerative diseases (Pickrell and Youle, 2015). Research during the last several years has defined a signal transduction pathway involving the mitochondrial protein kinase PINK1 and the cytoplasmic ubiquitin ligase PARKIN, both of which are mutated in certain forms of Parkinson’s Disease, in the surveillance of mitochondrial health (Pickrell and Youle, 2015). In response to mitochondrial damage, PINK1 is stabilized on the mitochondrial outer membrane (MOM) where it promotes phosphorylation of PARKIN on S65 in its N-terminal UB-like domain, as well as the conserved S65 within UB on mitochondria (Kane et al., 2014; Kazlauskaite et al., 2014; Kondapalli et al., 2012; Koyano et al., 2014; Ordureau et al., 2014). PARKIN phosphorylation and its binding to p-S65 UB promotes its assembly of K6, K11, K48, and K63 chains on numerous MOM proteins and PARKIN retention on the MOM (Cunningham et al., 2015; Ordureau et al., 2014; Sarraf et al., 2013). UB chains assembled by PARKIN appear to be a major form of UB targeted by PINK1, allowing PARKIN retention on mitochondria (Ordureau et al., 2015a; Ordureau et al., 2014). Together these events constitute a feed-forward amplification mechanism to promote mitophagy (Ordureau et al., 2014).
Targeting of ubiquitylated mitochondria to autophagosomes is a critical step in mitophagy (Stolz et al., 2014). Selective autophagy involves the action of cargo adaptor proteins such as SQSTM1 (also called p62), NBR1, NDP52 (also called CALCOCO2), Optineurin (OPTN), TAX1BP1, and NIX, each of which associate with ATG8 proteins via an LC3 interacting region (LIR) motif and with cargo via other domains (Stolz et al., 2014; Weidberg and Elazar, 2011). Importantly, the ability of SQSTM1 and OPTN to associate with ATG8 in vitro is increased upon phosphorylation of residues adjacent to the LIR by the TBK1 protein kinase (Matsumoto et al., 2011; Wild et al., 2011), a known binding partner of these adaptors (Matsumoto et al., 2011; Morton et al., 2008). Recognition of ubiquitylated cargo occurs through UBA domains in SQSTM1, UBAN and ZNF domains in OPTN, and UBZ domains in NDP52, which preferentially bind K63 chains as well as linear chains in the case of OPTN (Gleason et al., 2011; Laplantine et al., 2009; Matsumoto et al., 2011; Ordureau et al., 2015a; Sims et al., 2012; van Wijk et al., 2012). Early studies suggested that SQSTM1 is responsible for directing damaged mitochondria to the autophagosome (Geisler et al., 2010), but subsequent work revealed that SQSTM1 is required for aggregation of damaged mitochondria but not for mitophagy itself (Narendra et al., 2010). More recently, PARKIN-dependent mitophagy in HeLa cells has been linked to the function of OPTN (Wong and Holzbaur, 2014). Interestingly, the finding that SQSTM1 and OPTN, as well as TBK1, are mutated in amyotrophic lateral sclerosis (ALS) and that OPTN patient-derived mutants are defective in mitophagy implicates this arm of the autophagy system with this neurodegenerative disease (Cirulli et al., 2015; Fecto et al., 2011; Freischmidt et al., 2015; Maruyama et al., 2010; Wong and Holzbaur, 2014).
Here, we report that mitochondrial depolarization leads to PINK1 and PARKIN-dependent phosphorylation of S172 in TBK1, an activating modification (Kishore et al., 2002). As seen previously in other signaling contexts (Clark et al., 2009), TBK1 activation upon depolarization did not require TBK1 activity. Interestingly, TBK1 activation involves both the two autophagy adaptors OPTN and NDP52, and the ability of OPTN to bind poly-UB chains. Furthermore, TBK1 activation in response to mitochondrial depolarization promotes phosphorylation of SQSTM1, OPTN, and NDP52, and TBK1 activity is required for efficient recruitment of OPTN, NDP52, and SQSTM1 to depolarized mitochondria, while TAX1BP1 recruitment is PINK1-dependent but TBK1 independent. These adaptors are recruited to specific puncta on damaged mitochondria in a pattern that is distinct from p-S65 UB, present throughout damaged mitochondria. Cells lacking both OPTN and NDP52 or TBK1 are deficient in depolarization-dependent mitophagy. Using quantitative proteomics, we found depolarization-dependent OPTN phosphorylation on S177, which is known to promote ATG8 recruitment (Wild et al., 2011), as well as phosphorylation of S473 and S513 near the UB binding UBAN domain. Dual phosphorylation of S473 and S513 activates poly-UB chain binding in vitro, and promotes TBK1 activation, OPTN retention on damaged mitochondria, and mitophagy in vivo. Thus, UB chain synthesis on mitochondria sets in motion concerted adaptor protein recruitment, TBK1 activation, and self-reinforcing TBK1-dependent phosphorylation of autophagy adaptors to facilitate mitochondrial capture by autophagosomes. This work mechanistically links genes found mutated in Parkinson’s Disease and ALS, and is consistent with a prominent role for autophagy and mitochondrial quality control in both diseases.
RESULTS
PINK1-PARKIN activation upon mitochondrial depolarization promotes TBK1 phosphorylation on S172
Previous studies suggested a role for mitofusins and mitochondrial membrane potential in the response of cells to viral infection via the MAVS/TBK1 pathway (Koshiba et al., 2011). We previously identified 8 major ubiquitylation sites in MFN2 (K406, K416, K420, K460, K719, K720, K730 and K737) (Sarraf et al., 2013), the replacement of which to arginine largely blocked PARKIN and PINK1-dependent MFN2 ubiquitylation (Figure 1A). During analysis of PARKIN-dependent MFN2 ubiquitylation in the MAVS/TBK1 pathway, we made the surprising finding that mitochondrial depolarization with Antimycin A and Oligomycin A (AO) resulted in rapid TBK1 activation as judged by phosphorylation on its activation loop site (S172) independently of MFN2 ubiquitylation and phosphorylation of the TBK1 substrate Interferon Regulatory Factor 3 (IRF3), as measured using phospho-specific antibodies (Figure 1B). TBK1 activation and IRF3 phosphorylation is known to occur in response to the antiviral RIG-I/MDA5/MAVS pathway, mimicked here by the use of Poly(I:C) (Figure 1B).
Figure 1. Activation of TBK1 upon mitochondrial depolarization requires PARKIN and PINK1.

(A,B) HFT-MFN2−/− cells expressing an inducible MFN2 (WT or 8KR mutant)-IRES-PARKIN (WT or CS mutant) were depolarized (1h) and purified mitochondria (20μg) subjected to immunoblotting with MFN2 or TOMM20 antibodies (Panel A). In parallel experiments, cells were either left untreated, exposed to AO (1h) or transfected with Poly(I:C) (10μg/ml, 1.5h) and lysated subjected to immunoblotting with the indicated antibodies (Panel B).
(C) Schematic of cell system used in this work. HeLa Flp-in T-Rex (HFT) cells harboring PARKINWT or catalytically inactive PARKINCS were created in the context of PINK1+/+ or PINK1−/− generated using gene-editing with a TALEN targeting PINK1.
(D,E) HFT or HFT-PARKINWT cells were treated with AO (2h) to depolarize mitochondria or Poly(I:C) (2h) to activate the RIG-I/MDA5/MAVS pathway. Lysates were subjected to immunoblotting with the indicated antibodies (Panel D). Quantification of relative p-TBK1S172/TBK1 levels for biological triplicate experiments (Panel E). Error bars represent SEM from triplicate experiments.
(F,G) HFT cells with the indicated PARKIN and PINK1 genotypes were depolarized with AO (1h) and lysates subjected to immunoblotting with the indicated antibodies (Panel F). Quantification of relative p-TBK1/TBK1 levels for biological triplicate experiments (Panel G). Error bars represent SEM from triplicate experiments.
(H) U2OS cells stably expressing PARKINWT or SH-SY5Y cells were left untreated or depolarized with AO (1h) prior to immunoblotting of lysates. The p-TBK1S172/TBK1 ratios are indicated.
Panels E and G analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (n=3 biological replicates). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. n.s., not significant. See also Figure S1.
To understand the basis of TBK1 activation via mitochondrial depolarization, we used HeLa Flp-in T-REx (HFT) cells naturally deficient in PARKIN to examine the activation state of TBK1 with or without inducing low level expression of wild-type (WT) PARKIN (Ordureau et al., 2015a) (Figure 1C). Addition of AO for 2 h resulted in a 2-fold increase in TBK1S172 phosphorylation in the absence of PARKIN but a 5-fold increase in the presence of PARKINWT (Figure 1D,E). The increase in TBK1S172 phosphorylation in the presence of PARKINWT was similar to that seen with Poly(I:C) treatment (2h) with or without PARKINWT expression. As expected (Fitzgerald et al., 2003), Poly(I:C) treatment resulted in robust IRF3 phosphorylation, but interestingly, AO treatment did not induce IRF3 phosphorylation (Figure 1D,E). Thus, activation of TBK1 through a mitochondrial depolarization dependent process does not lead to cross-talk with the innate immunity pathway, suggesting strict compartmentalization of TBK1 function. AO-dependent TBK1 activation was time-dependent with maximum activation (~10-fold) observed at 1h post-depolarization (Figure S1A), and was abrogated in gene-edited HFT-PINK1−/− cells (Ordureau et al., 2014) (Figure 1A,F,G). In addition, PARKINCS, in which the catalytic cysteine C431 is replaced by serine, failed to promote TBK1 activation (Figure 1F,G, S1A). TBK1 phosphorylation was also observed in PARKIN-deficient U2OS cells stably expressing PARKINWT, and in SH-SY5Y and 293T cells expressing endogenous PARKIN (Figure 1H). Depletion of PINK1 by shRNA in SH-SY5Y blocked depolarization-dependent TBK1 phosphorylation (Figure S1B). Thus, mitochondrial depolarization promotes PARKIN and PINK1 dependent TBK1 activation, the regulation of which is elaborated below.
TBK1-dependent recruitment of the mitophagy receptors OPTN, NDP52, and SQSTM1 to depolarized mitochondria
TBK1 is known to associate with the selective autophagy adaptors OPTN, NDP52 and SQSTM1, and previous studies indicate that OPTN, NDP52, and SQSTM1 are recruited to ubiquitylated bacteria (Thurston et al., 2009; Wild et al., 2011). Moreover, OPTN has been shown to be recruited to damaged mitochondria in HeLa cells in a PINK1-dependent manner (Wong and Holzbaur, 2014). As such, we systematically examined recruitment of OPTN, NDP52, SQSTM1, and the related autophagy adaptor TAX1BP1 to depolarized mitochondria in the presence and absence of a specific small-molecule inhibitor of TBK1 (MRT67307, referred to MRT) (Clark et al., 2011) in PARKINWT expressing HFT cells with or without PINK1 using confocal microscopy and image analysis as described under Supplemental Experimental Procedures (Figure 2A–D, S2A–E). All four autophagy adaptors were localized diffusely in the cytoplasm in untreated cells. One hour post AO treatment, all four autophagy adaptors were recruited to puncta that co-localized with mitochondria, as visualized using α-TOMM20 antibodies. We used image analysis via Mander’s overlap coefficient (MOC) to measure the overlap of adaptor puncta with depolarized mitochondria, and normalized for the fraction of cell volume occupied by mitochondria to account for diffuse cytoplasmic localization of autophagy adaptors in untreated cells. In response to depolarization, the normalized MOC for OPTN increased from 1.0 to 3.0, but this increase was largely abolished in the presence of MRT or in the absence of PINK1 (Figure 2B). Similar results were obtained with NDP52 and SQSTM1 (Figure 2C,D and S2A–C). However, while TAX1BP1 co-localization with depolarized mitochondria was dependent upon PINK1, it was not inhibited by MRT (Figure S2D,E).
Figure 2. Recruitment of mitophagy adaptors OPTN NDP52, and SQSTM1, but not TAX1BP1, to depolarized mitochondria requires TBK1.<.

br>(A–D) The indicated HFT cells stably expressing FLAG-HA-OPTN (Panel A) or FLAG-HA-NDP52 (Panel C) were left untreated or depolarized for 1h with AO in the presence or absence of the TBK1 inhibitor MRT (2μM, pre-treatment time 1h). Cells were imaged by confocal microscopy after staining with α-HA (green), α-TOMM20 (red), and Hoechst to detect DNA (blue) (scale bar, 20 microns). Normalized MOC values for co-localization of α-HA and α-TOMM20 were determined from >50 cells as described in Experimental Procedures. Panels B and D analyzed by one-way ANOVA with Dunnett’s multiple comparisons test. *p<0.05, **p<0.01, ****p<0.0001. n.s., not significant. Error bars represent SEM.
(E,F) TBK1 is required for efficient recruitment of OPTN, NDP52, and SQSTM1, but not TAX1BP1 to depolarized mitochondria. Assays were performed as in Panel A in HFT-PARKINWT;TBK1−/− cells (scale bar, 20 microns). Panel F displays MOC values for SQSTM1 and TAX1BP1 co-localization with TOMM20 (error bars represent SEM).
(G) Co-localization of p-TBK1S172 (green) with TOMM20-positive mitochondria (red) in depolarized HFT-PARKINWT cells was examined by immunofluorescence and confocal microscopy prior to image analysis to determine the MOC. Error bars represent SEM from triplicate experiments. (scale bar, 20 microns).
(H) FLAG-HA-OPTN localizes to a subset of mitochondrial domains occupied by p-S65 UB in response to depolarization. HFT-PARKINWT cells stably expressing FLAG-HA-OPTN were depolarized for 75 min with AO and subjected to immunofluorescence with α-HA, α-p-S65 UB, or Hoechst to detect nuclei. Scale bar, 20 microns.
(I) MOC for localization of HA-OPTN with p-S65 UB and for localization of p-S65 UB puncta with HA-OPTN puncta. Error bars are SEM of MOCs obtained from the indicated number of cell.
(J) Localization of p-S65 UB to mitochondrial domains requires PARKIN. HFT cells lacking PARKIN were depolarized for 75 min with AO and subjected to immunofluorescence with α-TOMM20, α-p-S65 UB, or Hoechst to detect nuclei. Scale bar, 20 microns. See also Figure S2.
To rigorously examine the TBK1-dependence of mitophagy adaptor recruitment to damaged mitochondria, we deleted TBK1 using CRISPR-Cas9 in HFT cells that inducibly express PARKINWT. While TBK1+/+ cells displayed focal localization of OPTN, NDP52, SQSTM1, and TAX1BP1 as well as co-localization with mitochondria in response to depolarization, OPTN, NDP52, and SQSTM1 remained diffusely localized in the cytoplasm and failed to undergo puncta formation in TBK1−/− cells (Figure 2E). Normalized MOC values for OPTN, NDP52, and SQSTM1 were comparable to that seen in PINK1−/− cells (Figure 2B,D,F). Thus, TBK1 promotes recruitment of OPTN, NDP52, and SQSTM1, but not TAX1BP1, to depolarized mitochondria.
Focal localization of p-TBK1S172 with OPTN on depolarized mitochondria
Given the known association between TBK1 and OPTN (Gleason et al., 2011; Morton et al., 2008; Wild et al., 2011), as well as our finding that mitochondrial depolarization leads to TBK1 activation and phosphorylation on S172, we examined whether p-TBK1S172 is recruited to depolarized mitochondria and co-localized with OPTN. p-TBK1S172 was essentially undetectable in untreated cells, but in depolarized cells, we observed p-TBK1S172-positive puncta that co-localized with α-TOMM20-positive mitochondria with a MOC of 0.79 (Figure 2G), indicating that activated TBK1 is recruited to depolarized mitochondria. Moreover, p-TBK1S172 puncta overlapped OPTN puncta with a MOC of 0.50 while almost all OPTN puncta were p-TBK1S172 positive (MOC = 0.85) (Figure S2F,G).
Given that OPTN, NDP52, SQSTM1, and TAX1BP1 are all recruited to puncta that co-localize with mitochondria, we also examined the extent to which NDP52, SQSTM1, and TAX1BP1 co-localize with OPTN puncta. HFT-OPTN−/− cells stably reconstituted with FLAG-HA-OPTN were depolarized with AO (1h) and co-localization of OPTN with endogenous NDP52, SQSTM1, or TAX1BP1 examined in a pairwise manner. OPTN puncta were often co-incident with puncta formed by NDP52, SQSTM1, and TAX1BP1, with ~50% of OPTN puncta staining positive for other autophagy adaptors (MOC values ~0.5–0.6) (Figure S2F,G). These results indicate distinct types of recruitment sites, including some in which multiple mitophagy receptors including OPTN coalesce on depolarized mitochondria, and other sites that appear to contain a subset of autophagy adaptors, although we cannot rule out antibody sensitivity or kinetic differences in detection of individual adaptors in puncta.
Given the role of p-S65 UB in the PINK1-PARKIN pathway, we considered the possibility that puncta co-localize with sites of mitochondrial UB enriched for p-S65. However, we found that the domains of mitochondria associated with p-S65 UB chains were much more broadly localized than adaptor puncta (Figure 2H) in response to AO treatment (75 min). While OPTN localized with p-S65 UB with a MOC of 0.7, p-S65 UB co-localization with OPTN was much lower (MOC~0.2) (Figure 2I). As expected, cells lacking PARKINWT failed to generate detectable p-S65 UB positive puncta on mitochondria in response to depolarization as measured using immunofluorescence (Figure 2J). These data suggest that p-S65 UB alone does not specify sites of adaptor protein recruitment.
OPTN poly-UB binding activity promotes TBK1 activation in response to mitochondrial depolarization
OPTN is known to interact with linear and K63 poly-UB chains via its UBAN domain (Gleason et al., 2011). This raised the possibility that recruitment and binding of OPTN to poly-UB chains on depolarized mitochondria contributed to TBK1 activation. To examine this question, we stably expressed a poly-UB chain binding defective OPTN mutant in which a key residue D474 in the UBAN domain (Gleason et al., 2011) was mutated to Asn in HFT-PARKINWT;OPTN−/− cells (Figure 3A). This mutation is equivalent to the D311N mutation in NEMO that disrupts binding to poly-UB chains (Wu et al., 2006) and is near the E478G and A481V patient based mutants (Belzil et al., 2011; Maruyama et al., 2010) also located in the UBAN domain of OPTN that also disrupt the interaction with poly-UB chains (Gleason et al., 2011). While depolarization of OPTN−/− cells reconstituted with OPTNWT led to ~16-fold increase in the ratio of p-TBK1S172/TBK1, the OPTND474N mutant displayed only a 3-fold increase (Figure 3B). The extent of TOMM20 ubiquitylation was equivalent in both OPTNWT and OPTND474N-expressing cells, indicating that upstream pathways involving PINK1 and PARKIN activation are intact. These data indicate that OPTN binding to poly-UB chains on mitochondria is important for TBK1 activation.
Figure 3. TBK1 activation by mitochondrial depolarization requires OPTN and NDP52 and the ability of OPTN to bind poly-UB.
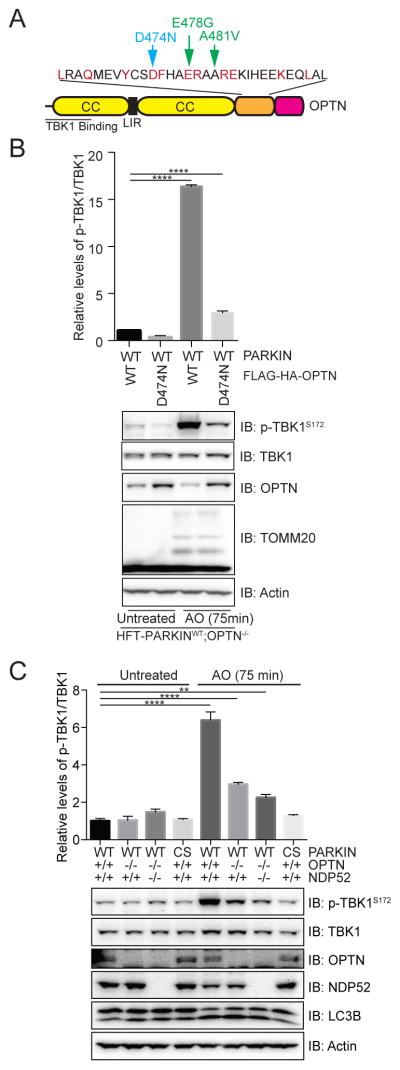
(A) Domain structure of OPTN showing the location of the poly-UB chain defective mutation (D474N) as well as 2 patient-based mutations in the UBAN motif.
(B) TBK1 phosphorylation on S172 in response to mitochondrial depolarization is defective in HFT-PARKINWT;OPTN−/− cells reconstituted with an OPTND474N mutant that lacks poly-UB binding. The results of triplicate experiments for p-TBK1S172/TBK1 levels are shown in the histogram. Error bars represent SEM from triplicate experiments.
(C) HFT PARKIN (WT or CS mutants) with the indicated genotypes for OPTN and NDP52 were left untreated or treated for 75 min with AO and protein extracts subjected to immunoblotting and analysis as described in panel B. Analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (n=3 biological replicates). **p<0.01, ****p<0.0001.
TBK1 activation redundantly requires OPTN and NDP52 in HeLa cells
The finding that both OPTN and NDP52 are recruited to depolarized mitochondria in a TBK1-dependent manner, together with the finding that inactivation of OPTN poly-UB binding did not fully abolish TBK1 activation, led us to examine TBK1 activation in HFT-PARKINWT cells lacking OPTN or both OPTN and NDP52 generated by using CRISPR-Cas9. Depolarization led to a 6-fold increase in the ratio of p-TBK1S172/TBK1 in WT cells, as measured in biological triplicate experiments (Figure 3C). In contrast, depolarization in OPTN deficient cells resulted in only a 2.5-fold increase in the ratio of p-TBK1S172/TBK1. The level of TBK1 activation was further reduced upon deletion of both OPTN and NDP52, although not fully eliminated relative to the activity seen with PARKINCS (Figure 3C). Thus, OPTN and NDP52 account for the majority of TBK1 activation in HeLa cells upon mitochondrial depolarization, indicating that their functions are partially redundant.
Mitochondrial depolarization promotes phosphorylation of autophagy adaptors in a PINK1- and TBK1-dependent manner
TBK1 is known to phosphorylate S403 in SQSTM1 and S177 in OPTN in vivo to promote their association with UB chains or ATG8 proteins, respectively (Matsumoto et al., 2011; Wild et al., 2011). To examine whether PINK1 and PARKIN promote depolarization dependent phosphorylation of OPTN, NDP52, SQSTM1, or TAX1BP1, we initially employed HFT-PARKINWT cells with or without PINK1 in the presence or absence of MRT to inhibit TBK1 activity. As expected, TBK1 was phosphorylated and TOMM20 was ubiquitylated in the presence, but not absence, of PINK1 as assessed by SDS-PAGE and immunoblotting (Figure 4A). Importantly, the accumulation of p-TBK1S172 was not affected by pre-treatment of cells with the MRT TBK1 inhibitor, indicating that TBK1 activation was not autocatalytic, as previously found in innate immunity pathways (Clark et al., 2011) (Figure 4A, S3B). In parallel, these extracts were also separated on Phos-tag gels, which separate proteins based on both size and the extent of phosphorylation. Multiple forms of NDP52, OPTN, and SQSTM1 were observed in PINK1+/+ cells in the absence of AO and additional forms of lower mobility were observed upon AO treatment in PINK1+/+ cells but not in PINK1−/− cells (Figure 4B). These lower mobility forms were absent when depolarization was performed in the presence of MRT inhibitor (Figure 4B), and were also absent in depolarized HFT-PARKINWT cells engineered to lack TBK1 (Figure 4C,D) or cells transfected with 4 independent siRNAs targeting TBK1 (Figure S3), ruling out a possible non-specific effect of MRT. Moreover, as expected, PARKINCS-expressing cells also failed to produce the more slowly migrating species for OPTN, NDP52, and SQSTM1 (Figure 4C,D), further supporting the importance of PARKIN UB ligase activity for this pathway. TAX1BP1, a known target of the PARKIN-PINK1 pathway (Sarraf et al., 2013), underwent a mobility shift consistent with ubiquitylation as assessed by SDS-PAGE, but did not produce detectable discrete more slowly migrating species upon separation in Phos-tag gels under these conditions (Figure 4C,D). These data suggest that the PINK1-PARKIN pathway promotes OPTN, NDP52, and SQSTM1 phosphorylation via TBK1.
Figure 4. Phosphorylation of OPTN, NDP52, and SQSTM1 in response to mitochondrial depolarization is dependent upon PINK1, PARKIN, and TBK1.

(A,B) HFT-PARKINWT cells with or without PINK1 were depolarized with AO (75 min) in the presence or absence of the TBK1 inhibitor MRT (2μM, pre-treatment time 1h) and extracts subjected to SDS-PAGE or electrophoresis on Phos-tag gels prior to immunoblotting with the indicated antibodies. The position of depolarization dependent phosphorylated forms of mitophagy adaptors are indicated by arrows (Panel B).
(C,D) As in panels A and B except that HFT cells expressing either PARKINWT or PARKINCS in the presence or absence of TBK1 were employed. See also Figure S3.
Quantitative proteomics identifies sites of depolarization-dependent OPTN phosphorylation by TBK1
To examine depolarization-dependent OPTN phosphorylation, we initially used LC-MS2 to identify phosphorylation sites in OPTN in PINK1WT or PINK1−/− HFT-PARKINWT cells with or without AO treatment, detecting two major phosphorylation sites (S473 and S513) that increased in abundance after AO treatment, based on spectral counting (Figure 5A,B and S4A,B). We also identified p-S177 (Gleason et al., 2011; Wild et al., 2011) in the presence or absence of depolarization. None of these sites were detected in PINK1−/− cells (Figure 5B). We also found that recombinant TBK1 phosphorylated recombinant OPTN in vitro on S177, S473, and S513 (Figure S4C). OPTN was previously shown to be phosphorylated in vitro on S177 and S513 by TBK1 (Gleason et al., 2011).
Figure 5. Quantitative proteomics identifies depolarization-dependent phosphorylation sites in OPTN that promote poly-UB binding.<.

br>(A) Domain structure of OPTN showing the major sites of phosphorylation as well as patient-based mutations in the UBAN motif.
(B) Identification of phosphorylation sites in OPTN upon mitochondrial depolarization by LC-MS2. HFT-PARKINWT cells expressing FLAG-HA-OPTN were depolarized (75 min) and the FLAG-HA-OPTN protein isolated by immunoprecipitation prior to trypsinization and analysis by LC-MS2.
(C) Quantification of mitochondrial depolarization-dependent OPTN phosphorylation on S177 and S513 using TMT-based LC-MS3 proteomics. Error bars represent SEM from triplicate experiments.
(D–F) Quantification of mitochondrial depolarization-dependent OPTN phosphorylation on S177, S473 and S513. OPTNS473 and S513 phosphorylation quantification from immune complexes was performed using AQUA proteomics (D–E) with heavy reference synthetic peptides (Table S1). Normalization for total OPTN levels was performed by also subjecting the same tryptic digest to TMT analysis (F) and quantifying all OPTN peptides identified. Analysis of OPTNS177 phosphorylation (F) was performed using TMT. N.D. - not determined due to mutation of a residue in the peptide of interest.
(G,H) 6His-GST-OPTN and its site-specific phosphorylated forms (p-S177, p-S473, p-S513, and p-S473/p-S513) were expressed in bacteria using a tRNAp-Ser suppressor system (G) and purified to near heterogeneity, as seen by staining of SDS-PAGE gels with Coomassie Blue (H).
(I) Quantification of phosphorylated recombinant 6His-GST-OPTN proteins using TMT-based proteomics.
(J) The indicated unphosphorylated or p-S65 phosphorylated K48 or K63 UB(2–7) chains were incubated with the indicated phosphorylated forms of GST-OPTN, and after washing, bound proteins were released and subjected to SDS-PAGE and immunoblotting with α-UB. See also Figure S4.
To examine phosphorylation dynamics, we first employed a Tandem Mass Tagging (TMT)-based approach using Multi-Notch detection of reporter ions in an LC-MS3 experiment performed in biological triplicate (see Experimental Procedures) (Figure S4A). This allowed robust detection and relative quantification of S177 and S513 phosphorylation with stably expressed FLAG-HA-OPTN in a mitochondrial depolarization dependent manner, with 4-fold and 7-fold increases in the abundance of p-S177 and p-S513 peptides observed upon AO treatment (Figure 5C). This increase was absent in PINK1−/− cells. For technical reasons, we were unable to quantify S473 phosphorylation in this experiment. To examine the dynamics of S473 phosphorylation, we employed a more sensitive Absolute Quantification (AQUA) approach (Figure S4A). An isotopically heavy reference peptide corresponding to the p-S473 peptide (AQMEVYCpSDFHAER) was added in known amounts to immune complexes of OPTN, OPTNS177A, OPTNS513A, or OPTND474N purified from HFT-PARKINWT or PARKINCS cells with or without depolarization. Based on AQUA, there was a ~42.5-fold PARKIN-dependent increase in the abundance of S473 phosphorylation (Figure 5D). Analogous experiments examining S513 phosphorylation revealed a ~14.6-fold increase in response to depolarization (Figure 5E). AQUA indicated a phosphorylation stoichiometry of ~1% for S473 and ~10% for S513. The increase in S473 phosphorylation was reduced to a ~4.75-fold increase in the context of OPTNS513A, suggesting coordinated phosphorylation. In a parallel TMT experiment, we observed a 4.35-fold increase in OPTN S177 phosphorylation that was absent in the context of PARKINCS or the OPTND474N mutant that cannot bind poly-UB (Figure 5F). Thus, mitochondrial depolarization promotes OPTN phosphorylation at S177, S473, and S513 in a PARKIN UB-ligase dependent manner.
Phosphorylation of OPTN’s UBAN domain by TBK1 enhances binding to poly-UB chains
Previous studies (Gleason et al., 2011) have shown that OPTN binds preferentially to linear as well as K63 UB chains. This interaction is abolished in the context of either the D474N mutation or the patient mutant E478G (Gleason et al., 2011), both of which are located in the UB-binding UBAN domain (Figure 5A). The identification of OPTNS473 and OPTNS513 phosphorylation in response to TBK1 activation was interesting, given that these sites are within and adjacent to the UB-binding UBAN domain, respectively (Figure 5A). To examine whether phosphorylation of S473 and S513 adjacent to the UBAN motif affect poly-UB binding, we employed a tRNAp-Ser amber codon suppressor system to synthesize site-specifically phosphorylated GST-OPTN proteins in E. coli (Pirman et al, 2015) (Figure 5G), thus avoiding the use of phospho-mimetics. Purified GST-OPTN phosphoproteins (Figure 5H) were found to be quantitatively phosphorylated at the desired position(s) using TMT-based quantitative proteomics (Figure 5I).
We next examined the ability of unphosphorylated and site-specifically phosphorylated GST-OPTN proteins to bind to K48 or K63 chains in vitro (Figure 5J), two major chain types found on depolarized mitochondria (Ordureau et al., 2014). As expected, GST-OPTNWT associated with K63, but not K48 linked UB chains (Gleason et al., 2011; Ordureau et al., 2015a), and the extent of interaction was similar with GST-p-OPTNS177 and GST-p-OPTNS513 and as shown previously, OPTN cannot bind mono-UB with or without p-S65 phosphorylation (Figure 5J) (Ordureau et al., 2015a). In contrast, association of K63 poly-UB chains with GST-p-OPTNS473 was dramatically increased, and was maintained with dual phosphorylation of S473 and S513 (Figure 5J). Interestingly, while GST-OPTNWT did not associated with K48 UB chains, binding was detectable when S473 or S473 and S513 in OPTN were phosphorylated.
Given that S65 in mitochondrial poly-UB chains is phosphorylated via PINK1 in vivo (stoichiometry of ~0.2) (Ordureau et al., 2014; Ordureau et al., 2015a), we explored the possibility that the combination of OPTNS473 and/or OPTNS513 phosphorylation and K63 UB chain phosphorylation would augment the extent of binding. As found previously (Ordureau et al., 2015a), association between p-S65 poly-UBK63 chains (~0.5 stoichiometry) was reduced with all forms of GST-OPTN tested (Figure 5J). However, both GST-p-OPTNS473 and GST-p-OPTNS473/S513 maintained an ability to interact with p-S65 poly-UBK63 chains, albeit less efficiently than with unphosphorylated K63 UB chains (Figure 5J). Although linear chains are found in only very low abundance on depolarized mitochondria (Ordureau et al., 2014; Cunningham et al., 2015), we found, as expected (Gleason et al., 2011), that GST-OPTN associated tightly with M1 poly-UB chains compared to K48 or K63 (Figure S4E) and this was slightly enhanced when S473 and S473/S513 were phosphorylated (Figure S4D). As with K63 chains, interaction was impaired when poly-UB M1 chains were phosphorylated on S65 with a stoichiometry of 0.7, as determined by AQUA proteomics (Figure S4D,E,F). Thus, phosphorylation of S473 and dual phosphorylation of S473 and S513 facilitate binding of OPTN to poly-UB chains in vitro and can partially rescue the loss of binding due to S65-UB phosphorylation.
Phosphorylation of OPTN on S473 and S513 promotes TBK1 activation and recruitment of OPTN to depolarized mitochondria
Given that TBK1 can phosphorylate OPTN on S177, S473 and S513 in response to mitochondrial depolarization, we explored the function of these events in vivo. Initially, we found that mutation of none of these sites to Ala affected the ability of OPTN to associate with TBK1, as assessed by immunoprecipitation and immunoblotting with or without mitochondrial depolarization (Figure 6A). Interestingly, the depolarization-dependent mobility shift seen in Phos-tag gels for OPTN was abolished in the S513A and S473A/S513A mutants (Figure S5A). However, depolarization-dependent phosphorylation of NDP52 and SQSTM1 was unaffected (Figure S5A). We then examined TBK1 activation in gene-edited OPTN−/− cells stably reconstituted with GFP-OPTN proteins in which phosphorylation sites were mutated to Ala either singly or in combination (Figure 6B). The level of GFP-OPTN in these cells was lower than endogenous OPTN in parental HFT cells (Figure 6B). In cells reconstituted with OPTNWT, a 4.2-fold increase in the ratio of p-TBK1S172/TBK1 was observed upon depolarization, and this was reduced to 2.3-fold in OPTNS473/S513-expressing cells. In contrast, individual mutation of S177, S473, or S513 had little effect TBK1 activation (Figure 6B). Reduced TBK1 activation was also observed in HFT-OPTN−/−;NDP52−/− cells reconstituted with GFP-OPTNWT and mutant proteins in that the S473A/S513A mutant had similar activity as OPTN D474N and E478G mutants that cannot associate with poly-UB (Figure S5B).
Figure 6. Role of OPTN phosphorylation in TBK1 activation and OPTN recruitment to depolarized mitochondria.

(A) Phosphorylation site mutants in OPTN do not affect association of TBK1 with OPTN. The indicated FLAG-HA-OPTN mutants in phosphorylation sites or in D474 required for UB binding were expressed in HFT-PARKIN (WT or CS mutants) cells engineered to lack OPTN using CRISPR-Cas9. Cells were either left untreated or treated with AO for 75 min and α-FLAG immunoprecipitates (OPTN) probed with antibodies against endogenous TBK1. Lysates were also immunoblotted to demonstrate loading.
(B) Phosphorylation of S473 and S513 is required for efficient TBK1 activation in response to mitochondrial depolarization. The indicated GFP-OPTN mutants in phosphorylation sites were expressed in HFT-PARKIN (WT or CS mutants) cells engineered to lack OPTN using CRISPR-Cas9. Cells were either left untreated or treated with AO for 90 min lysates subjected to immunoblotting with the indicated antibodies. Analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (n=3 biological replicates). **p<0.01, ****p<0.0001, n.s., not significant.
(C,D) OPTNS473A/S513A is defective in retention on depolarized mitochondria. In Panel C, HFT-PARKINWT;OPTN−/− cells stably expressing the indicated GFP-OPTN mutants were depolarized with AO (90 min) and subjected to immunofluorescence using the indicated antibodies or direct observation of GFP-OPTN proteins. Scale bar, 20 microns. In Panel D, MOC values were obtained for >50 cells from triplicate experiments as described under Experimental Procedures. Analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (n=3 biological replicates). ****p<0.0001, n.s., not significant. See also Figure S5.
We then examined the effect of these phosphosite mutations on recruitment of GFP-OPTN to depolarized mitochondria (90 min), and compared the phenotypes with that observed with GFP-OPTND474N, which is defective in poly-UB binding (Figure 6C). While WT, S177A, S473A, and S513A single mutants formed punta that co-localized with TOMM20-positive mitochondria in response to depolarization, the S473/S513 double mutant was as defective as the GFP-OPTND474N mutant analyzed in parallel, as determined by normalized MOC analysis (Figure 6C,D). We cannot rule out a subtle or kinetic defect in the S473A and S513A single mutants, as the abundance of diffusely localized OPTN in these cells was higher than that seen in OPTNWT cells (Figure 6C). Taken together, these data indicate a role for phosphorylation of both S473 and S513 in OPTN in the ability to facilitate TBK1 activation, possibly due to a reduction in the ability of these OPTN mutants to be effectively retained on poly-UB chains tethered to depolarized mitochondria.
TBK1 and OPTN/NDP52 are required for efficient mitophagy
Given the relationship between OPTN/NDP52 and TBK1, as well as the ability of OPTN and NDP52 to interact with poly-UB chains (Ordureau et al., 2015a), we examined mitophagy efficiency in the context of defects in the pathway. As expected, ~93% of HFT-PARKINWT cells displayed cleared (~60%) or aggregated (~33%) mitochondria as detected by α-DNA staining 31h post-AO treatment and this required active PARKIN, as PARKINCS cells were devoid of aggregated or cleared mitochondria (Figure 7A), indicating that PINK1 alone isn’t sufficient for detectable mitophagy in this system. In contrast, HFT-PARKINWT cells engineered to lack TBK1 using CRISPR-Cas9 displayed only ~25% fully cleared mitochondria, indicating a significant delay in mitophagy (Figure 7A). In this context, ~61% of cells displayed aggregated mitochondria, indicating that TBK1 isn’t required for depolarization-dependent aggregation. Defects in mitochondrial clearance were also seen at 24h in TBK1−/− cells as assessed by both α-DNA staining and immunoblotting of cell extracts using α-TOMM20 antibodies as an independent measure of mitophagy (Figure S6A–C).
Figure 7. A TBK1-OPTN-NDP52 network is required for efficient mitophagy and recruitment of LC3B to depolarized mitochondria.

(A) HFT cells expressing the indicated PARKIN proteins and harboring the indicated TBK1, OPTN, and NDP52 genotypes were treated with AO and stained for α-DNA to detect mitochondria after 31h. Image analysis was used to determine the fraction of cells displaying unaggregated, aggregated, and cleared mitochondria as described (Ordureau et al., 2015a).
(B) HFT-PARKINWT;TBK1−/− cells alone or reconstituted with the indicated TBK1 protein were subjected to AO treatment (30h) and mitochondria monitored and quantified as in panel A.
(C) HFT-PARKINWT;OPTN−/−;NDP52−/− cells alone or reconstituted with the indicated FLAG-HA-OPTN proteins were subjected to AO treatment (30h) and mitochondria monitored and quantified as in panel A.
(D) OPTN and NDP52-dependent recruitment of the ATG8 protein LC3B to damaged mitochondria. Cells were stained with α-LC3B to mark autophagosomes (green), α-TOMM20 to mark mitochondria, and Hoechst (blue) to mark nuclei and imaged by confocal microscopy (scale bar, 20 microns). The fraction of cells showing co-localization of LC3B and TOMM20 staining was determined by image analysis examining 24,455 cells (WT) and 17,343 cells (OPTN−/−;NDP52−/−).
(D) Schematic of a positive feedback mechanism describing the role of UB chains, mitophagy adaptor phosphorylation, and TBK1 phosphorylation in ATG8 recruitment and mitophagy. In Step 1, PINK1 and PARKIN are activated and collaborate to generate UB chains on the MOM in Step 2, including chains with K63 linkages. In Step 3, mitophagy adaptors are recruited to various poly-UB chains, and in this context, TBK1 is recruited to mitochondria and activated by phosphorylation on S172 (Step 4) in a manner that depends primarily upon OPTN and NDP52 recruitment to poly-UB chains. The kinase required for this is unknown. In Step 5, activated TBK1 phosphorylates OPTN and NDP52. Steps 4 and 5 may be concerted. Phosphorylation of OPTN on S473 stabilizes its binding to various poly-UB chains while phosphorylation of S177 promotes association with LC3B (Step 6).
Analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (n=3 biological replicates). ***p<0.001, ****p<0.0001. n.s., not significant. See also Figure S6.
Given that OPTN and NDP52 are required for near maximal TBK1 activation, we directly examined their involvement in mitophagy. HFT-PARKINWT cells lacking OPTN displayed a modest reduction in the percentage of cells displaying cleared mitochondria; 60% cleared mitochondria in the presence of OPTN and 45% clearance in the absence of OPTN (Figure 7A). In contrast, cells engineered to lack both OPTN and NDP52 displayed only 28% clearance at 31h, comparable to that seen with TBK1 deletion (Figure 7A). Similar results for OPTN/NDP52 deletion cells were obtained at 24h post-depolarization using both α-DNA staining and TOMM20 abundance by immunoblotting (Figure S6B,C).
In order to test the involvement of TBK1 kinase activity and its ability to bind to OPTN in mitophagy, HFT-PARKINWT;TBK1−/− cells were reconstituted with TBK1WT, a catalytically defective TBK1K38A mutant, and the ALS patient-derived mutant TBK1E696K that fails to bind OPTN (Freischmidt et al., 2015) (Figure 7B). While TBK1WT partially rescued the mitophagy defects observed in TBK1−/− cells, consistent with its expression level being lower than that found for endogenous TBK1 in these cells (Figure S6D), neither the K38A or E696K mutants rescued the defect seen in the TBK1−/− cells despite expression levels similar to or higher than re-introduced TBK1WT (Figure 7B, S6D). Similar experiments using OPTN−/−;NDP52−/− cells reconstituted with OPTN phosphosite mutants demonstrated a partial (~20%) defect in mitophagy with the S473 and S513 double Ala point mutant (Figure 7C). Taken together, these data indicate involvement of TBK1, OPTN, and NDP52 in depolarization-dependent mitophagy. Consistent with this, cells lacking both OPTN and NDP52 were defective in the ability to recruit LC3B to damaged mitochondria, as determined by high-content imaging to measure mitochondrially localized LC3B puncta upon AO treatment (Figure 7D), potentially explaining the defects in mitophagy observed.
DISCUSSION
Recent reports have placed a central focus on a network of proteins including the autophagy receptor OPTN and its regulatory kinase TBK1, in part due to links between these proteins and neurodegenerative diseases, and the parallels between mitophagy and xenophagy. First, OPTN was found to promote mitophagy in HeLa cells (Wong and Holzbaur, 2014), as well as restrict Salmonella infection in tissue culture cells, where its phosphorylation on S177 by TBK1 is thought to promote association with ATG8 proteins (Wild et al., 2011). In parallel, an additional autophagy adaptor NDP52 has also been linked with xenophagy (Thurston et al., 2009; Verlhac et al., 2015; von Muhlinen et al., 2012). Second, both OPTN and TBK1 are mutated in ALS and related diseases, although the molecular defects found in various alleles are poorly understood outside of the finding that mutations near the C-terminal coiled-coil domain of TBK1 abolish OPTN binding (Cirulli et al., 2015; Freischmidt et al., 2015). Here, we find that mitochondrial ubiquitylation by PARKIN promotes a program of autophagy adaptor recruitment that is critical for activation of a pool of TBK1 associated with autophagy adaptors. Activation of TBK1 in this context promotes phosphorylation of autophagy adaptors and we demonstrate that depolarization-dependent OPTN phosphorylation by TBK1 enhances its ability to bind to poly-UB chains in vitro and maintain OPTN association with depolarized mitochondria in cells.
A working model for this process is presented in Figure 7F. Mitochondrial depolarization promotes the assembly of K6, K11, K48, and K63 UB chains on numerous mitochondrial proteins via PINK1-activated PARKIN (Steps 1 and 2) (Cunningham et al., 2015; Ordureau et al., 2014). In this context, PINK1 functions to phosphorylate both PARKIN and UB chains on mitochondria, which forms the basis of a feed-forward mechanism involving PARKIN recruitment and retention to p-S65-UB chains, resulting in further UB chain synthesis (Ordureau et al., 2015b; Pickrell and Youle, 2015). Our data suggest that the accumulation of poly-UB chains drive multiple downstream events promoting mitophagy, possibly in a highly concerted manner. We find that OPTN, NDP52, SQSTM1, and TAX1BP1 are rapidly recruited to puncta on damaged mitochondria, and the recruitment of OPTN, NDP52, and SQSTM1 (but not TAX1BP1) in puncta is blocked by inhibition or deletion of TBK1 (Step 3). Recruitment of p-TBK1S172 and p62 to damaged mitochondria has also been recently reported (Matsumoto et al., 2015). This binding step in the context of OPTN and NDP52 is critical for TBK1 phosphorylation on its activation loop apparently within the pool of TBK1 that is associated with OPTN and NDP52 (Step 4). Indeed, deletion of OPTN alone reduces TBK1 phosphorylation on S172 by ~70% while deletion of both OPTN and NDP52 further reduces TBK1 phosphorylation, indicating that NDP52 and OPTN function partially redundantly in this process. The ability of OPTN to promote activation of TBK1 depends on its poly-UB binding activity and localization, as mutation of a single residue in the UB binding UBAN motif of OPTN largely abolishes TBK1 S172 phosphorylation upon mitochondrial depolarization in OPTN−/− cells reconstituted with OPTNWT or OPTND474N proteins. Further studies are required to understand the process underlying residual TBK1 activation in cells lacking both OPTN and NDP52. We cannot exclude a contribution of a SQSTM1-TBK1 complex in activation of TBK1 in some circumstances, but in the cells we have employed, the contribution of SQSTM1 is small, possibly reflecting the relative abundance of the mitophagy adaptors in the cell. Importantly, experiments using a small-molecule TBK1 inhibitor indicate strongly that TBK1 activation in this in vivo context requires an upstream kinase, and similar to previous reports (Clark et al., 2011; Clark et al., 2009), does not occur through a previously described in vitro auto-catalytic mechanism (Ma et al., 2012). Interestingly, OPTN puncta often but not always co-localize with other autophagy adaptor puncta. Further studies are required to understand whether “domains” of adaptor recruitment are distinct and what underlies the array of adaptors that are recruited to individual puncta.
Using quantitative proteomics, we found that mitochondrial depolarization leads to phosphorylation of S177 in OPTN, which is known to be phosphorylated by TBK1 (Wild et al., 2011), as well as phosphorylation of two sites (S473 and S513) located near the UBAN domain. Surprisingly, phosphorylation of S473 led to a dramatic increase in binding to K48 and K63 poly-UB chains in vitro, and this was maintained upon dual phosphorylation of S473 and S513, while phosphorylation of S513 alone had no effect. Association with M1 poly-UB chains was also increased but to a lesser extent. Moreover, phosphorylation of S177 had no effect on poly-UB binding in vitro, consistent with the idea that its primary role is in LC3 recruitment (Wild et al., 2011). Given that UB chains assembled on mitochondria by PARKIN are phosphorylated with a stoichiometry of ~0.2 (Ordureau et al., 2014), we explored the interaction of these various phosphorylated forms of OPTN with phosphorylated poly-UB chains. Overall, phosphorylation with a stoichiometry of ~0.5–0.7 had an inhibitory effect on the overall efficiency of binding with all three chain types (Ordureau et al., 2015a), but the extent of binding between p-S65 K63 poly-UB chains and p-OPTNS473 or p-OPTNS473/S513 was comparable to that seen between unphosphorylated OPTN and unphosphorylated K63 chains. Modeling studies (not shown) of the OPTN UBAN domain using the NEMO-diK63 UB complex as a template (pdb: 2ZVO) (Rahighi et al., 2009) indicate a potential interaction between p-S473 and Arg42 in the proximal UB molecule, possibly explaining the higher affinity interaction with p-OPTNS473, but structural studies with UB polymers are required to fully understand the basis for increased affinity. Interestingly, while p-S65 UB is localized broadly on the majority of mitochondria in depolarized cells, OPTN is localized within puncta and co-localizes with only a small subset of p-S65 UB decorated mitochondria. This suggests that p-S65 UB within chains on depolarized mitochondria does not exclusively dictate the sites of recruitment of autophagy adaptors. Further studies are required to understand any relationship with p-S65 UB within poly-UB chains.
Our data present an apparent paradox. On one hand, OPTN and NDP52 are required for efficient TBK1 activation in response to mitochondrial depolarization. On the other hand, TBK1 activity is required for efficient retention of OPTN, NDP52, and SQSTM1 on damaged mitochondria. The simplest explanation for this is that the ability of unphosphorylated OPTN to bind to poly-UB chains is sufficient for a transient encounter that promotes activation of TBK1 through an unknown mechanism (Figure 7F, Steps 3 and 4), but activated TBK1 in a concerted manner phosphorylates the mitophagy adaptors to facilitate their retention on mitochondria, binding to ATG8 proteins, and possibly other functions. Although this may involve enhanced affinity for poly-UB chains, it may also involve other factors that help to stabilize mitochondrial “domains” with a high concentration of mitophagy adaptors coalescing into structures that promote mitophagy (Figure 7F, Steps 5 and 6), an idea that is suggested by the localization patterns of autophagy adaptors on damaged mitochondria. Further analysis of this system will facilitate an understanding of how mutations found in neurodegenerative diseases alter the efficiency of the pathway and impact the autophagic targeting of other types of cargo (e.g. protein aggregates) linked with disease.
EXPERIMENTAL PROCEDURES
Cell Lines
Inducible HeLa Flp-In T-Rex (HFT) cells expressing with or without expression of PARKINWT or PARKINCS proteins (Ordureau et al., 2015a) were depolarized were gene edited (Ran et al., 2013) to ablate OPTN, NDP52, TBK1, of PINK1 and in some cases, reconstituted with the indicated proteins using a Flp-in construct in which PARKIN is expressed via an Internal Ribosomal Entry Site. Cells were depolarized with antimycin A (10 μM) and oligomycin A (5 μM) prior to analysis by immunoblotting using SDS-PAGE or Phos-Tag (300-93523, Wako) gels and confocal microscopy.
Analysis of Phosphorylation
HA-FLAG-OPTN immune complexes from untreated or AO treated HFT cells were isolated and trypsinized prior to analysis by mass spectrometry using either a Thermo Q-Exactive or Thermo fusion instruments (see Supplemental Experimental Procedures).
Binding of OPTN to poly-UB chains
The indicated GST-OPTN phosphoproteins proteins were expressed using suppressor tRNA system in bacteria, allowing for direct incorporation of phosphoserine at a desired residue position (Pirman et al., 2015). Binding of GST-OPTN to poly-UB chains with or without phosphorylation with PINK1 was performed as described (Ordureau et al., 2015a).
Mitophagy assays
The indicated cells were treated with AO for 24 or 30 h and stained with either anti-DNA antibodies or anti-TOMM20 antibodies prior to quantification of mitochondria using confocal microscopy as described (Ordureau et al., 2015a).
Supplementary Material
Acknowledgments
We thank the Nikon Imaging Center and the Imaging and Data Analysis Core at Harvard Medical School for imaging support, Steve Gygi, Richard Youle and Ivan Dikic for helpful discussions, and Sean Beausoleil for AQUA peptides. This work was supported by research grants to J.W.H. from the NIH (R37 NS083524) and Biogen. A.O. was supported by an Edward R. and Anne G. Lefler Center Postdoctoral Fellowship. J.W.H. is a consultant for Millennium: the Takada Oncology Company and Biogen.
Footnotes
AUTHOR CONTRIBUTIONS
J.-M.H., A.O., and J.W.H. conceived the study. J.-M.H. and A.O. performed experiments. J.A.P. provided proteomics support. J.R. provided reagents. J.W.H., J.-M.H. and A.O. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Belzil VV, Daoud H, Desjarlais A, Bouchard JP, Dupre N, Camu W, Dion PA, Rouleau GA. Analysis of OPTN as a causative gene for amyotrophic lateral sclerosis. Neurobiol Aging. 2011;32:555 e513–554. doi: 10.1016/j.neurobiolaging.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Peggie M, Plater L, Sorcek RJ, Young ER, Madwed JB, Hough J, McIver EG, Cohen P. Novel cross-talk within the IKK family controls innate immunity. Biochem J. 2011;434:93–104. doi: 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]
- Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol. 2015;17:160–169. doi: 10.1038/ncb3097. [DOI] [PubMed] [Google Scholar]
- Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, Hubers A, Weydt P, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6:871–878. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem. 2011;286:35663–35674. doi: 10.1074/jbc.M111.267567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–39. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, Lange G, Caroll J, Reitz B, Donnelly A, et al. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J Biol Chem. 2002;277:13840–13847. doi: 10.1074/jbc.M110474200. [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal. 2011;4:ra7. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Veron M, Agou F, Israel A. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009;28:2885–2895. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A. 2012;109:9378–9383. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24:4429–4442. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008;582:997–1002. doi: 10.1016/j.febslet.2008.02.047. [DOI] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, Harper JW. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A. 2015a;112:6637–6642. doi: 10.1073/pnas.1506593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Munch C, Harper JW. Quantifying Ubiquitin Signaling. Mol Cell. 2015b;58:660–676. doi: 10.1016/j.molcel.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirman NL, Bareber KW, Ma NJ, Haimovich AD, Rogulina S, Isaacs FJ, Rinehart J. A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat Commun. 2015 doi: 10.1038/ncomms9130. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JJ, Scavone F, Cooper EM, Kane LA, Youle RJ, Boeke JD, Cohen RE. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat Methods. 2012;9:303–309. doi: 10.1038/nmeth.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I. Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol Cell. 2012;47:797–809. doi: 10.1016/j.molcel.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, Faure M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe. 2015;17:515–525. doi: 10.1016/j.chom.2015.02.008. [DOI] [PubMed] [Google Scholar]
- von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell. 2012;48:329–342. doi: 10.1016/j.molcel.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal. 2011;4:pe39. doi: 10.1126/scisignal.2002355. [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111:E4439–4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.