

Abstract
In chronic lymphocytic leukemia (CLL), strategies to overcome drug resistance due to p53 dysfunction are highly needed. Platinum-based compounds such as cisplatinum (CDDP) are active in fludarabine-refractory CLL through a largely unknown mechanism. We analyzed the mechanism of action of CDDP in the context of p53 dysfunctionality. In vitro treatment with CDDP did not induce death in quiescent CLL cells, but did induce apoptosis in CD40-ligand (and CpG) stimulated and proliferating cells, irrespective of p53 function. In the p53 dysfunctional prolymphocytic cell-line MEC1, CDDP treatment resulted in apoptosis, cell cycle arrest and ABL1-dependent expression of TAp73, CDKN1A, PUMA and BID. TAp73 RNA-interference decreased sensitivity to CDDP. Finally, both in vitro stimulated CLL cells and lymph node (LN) derived CLL cells showed increased TAp73 expression in comparison with quiescent peripheral blood derived cells. Activity of CDDP may therefore be mediated by TAp73, especially in the context of activation such as occurs in the LN microenvironment.
Keywords: Chronic lymphocytic leukemia, drug resistance, p53, TAp73, CDDP
Introduction
Although considerable advances have been made in upfront treatment strategies for chronic lymphocytic leukemia (CLL), at present virtually all patients experience relapsing disease requiring renewed treatment. Most cytotoxic drugs, including fludarabine (F-ara-A), depend on intact p53 function for their activity, and repeated cycles of therapy may eventually result in drug resistance due to outgrowth of clones with cytogenetic alterations affecting genes involved in the p53 response [1]. Fludarabine-refractory disease infers a very poor prognosis with, until recently, a median life expectancy of less than 1 year [2,3]. Recent advances using B-cell receptor (BCR) signaling-targeted drugs (e.g. ibrutinib and idelalisib) have caused a shift toward a non-chemotherapy treatment era (reviewed by Jones and Byrd) [4]. However, available data suggest that these drugs do not induce deep remissions, and should be taken continuously to prevent relapse.
So far, the only treatment modality resulting in long-lasting remissions and perhaps even cure in these patients is reduced intensity allogeneic hematopoietic stem cell transplant (RIST). Response status prior to RIST is found to be an important determinant of long-term outcome, as patients with high disease burden, particularly bulky lymphadenopathy at the time of transplant or poor response to last treatment, have the tendency to relapse more often, whereas patients with progressive disease uniformly do poorly [5]. Currently, no optimal induction regimen has been established for patients with chemorefractory disease. Therefore, the identification of alternative drug targets, independent of p53 function, is urgently needed.
Platinum-based compounds such as cisplatinum (CDDP) are among the most active antitumor agents. These compounds are mainly used in the treatment of solid malignancies. In relapsed and refractory B cell non-Hodgkin lymphoma, platinum-based compound containing regimens have been successfully applied as salvage treatment and also as induction therapy prior to (autologous) stem cell transplant [6–8]. Evidence for the clinical activity of platinum-based compounds in refractory CLL comes from several studies. In a phase I–II trial the OFAR regimen (oxaliplatin, fludarabine, cytarabine and rituximab) resulted in a response in 33% of patients with fludarabine-resistant CLL (and in 37% of patients with documented deletion of 17p) [9]. In a subsequent phase II clinical trial using a slightly modified OFAR regimen, the overall response rate in patients with relapsed or refractory CLL was 55% [10]. We recently found marked activity of the R-DHAP regimen (rituximab, dexamethasone, cytarabine and CDDP) in eight of 10 patients with fludarabine-refractory disease, including six of eight patients with bulky disease [11]. Based on these data, R-DHAP was chosen as induction regimen in a prospective Dutch/Belgian phase II trial (HOVON 88) studying the efficacy and safety of salvage chemotherapy followed by RIST in patients with fludarabine-refractory and relapsed high-risk CLL.
The observation that platinum-based compounds are active in patients with fludarabine-refractory disease and/or with del17p suggests that, in contrast to most drugs used in the treatment of CLL, the mechanism of action does not depend on functional p53. Knowledge about p53 independent death pathways could aid in the development of novel treatment strategies for the expanding group of patients with refractory CLL. Therefore, to elucidate the mechanism of action of CDDP, we performed in vitro and ex vivo studies in p53 dysfunctional CLL samples as well as in vitro studies in the p53 dysfunctional cell-line MEC1.
Materials and methods
Patient material
Peripheral blood mononuclear cells (PBMCs) from patients with CLL were isolated, frozen and stored as previously described [12]. Approval for these side-studies was obtained from the Amsterdam Academic Medical Center Medical Ethical Committee. Informed consent was separately obtained in accordance with the Declaration of Helsinki. All included samples contained > 90% CD5 + CD19+ cells. p53 dysfunction was assessed by reverse transcription-multiplex ligation-dependent probe amplifi (RT-MLPA) as previously described [13]. Lymph node (LN) material, diffusely infiltrated by CLL (>90%), was frozen in liquid nitrogen directly after surgical removal. These samples were taken before the start of (a new line of) treatment and after a treatment-free interval of at least 1 year.
Cell culture and drugs
MEC1 cells were provided by Dr. M. Hallek (Uniklinik Koeln). The immunophenotype matched the original description and the complex karyotype was highly similar to what was previously described [14]. p53 dysfunction was confirmed by the absence of p53 protein (Western blot) and absence of up-regulation of PUMA mRNA (RT-MLPA) and CD95 (FAS; fluorescence activated cell sorting [FACS]-staining) upon radiation (Supplementary Figure 2 to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.996751). Cells were cultured in Iscove modified Dulbecco medium (IMDM; Gibco Life Technology, Paisley, UK) supplemented with 10% (vol/vol) heat inactivated fetal calf serum (FCS; ICN Biomedicals, Meckenheim, Germany), 100 μg/mL gentamycin and 5 mM L-glutamine (Invitrogen, Carlsbad, CA) at 37°C.
For CD40 stimulation, the CD40-ligand transfected cell line HeLa-CD154 (for control: mock-transfected HeLa cells) was used (American Type Culture Collection [ATCC], Manassas, VA) as described previously [15]. In the synergy experiments the following drugs and reagents were used: CDDP (Mayne Pharma, Brussels, Belgium), CpG oligonucleotide type B-Human TLR9 ligand (ODN2006; Invivogen, San Diego, CA), F-ara-A (Sigma-Aldrich, St Louis, MO), imatinib (Novartis, Basel, Switzerland) and CH11 (Beckham Coulter Company, Marseille, France).
Analysis of apoptosis, proliferation, cell cycle and surface marker expression
For assessment of apoptosis, cells were stained with 200 nM MitoTracker Orange (Molecular Probes, Leiden, The Netherlands) at 37°C for 30 min.
Carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) was used to analyze proliferation. Before treatment, cells were resuspended in phosphate buffered saline (PBS) at 1.0 × 107/mL in 0.5 μM CFSE for 15 min at 37°C and washed.
Cell cycle progression was analyzed by propidium iodide (PI) staining. After treatment, cells were washed in PBS and fixated in cold 70% ethanol. After 30 min on ice, the cells were washed twice in phosphate–citrate buffer (192 parts 0.2 M disodium phosphate, eight parts 0.1 M citric acid with pH 7.8) and ribonuclease (Sigma-Aldrich) was added. Lastly, PI was added (50 μg/mL).
For analysis, cells were acquired using a FACSCalibur flow cytometer and analyzed with CellQuest software (Becton Dickinson [BD] Biosciences, San Jose, CA).
The interaction between drugs was analyzed as described previously [16]. For each condition, expected viability after treatment with a combination of drugs was calculated by multiplication of the measured viability of the samples treated with the drugs separately (corrected for baseline viability). Measured viability (after treatment with a combination of drugs) lower than expected (calculated) viability indicates synergy between the drugs.
Protein isolation and Western blot
CLL cells were collected, washed in ice-cold PBS and lysed by sonification in radioimmunoprecipitation (RIPA) buffer as previously described [15]. For separated nucleus/cytoplasm samples, cells were lysed in NP40 buffer and the remaining cell-pellet (mainly nuclear components) in Leammli buffer, as previously described [17]. Protein content was measured using a bicinchoninic acid assay (BCA) protein assay kit. Thirty to 100 μM of protein lysate was loaded onto each lane of a 7.5%, 10% or 13% gradient sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) gel (Biorad, Hercules, CA) and transferred onto a polyvinylidene fluoride microporous membrane (PVDF-FL; Millipore, Billerica, MA). Membranes were probed with antibodies against β-actin (clone I-19; Santa Cruz Biotechnology, Santa Cruz, CA), polyclonal anti-α-tubulin (Cell Signaling, Beverly, MA), polyclonal anti-CDKN1A (p21, Santa Cruz Biotechnology), polyclonal anti-PUMA (Cell Signaling), polyclonal anti-BID (Cell Signaling) or mouse anti-ABL1 (c-Abl, clone 8E9; BD Pharmingen, San Jose, CA). For detection of the pro-apoptotic TAp73 isoform, an antibody was chosen which reacts with an epitope located on the N-terminal region of human TP73 (IMG-246; Imgenex, San Diego, CA). Hence this antibody only reacts with TAp73 isoforms but not with deltaNp73 [18]. IRDye 680 donkey anti-rabbit immunoglobulin (IgG), IRDye 800 donkey anti-goat IgG or IRDye 800 donkey anti-mouse IgG (Westburg, Leusden, The Netherlands) was used as secondary antibody, and blots were scanned on an Odyssey imager (LI-COR Biosciences, Lincoln, NE).
Lentivirus-mediated RNA interference targeting ABL1 and TAp73 (TP73)
One shRNA target set designed for ABL1 (sequence: CCGGGCTGAAATCCACC-AAGCCTTTCTCGAGAAA GGCTTGGTGGATTTCAGCTTTTTG) and two designed for TP73 (sequence: CCGGCCCGCTCTTGAAGAAACTCTACT CGAGTAGAGTTTCTTCAAGAGCGGGTTTTT and sequence: CCGGCCAAGGGTTACAGAGCATTTACTCGAGTAAATGCTC-TGTAACCCTTGGTTTTT, respectively) were inserted in the pLKO.1 lentiviral vector. As control for transduction efficiency, pLKO-TurboGFP was used. The plasmids were prepared to produce lentiviral particles as previously described [19]. Lentivirus titer determination was performed according to the manufacturer’s instructions (Open Biosystems, Huntsville, AL). Primary cultures of MEC1 cells were infected with concentrated lentiviral stocks at 20 multiplicity of infection (MOI) in the presence of 8 μg/mL polybrene. After 24 h cells were washed and suspended in IMDM supplemented with puromycin (1 μg/mL) for 6 days to select for stably infected cells. The transfected MEC1 cells were cultured in fresh medium for 5 days after puromycin selection before quantitative reverse transcription-polymerase chain reaction (qRT-PCR), immunoblotting and functional experiments.
mRNA extraction and qRT-PCR
Total mRNA was extracted using an RNAeasy Mini Kit (Qiagen, Hilden, Germany) and reverse transcribed using a SuperScript II kit (Invitrogen) according to the manufacturers’ instructions. PCR containing first-strand cDNA (1:10 dilution), SYBR Green PCR Master Mix and forward and reverse primers was performed using a 79HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Cycling conditions were as follows: initial step 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 min and 60°C for 1 min. All reactions were run in triplicate. The primers for the PCR amplification of TAp73, ABL1, CDKN1A, BID and PUMA were designed using Primer Express software (version 2.0). The relative expression levels of genes were expressed in arbitrary units, where the Ct value of the gene of interest was normalized to that determined for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA, a housekeeping gene, to correct for differences in concentrations of the cDNA templates. Then, fold induction was calculated by dividing the value for CDDP-treated samples by that for medium control.
Statistical analysis
The d’Agostino and Pearson omnibus normality test was performed to assess normal distribution of data. In the case of Gaussian distribution of the data, a paired or unpaired two-sided t-test was used to analyze differences between groups. If there was no Gaussian distribution, a two-sided Mann–Whitney U-test was applied to assess the differences. A p-value of < 0.05 was considered statistically significant.
Role of the funding source
The Dutch Society for Scientific Research (NWO), which provided a grant to Arnon P. Kater, had no role in the design of this study, nor in the collection, analysis or interpretation of data or in the writing or submission of the manuscript.
Results
CDDP induces cell death in dividing, but not in resting CLL cells
Irrespective of p53 functional status, peripheral blood (PB) derived CLL cells were poorly sensitive to CDDP treatment [Figure 1(A)], even when used in doses greater than 20 μM, which exceeds the concentration used in vivo (data not shown). In addition, we have previously shown that treatment of resting (p53 functional) CLL cells with CDDP does not induce a p53 response (as defined by up-regulation of mRNA levels of PUMA, BAX and CDKN1A), in contrast to the response to treatment with F-ara-A [20]. These data seem to be in conflict with the observed clinical efficacy of CDDP-based therapy in vivo.
Figure 1.
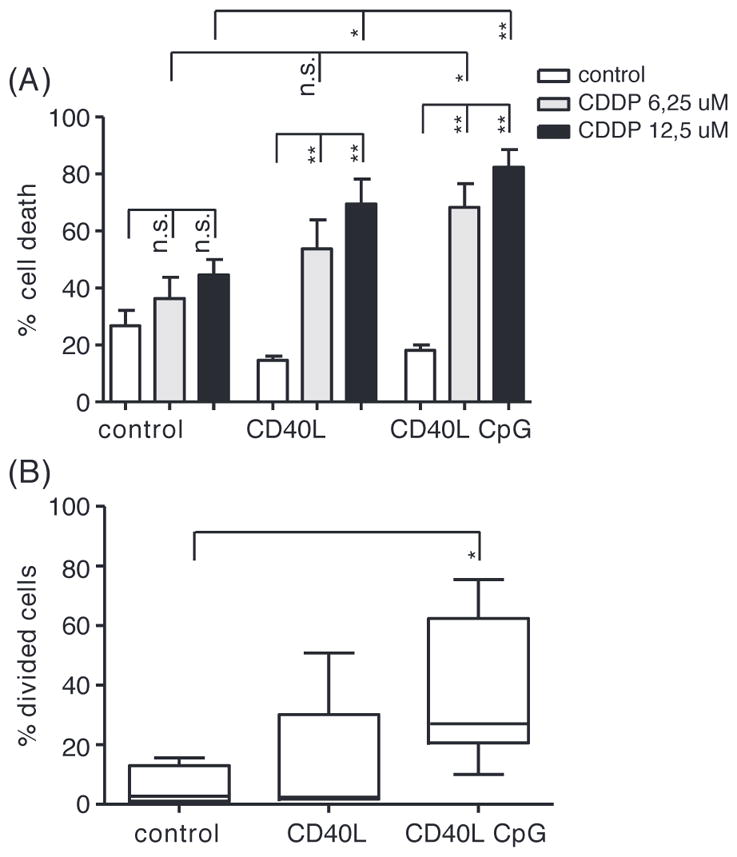
CDDP treatment of resting and dividing CLL cells. (A) Apoptosis after 48 h of treatment with CDDP as indicated in PB derived CLL cells stimulated with CD40L ± GpC for 4 days, assessed by MitoTracker staining (n = 6); mean ± standard error of mean (SEM); *p < 0.05, **p < 0.01 (Mann–Whitney test). (B) Percentage of CLL cells proliferating upon stimulation with CD40L and CpG for 4 days (n = 6) as indicated (and as described in “Materials and methods” section); mean, whiskers min to max; *p < 0.05 (Mann–Whitney test).
The activity of platinum-based compounds is usually described to be based on infliction of DNA damage [21] and the subsequent activation of DNA repair machineries in which p53 has a key role. CLL cells derived from peripheral blood are in G0/G1 arrest [22]. As a consequence, active DNA-replication does not occur, and the PB compartment may therefore be less suitable to study the activity of DNA-damaging compounds such as CDDP. In lymphoid tissue, so-called pseudofollicles (PF) are found, a hallmark in CLL histopathology. These are considered to represent the proliferative compartment of the disease. Here CLL cells interact with various non-leukemic cell types (such as activated T cells and follicular dendritic cells), which results in pro-survival and anti-apoptotic signaling [23,24]. Stimulation of CLL cells with CD40L ± CpG, as a model for the LN environment [25], rendered the cells sensitive to CDDP induced apoptosis [Figure 1(A)]. This coincided with proliferation, especially when CD40-ligation was combined with CpG stimulation [Figure 1(B) and Supplementary Figure 1 to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.996751].
CDDP induces p53 independent cell death and cell cycle arrest in MEC1 cells
Further studies into the mechanisms of action of CDDP were performed in the human prolymphocytic cell line MEC1. This cell line, originally derived from a patient with CLL, is p53-dysfunctional and has an activated phenotype (Supplementary Figure 2 to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.996751). Hence this cell line was used as a model for dividing p53 dysfunctional CLL cells in the LN environment [14] (Supplementary Figure 2 to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.996751).
First, the sensitivity of MEC1 cells to CDDP was tested in dose–response experiments [Figure 2(A)].
Figure 2.

Functional consequences of treatment of MEC1 cells with CDDP. (A) Apoptosis after 48 h of treatment with increasing doses of CDDP as indicated assessed by MitoTracker staining; mean ± SEM of three experiments. (B) Viability of MEC1 cells after treatment with CDDP (5 μM open symbols; 10 μM solid symbols) and F-ara-A (▾ 5 μM, ▴ 10 μM, ●; 25 μM, ▪ 50 μM). Measured viability is plotted against expected viability (based on activity of the separate drugs; see “Materials and methods” section); the area below the diagonal represents synergism. (C) Analysis of cell cycle transition by propidium iodide (PI) staining after 8, 24 and 48 h of CDDP treatment in doses as indicated.
Next, the potency of CDDP to enhance the intrinsic (mitochondrial) and extrinsic (death receptor mediated) apoptosis pathway was studied. In accordance with the lack of functional p53, MEC1 cells were not sensitive to F-ara-A (data not shown). However, additive to synergistic activity was observed of treatment with low-dose CDDP in combination with F-ara-A [Figure 2(B)]. In order to analyze the effects on the extrinsic apoptosis pathway, cells were treated with CDDP and CH11 (soluble FAS-ligand), which resulted in increased surface expression of FAS (data not shown) and enhanced soluble FAS-mediated cell death [Supplementary Figure 3(A) to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.996751].
Finally, we analyzed CDDP mediated changes in cell cycle transition. As shown in Figure 2(C), treatment with CDDP at low doses caused G2/M phase cell cycle arrest after 48 h (preceded by a transient accumulation in S phase after 24 h). At higher concentrations of CDDP, cells accumulated in the S phase, but this may reflect a non-specific effect due to the formation of DNA-adducts [Supplementary Figure 3(B) to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.996751].
ABL1 dependent up-regulation of TAp73 upon treatment with CDDP
In human solid tumor cell lines, treatment with CDDP results in up-regulation of the p53 family member TAp73 [26–28]. In fact, the activity of this drug was found to be mediated primarily by TAp73, especially in the absence of p53. The transcription factor TAp73 shares many functions with p53, but is rarely mutated in cancer [29]. We therefore studied whether the observed effects of CDDP in p53-deficient MEC1 cells are mediated through activation of TAp73. After 24 h of CDDP treatment of MEC1 cells, induction of TAp73, and its downstream targets CDKN1A (p21) [30] and BID [15], could be detected [Figure 3(A)]. In accordance with previous data on the regulation of TAp73 [31], induction was regulated at the post-transcriptional level as mRNA expression of TAp73 was not increased (data not shown). Treatment with CDDP also led to up-regulation of PUMA in MEC1 cells, which in the absence of p53 may be mediated by TAp73.
Figure 3.
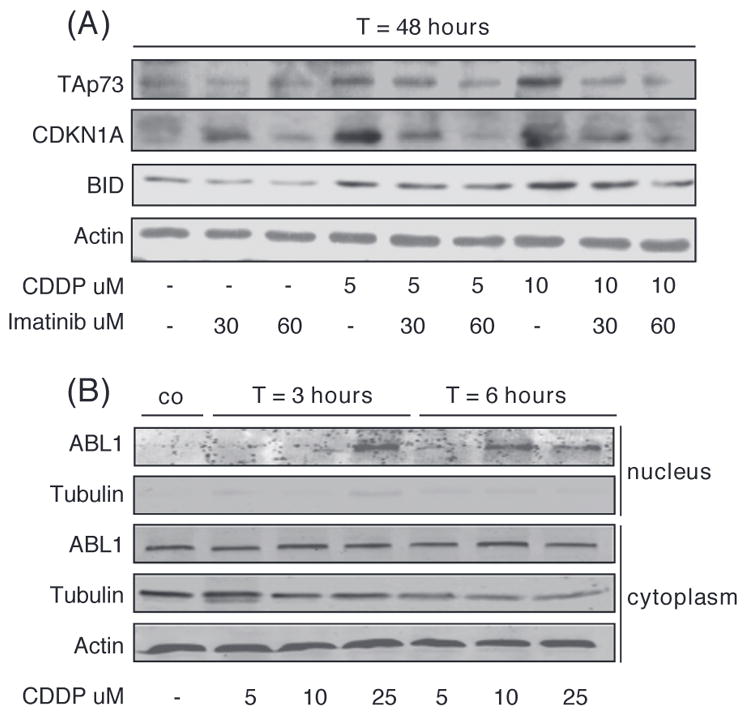
ABL1-dependent up-regulation of TAp73 upon treatment of MEC1 cells with CDDP. (A) Protein levels of TAp73, CDKN1A and BID analyzed by Western blot after 48 h of treatment with CDDP and/or imatinib as indicated. Actin was used as a loading control. (B) Nuclear and cytoplasmatic protein levels of ABL1 after 3 and 6 h of treatment with CDDP as indicated. Actin was used as a loading control. Tubulin was used as control for adequate division of the nuclear and cytoplasmatic compartment.
Induction of TAp73 after CDDP-induced DNA damage in solid tumor cell lines depends on post-translational modification mediated by ABL1 [26]. We repeated the experiment in the presence of imatinib, a specific ABL1 kinase inhibitor. Indeed, the up-regulation of TAp73, CDKN1A and BID was inhibited in the presence of imatinib [Figure 3(A)]. It has recently been shown that nuclear entry of ABL1 contributes to DNA damage-induced activation of the intrinsic apoptotic pathway [32]. As shown in Figure 3(B), following CDDP treatment ABL1 indeed translocated to the nucleus, further supporting a role for ABL1 (and TAp73) in CDDP-induced apoptosis in MEC1 cells.
Knockdown of both ABL1 and TAp73 results in impaired CDDP-induced up-regulation of CDKN1A, PUMA and BID
To confirm an essential role of ABL1 in CDDP-induced TAp73 induction, protein expression levels of TAp73 following CDDP treatment were measured in mock transfected MEC1 cells and MEC1 cells transfected with an ABL1-specific shRNA construct. As presented in Figure 4(A), knockdown of ABL1 prevented up-regulation of TAp73. mRNA expression levels of the reported TAp73 target genes BID, CDKN1A and PUMA were measured after knockdown of ABL1 and TAp73 (TP73), respectively. Using densitometry, the TAp73/actin ratios in the CDDP treated TP73-knockdown (KD) cells relative to the ratio in MEC1-NO cells were 0.4 and 0.3, respectively. In ABL1-KD cells the TAp73/actin ratio was 0.8 relative to MEC1-NO cells. Although basal levels of TAp73 are very low in these cell lines (and therefore difficult to compare), it may well be that also in “steady state” ABL1 is required for basal stability of TAp73. Knockdown of either ABL1 or TAp73 prevented up-regulation of CDKN1A and PUMA after CDDP treatment [Figure 4(B)]. mRNA expression levels of BID are relatively high in MEC1 cells (data not shown); this could explain the observation that BID mRNA levels were only slightly up-regulated upon CDDP treatment.
Figure 4.

Knockdown of ABL1 or TP73 in MEC1 cells. (A) Protein levels of ABL1 and TAp73 analyzed by Western blot upon 48 h of treatment with 10 μM CDDP as indicated after lentiviral transduction of MEC1 cells with a shRNA targeting ABL1 or TP73 (two constructs) or a non-targeting shRNA. (B) mRNA expression levels, assessed by qRT-PCR, of CDKN1A, PUMA and BID upon treatment of lentiviral transduced MEC1 cells with CDDP 10 μM for 48 h. Plotted is fold induction compared to untreated cells; dotted line represents “no induction” (fold induction = 1). (C) Apoptosis after 48 h of treatment with CDDP as indicated, assessed by MitoTracker staining; mean ± SEM of three experiments. MEC-NO = MEC1 cell transduced with nonsense shRNA; *p < 0.05 (paired t-test).
These data not only demonstrate that CDKN1A and PUMA are downstream targets of TAp73 (and ABL1), but also confirm that in the absence of functional p53, transcription is mediated by TAp73. At the functional level, apoptosis upon treatment with CDDP was markedly diminished in TAp73-KD cells [Figure 4(C)], indicating that the response to this drug depends, at least in part, on TAp73. In ABL1-KD cells, high rates of spontaneous apoptosis were observed (data not shown), which implies that ABL1 also has an important role in maintaining basal viability.
TAp73 is expressed in CLL cells residing in the lymph nodes
Data derived from the (proliferating) MEC1 cells suggest that CDDP induces p53 independent apoptosis mediated through TAp73. Resting CLL cells do not express TAp73 [Figures 5(A) and 5(C), left panel]. In accordance with the resistance to CDDP as shown in Figure 1(A), CDDP treatment did not induce expression of TAp73 nor of its downstream target BID. Even after 96 h of treatment with CDDP at concentrations up to 100 μM, no induction of TAp73 was seen (data not shown). In contrast, CD40-ligand stimulation of PB derived CLL cells did result in increased protein expression of TAp73 protein and BID [Figures 5(B) and 5(C), right panel], in line with our observation that CDDP is only active in CD40L (and CpG) stimulated cells [Figure 1(A)]. These data support the hypothesis that TAp73 induction and activity in primary CLL cells may be restricted to the activated compartment.
Figure 5.

Differential expression of TAp73 in PB versus LN derived CLL cells. (A) Protein levels of TAp73 and BID assessed by Western blot in CLL cells with dysfunctional p53 after CDDP treatment for 48 h as indicated. Actin was used as loading control. (B) Protein levels of TAp73, CDKN1A and BID in CLL cells cocultured with CD40-ligand expressing fibroblasts for 24 h with or without imatinib. Cells were washed and lysed after an additional 24 h of culture. Actin was used as loading control. (C) Fold induction in TAp73/actin ratio upon CDDP treatment for 48 h as indicated in p53 functional (n = 7) and p53 dysfunctional (n = 8) CLL (left panel) or after CD40-ligand stimulation in p53 functional (n = 6) and p53 dysfunctional (n = 5) CLL (right panel). (D) Protein levels of TAp73 and BID in peripheral blood (PB) and lymph node (LNN) derived CLL cells. Actin was used as a loading control.
In vitro stimulation of CLL cells with CD40-ligand is used as a model for the interactions between activated T cells and CLL cells residing in the LN [24]. Such interactions with the microenvironment are thought necessary for cell cycle induction [22,33]. To investigate whether induction of TAp73 expression upon CD40-ligation indeed relates to TAp73 expression in LN derived CLL cells, we compared TAp73 protein expression levels in PB derived CLL cells of untreated patients with expression levels in LN derived CLL cells. Strong TAp73 expression was detected in four of seven LN derived CLL samples, whereas very little or no expression was seen in PB derived CLL cells [Figure 5(D)]. Furthermore, the downstream target BID was consistently up-regulated in the LN samples, whereas no expression of BID was seen in PB derived cells.
In vivo induction of TAp73
In accordance with our earlier findings in a retrospective analysis of 10 patients with refractory CLL treated with R-DHAP [11], a recent interim analysis of a prospective Dutch/Belgian phase II trial (HOVON 88; studying the efficacy and safety of salvage chemotherapy followed by RIST in patients with fludarabine-refractory and relapsed high-risk CLL) showed promising activity of the R-DHAP regimen. After a median of three cycles of R-DHAP (range 1–6), the overall response rate (ORR) in 39 evaluable patients was 56% (with 15% complete response [CR] and 41% partial response [PR]). This regimen was also active in patients with del17p and patients with bulky lymphadenopathy, with an ORR of 67% and 53%, respectively, which is of particular importance as the presence of bulky lymphadenopathy at the time of transplant is the strongest independent prognostic factor for poor outcome after allogeneic stem cell transplant [5]. Twenty-six patients (67%) proceeded to RIST [34].
In order to study in vivo effects of this CDDP-containing treatment regimen, blood samples were obtained from a 35-year-old male with fludarabine-refractory CLL included in this trial, at baseline and after 24 and 48 h of R-DHAP treatment. Cytogenetic analysis at baseline revealed deletion of 17p in 87% of the cells. Furthermore, the leukemic cells proved to be p53-dysfunctional, as p53 and its downstream targets CDKN1A and PUMA were not induced following 5 Gy irradiation (Supplementary Figure 4 to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.996751). After initiation of R-DHAP combination therapy, the PB lymphocyte count decreased rapidly following an initial rise. The lymphadenopathy decreased significantly within 1 week. A similar pattern was seen in other patients with bulky lymphadenopathy treated with R-DHAP in this trial [Figure 6(A)]. Already 24 h after the start of treatment, an induction of PUMA and BID was seen. This correlated with an induction of TAp73 [1.6- and 2.5-fold after 24 and 48 h, respectively; Figure 6(B)]. These results support the notion that PUMA and BID can be regulated by TAp73 in the absence of p53 function.
Figure 6.
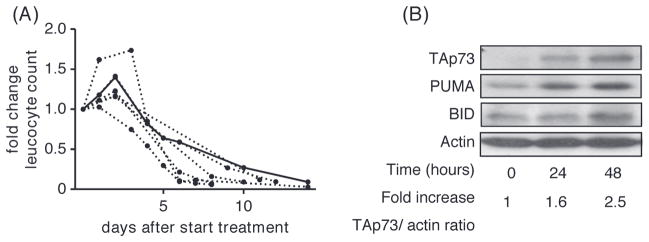
Induction of TAp73 protein expression in vivo. (A) Fold change in leukocyte number (× 109/L) following treatment with R-DHAP in seven patients with fludarabine-refractory and/or p53-dysfunctional CLL; the uninterrupted curve represents the index patient. (B) Protein levels of TAp73, PUMA and BID, assessed by Western blot, 24 and 48 h after treatment with CDDP in vivo. Actin was used as a loading control.
Discussion
We here demonstrate that the pro-apoptotic protein TAp73 has a role in apoptosis induction upon drug treatment in CLL cells. Both up-regulation of TAp73 and cell death after treatment with CDDP were only seen when resting CLL cells were stimulated with CD40L (± CpG). Increased expression of TAp73 was found in LN derived CLL cells when compared with PB derived CLL cells. Together, these data suggest that the transcription factor TAp73 may have a role in apoptosis regulation mainly in activated cells residing in the secondary lymphoid tissue. Indeed, clinical experience with the platinum-compound based regimen R-DHAP shows that this regimen is active at bulky, nodal disease sites.
The p53 family member TP73 was first described in 1997 [30]. Since then many isoforms have been identified, which are represented in two major groups of proteins with opposing functions. TAp73 induces cell cycle arrest and apoptosis, whilst the oncogenic (N-terminal truncated) deltaNp73 inhibits both TAp73 and p53 induced apoptosis [29,35,36]. The role of the TP73 family in hematological oncogenesis is increasingly appreciated. Specifically, inactivation of the TP73 gene by epigenetic silencing or deletion is a common finding in malignant lymphoproliferative disorders. TP73 is inactivated in about 35% of acute lymphoblastic leukemia (ALL) and about one-third of non-Hodgkin lymphomas [37–40].
A functional role of TAp73 in apoptosis regulation and response to drug treatment in CLL was first described by Dicker et al. [15]. Upon CD40-ligation, TAp73 was up- regulated in an ABL1-dependent manner, CD95 and BID were induced and cells were sensitized to F-ara-A treatment, notably independent of p53 function. Based on these and previous results, a phase I trial was initiated in which autologous CLL cells transduced to express CD40-ligand were infused in patients with CLL. This resulted in a comparable pattern of protein expression, including increased expression of TAp73. Furthermore, clinical responses were seen, also in patients with documented 17p deletion [41]. In addition, the activity in CLL of some novel drugs has recently been described to involve up-regulation of TAp73, including histone deacetylase (HDAC) inhibitors [42], lenalidomide [43] and forodesine [44].
Although we found clear expression of TAp73 in LN derived CLL cells, and in CD40-ligand stimulated PB derived CLL cells, no induction was seen upon CDDP treatment of quiescent PB derived CLL cells. The cause of this difference is not known, but it is conceivable that it relates to the proliferative state of the clone and interactions with the microenvironment as discussed below.
First, TAp73 regulation primarily takes place at the level of protein degradation. Normally, TAp73 is maintained at low levels because of constitutive activity of the ubiquitin E3 ligase Itch, which targets TAp73 for poly-ubiquitination and degradation via the ubiquitin–proteasome pathway. Upon DNA damage Itch is degraded and expression levels of TAp73 are stabilized [31]. Itch was recently found to be negatively regulated by the microRNA miR106b [42]. It was postulated that silencing of specific microRNA-regulated pathways may characterize quiescent tumors such as CLL [42,45], which in this case would imply impaired protein stabilization of TAp73.
Second, in contrast to p53, TAp73 has been shown to be regulated in a cell cycle-dependent fashion in various cell lines. TAp73 is a target of transcription factor E2F1 during the S phase of the cell cycle, and TAp73 proteins accumulate in cells in S phase [46]. E2F-1 regulates TAp73 expression either directly [46,47] or via microRNAs such as miR106b [42]. Furthermore, expression of E2F-1 was linked to CD40-ligation in a B cell lymphoma cell line and also in mouse B cells, as CD40-ligation allows for release of repression of this transcription factor [48,49].
It is highly likely that additional interactions with the environment contribute to the mechanisms available to the cell to respond to cytotoxic insults.
Although CD40 activation in vitro and presumably also in vivo (in the secondary lymphoid tissue) induces expression of TAp73 and its downstream targets, CD40 activation also results in nuclear factor-κB (NF-κB) mediated expression of anti-apoptotic molecules, such as Bcl-xL and A1/B3-1 [25], which probably tips the balance toward an anti-apoptotic, pro-survival profile. The observation of TAp73 expressing cells in the PB following CDDP and dexamethasone treatment might reflect a migration of recently activated CLL cells from the secondary lymphoid tissue into PB. There, in the absence of pro-survival stimuli, a shift in the apoptotic balance occurs, which results in (TAp73 mediated) apoptosis. This hypothesis is supported by the initial rise and subsequent decrease in the PB leukocyte count observed in patients who have been treated with R-DHAP and also by the major LN responses induced by this regimen.
As we found induction of TAp73 in both in vivo treated CLL cells and LN derived CLL cells, we expect that TAp73 has a role in the response to treatment in CLL. Further exploring the role and regulation of TAp73 in CLL may provide clues for the development of novel, less toxic, treatment strategies for p53-dysfunctional and drug-resistant CLL.
Supplementary Material
Acknowledgments
The authors would like to thank the patients for donating samples and the clinicians for providing clinical data. This work was partly funded by a personal grant from the Dutch Society for Scientific Research to A. P. Kater.
Footnotes
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article at www.informahealthcare.com/lal.
Supplementary material available online
Supplementary Figures 1–4 showing further data.
References
- 1.Rosenwald A, Chuang EY, Davis RE, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood. 2004;104:1428–1434. doi: 10.1182/blood-2003-09-3236. [DOI] [PubMed] [Google Scholar]
- 2.Seymour JF, Robertson LE, O’Brien S, et al. Survival of young patients with chronic lymphocytic leukemia failing fludarabine therapy: a basis for the use of myeloablative therapies. Leuk Lymphoma. 1995;18:493–496. doi: 10.3109/10428199509059650. [DOI] [PubMed] [Google Scholar]
- 3.Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood. 2002;99:3554–3561. doi: 10.1182/blood.v99.10.3554. [DOI] [PubMed] [Google Scholar]
- 4.Jones JA, Byrd JC. How will B-cell-receptor-targeted therapies change future CLL therapy? Blood. 2014;123:1455–1460. doi: 10.1182/blood-2013-09-453092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorror ML, Storer BE, Sandmaier BM, et al. Five-year follow-up of patients with advanced chronic lymphocytic leukemia treated with allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. J Clin Oncol. 2008;26:4912–4920. doi: 10.1200/JCO.2007.15.4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Velasquez WS, Cabanillas F, Salvador P, et al. Effective salvage therapy for lymphoma with cisplatin in combination with high-dose Ara-C and dexamethasone (DHAP) Blood. 1988;71:117–122. [PubMed] [Google Scholar]
- 7.Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med. 1995;333:1540–1545. doi: 10.1056/NEJM199512073332305. [DOI] [PubMed] [Google Scholar]
- 8.Witzig TE, Geyer SM, Kurtin PJ, et al. Salvage chemotherapy with rituximab DHAP for relapsed non-Hodgkin lymphoma: a phase II trial in the North Central Cancer Treatment Group. Leuk Lymphoma. 2008;49:1074–1080. doi: 10.1080/10428190801993470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsimberidou AM, Wierda WG, Plunkett W, et al. Phase I–II study of oxaliplatin, fludarabine, cytarabine, and rituximab combination therapy in patients with Richter’s syndrome or fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol. 2008;26:196–203. doi: 10.1200/JCO.2007.11.8513. [DOI] [PubMed] [Google Scholar]
- 10.Tsimberidou AM, Wierda W, Plunkett W, et al. Oxaliplatin, fludarabine, cytarabine, and rituximab combination therapy induces high response rates in aggressive chronic lymphocytic leukemia (CLL) and Richter’s syndrome (RS) Blood. 2009;114(Suppl 1):Abstract 3443. [Google Scholar]
- 11.Tonino SH, van Gelder M, Eldering E, et al. R-DHAP is effective in fludarabine-refractory chronic lymphocytic leukemia. Leukemia. 2010;24:652–654. doi: 10.1038/leu.2009.240. [DOI] [PubMed] [Google Scholar]
- 12.Hallaert DY, Jaspers A, van Noesel CJ, et al. c-Abl kinase inhibitors overcome CD40-mediated drug resistance in CLL: implications for therapeutic targeting of chemoresistant niches. Blood. 2008;112:5141–5149. doi: 10.1182/blood-2008-03-146704. [DOI] [PubMed] [Google Scholar]
- 13.Mous R, Jaspers A, Luijks DM, et al. Detection of p53 dysfunction in chronic lymphocytic leukaemia cells through multiplex quantification of p53 target gene induction. Leukemia. 2009;23:1352–1355. doi: 10.1038/leu.2009.71. [DOI] [PubMed] [Google Scholar]
- 14.Stacchini A, Aragno M, Vallario A, et al. MEC1 and MEC2: two new cell lines derived from B-chronic lymphocytic leukaemia in prolymphocytoid transformation. Leuk Res. 1999;23:127–136. doi: 10.1016/s0145-2126(98)00154-4. [DOI] [PubMed] [Google Scholar]
- 15.Dicker F, Kater AP, Prada CE, et al. CD154 induces p73 to overcome the resistance to apoptosis of chronic lymphocytic leukemia cells lacking functional p53. Blood. 2006;108:3450–3457. doi: 10.1182/blood-2006-04-017749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaspers GJ, Veerman AJ, Pieters R, et al. Drug combination testing in acute lymphoblastic leukemia using the MTT assay. Leuk Res. 1995;19:175–181. doi: 10.1016/0145-2126(94)00126-u. [DOI] [PubMed] [Google Scholar]
- 17.Mackus WJ, Lens SM, Medema RH, et al. Prevention of B cell antigen receptor-induced apoptosis by ligation of CD40 occurs downstream of cell cycle regulation. Int Immunol. 2002;14:973–982. doi: 10.1093/intimm/dxf065. [DOI] [PubMed] [Google Scholar]
- 18.Sayan AE, Paradisi A, Vojtesek B, et al. New antibodies recognizing p73: comparison with commercial antibodies. Biochem Biophys Res Commun. 2005;330:186–193. doi: 10.1016/j.bbrc.2005.02.145. [DOI] [PubMed] [Google Scholar]
- 19.Suo G, Jiang Y, Cowan B, et al. Platelet-derived growth factor C is upregulated in human uterine fibroids and regulates uterine smooth muscle cell growth. Biol Reprod. 2009;81:749–758. doi: 10.1095/biolreprod.109.076869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tonino SH, van Laar J, van Oers MH, et al. ROS-mediated upregulation of Noxa overcomes chemoresistance in chronic lymphocytic leukemia. Oncogene. 2011;30:701–713. doi: 10.1038/onc.2010.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali MS, Khan SR, Ojima H, et al. Model platinum nucleobase and nucleoside complexes and antitumor activity: X-ray crystal structure of [PtIV(trans-1R,2R-diaminocyclohexane)trans-(acetate) 2(9-ethylguanine)Cl]NO 3.H2O. J Inorg Biochem. 2005;99:795–804. doi: 10.1016/j.jinorgbio.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 22.Bertilaccio MT, Scielzo C, Muzio M, et al. An overview of chronic lymphocytic leukaemia biology. Best Pract Res Clin Haematol. 2010;23:21–32. doi: 10.1016/j.beha.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Munk Pedersen I, Reed J. Microenvironmental interactions and survival of CLL B-cells. Leuk Lymphoma. 2004;45:2365–2372. doi: 10.1080/10428190412331272703. [DOI] [PubMed] [Google Scholar]
- 24.Smit LA, Hallaert DY, Spijker R, et al. Differential Noxa/Mcl-1 balance in peripheral versus lymph node chronic lymphocytic leukemia cells correlates with survival capacity. Blood. 2007;109:1660–1668. doi: 10.1182/blood-2006-05-021683. [DOI] [PubMed] [Google Scholar]
- 25.Tromp JM, Tonino SH, Elias JA, et al. Dichotomy in NF-kappaB signaling and chemoresistance in immunoglobulin variable heavy-chain-mutated versus unmutated CLL cells upon CD40/TLR9 triggering. Oncogene. 2010;29:5071–5082. doi: 10.1038/onc.2010.248. [DOI] [PubMed] [Google Scholar]
- 26.Gong JG, Costanzo A, Yang HQ, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 27.Sedletska Y, Giraud-Panis MJ, Malinge JM. Cisplatin is a DNA-damaging antitumour compound triggering multifactorial biochemical responses in cancer cells: importance of apoptotic pathways. Curr Med Chem Anticancer Agents. 2005;5:251–265. doi: 10.2174/1568011053765967. [DOI] [PubMed] [Google Scholar]
- 28.Million K, Horvilleur E, Goldschneider D, et al. Differential regulation of p73 variants in response to cisplatin treatment in SH-SY5Y neuroblastoma cells. Int J Oncol. 2006;29:147–154. [PubMed] [Google Scholar]
- 29.Ozaki T, Nakagawara A. p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 2005;96:729–737. doi: 10.1111/j.1349-7006.2005.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 31.Rossi M, De Laurenzi V, Munarriz E, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–848. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Preyer M, Shu CW, Wang JY. Delayed activation of Bax by DNA damage in embryonic stem cells with knock-in mutations of the Abl nuclear localization signals. Cell Death Differ. 2007;14:1139–1148. doi: 10.1038/sj.cdd.4402119. [DOI] [PubMed] [Google Scholar]
- 33.Lanasa MC. Novel insights into the biology of CLL. Hematology Am Soc Hematol Educ Program. 2010:70–76. doi: 10.1182/asheducation-2010.1.70. [DOI] [PubMed] [Google Scholar]
- 34.Van Gelder M, Ghidey W, Chamuleau MED, et al. R-DHAP immunochemotherapy is an effective remission-induction treatment for CLL patients with fludarabine refractory disease with or without deletion 17p, enabling the majority to proceed to allogeneic stem cell transplantation [abstract] Blood. 2013;122(Suppl 1):Abstract 2883. [Google Scholar]
- 35.Grob TJ, Novak U, Maisse C, et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 2001;8:1213–1223. doi: 10.1038/sj.cdd.4400962. [DOI] [PubMed] [Google Scholar]
- 36.Ruffini A, Agostini M, Grespi F, et al. p73 in cancer. Genes Cancer. 2011;2:491–502. doi: 10.1177/1947601911408890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corn PG, Kuerbitz SJ, van Noesel MM, et al. Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt’s lymphoma is associated with 5′ CpG island methylation. Cancer Res. 1999;59:3352–3356. [PubMed] [Google Scholar]
- 38.Kawano S, Miller CW, Gombart AF, et al. Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood. 1999;94:1113–1120. [PubMed] [Google Scholar]
- 39.Martinez-Delgado B, Melendez B, Cuadros M, et al. Frequent inactivation of the p73 gene by abnormal methylation or LOH in non-Hodgkin’s lymphomas. Int J Cancer. 2002;102:15–19. doi: 10.1002/ijc.10618. [DOI] [PubMed] [Google Scholar]
- 40.Pluta A, Nyman U, Joseph B, et al. The role of p73 in hematological malignancies. Leukemia. 2006;20:757–766. doi: 10.1038/sj.leu.2404166. [DOI] [PubMed] [Google Scholar]
- 41.Wierda WG, Castro JE, Aguillon R, et al. A phase I study of immune gene therapy for patients with CLL using a membrane-stable, humanized CD154. Leukemia. 2010;24:1893–1900. doi: 10.1038/leu.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sampath D, Calin GA, Puduvalli VK, et al. Specific activation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation. Blood. 2009;113:3744–3753. doi: 10.1182/blood-2008-09-178707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lapalombella R, Andritsos L, Liu Q, et al. Lenalidomide treatment promotes CD154 expression on CLL cells and enhances production of antibodies by normal B cells through a PI3-kinase-dependent pathway. Blood. 2010;115:2619–2629. doi: 10.1182/blood-2009-09-242438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alonso R, Lopez-Guerra M, Upshaw R, et al. Forodesine has high antitumor activity in chronic lymphocytic leukemia and activates p53-independent mitochondrial apoptosis by induction of p73 and BIM. Blood. 2009;114:1563–1575. doi: 10.1182/blood-2009-02-207654. [DOI] [PubMed] [Google Scholar]
- 45.Seeliger B, Wilop S, Osieka R, et al. CpG island methylation patterns in chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50:419–426. doi: 10.1080/10428190902756594. [DOI] [PubMed] [Google Scholar]
- 46.Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407:645–648. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 47.Stiewe T, Putzer BM. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet. 2000;26:464–469. doi: 10.1038/82617. [DOI] [PubMed] [Google Scholar]
- 48.Lam EW, Choi MS, van der Sman J, et al. Modulation of E2F activity via signaling through surface IgM and CD40 receptors in WEHI-231 B lymphoma cells. J Biol Chem. 1998;273:10051–10057. doi: 10.1074/jbc.273.16.10051. [DOI] [PubMed] [Google Scholar]
- 49.Lam EW, Glassford J, van der Sman J, et al. Modulation of E2F activity in primary mouse B cells following stimulation via surface IgM and CD40 receptors. Eur J Immunol. 1999;29:3380–3389. doi: 10.1002/(SICI)1521-4141(199910)29:10<3380::AID-IMMU3380>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.