

Abstract
Blinatumomab is a CD19/CD3 bispecific T-cell engager (BiTE®) antibody construct for treatment of leukemia. Transient elevation of cytokines (interleukin (IL)-6, IL-10, interferon-gamma (IFN-γ)) has been observed within the first 48 hours of continuous intravenous blinatumomab infusion. In human hepatocytes, blinatumomab showed no effect on cytochrome P450 (CYP450) activities, whereas a cytokine cocktail showed suppression of CYP3A4, CYP1A2, and CYP2C9 activities. We developed a physiologically based pharmacokinetic (PBPK) model to evaluate the effect of transient elevation of cytokines, particularly IL-6, on CYP450 suppression. The predicted suppression of hepatic CYP450 activities was <30%, and IL-6–mediated changes in exposure to sensitive substrates of CYP3A4, CYP1A2, and CYP2C9 were <twofold and lasted <1 week. Model verification indicated that IL-6 was the key cytokine suppressing CYP450 activities; the duration of cytokine elevation was a major determinant of magnitude of suppression. This study shows the utility of PBPK modeling for risk assessment of cytokine-mediated drug interactions.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Blinatumomab immunotherapy mediates transient cytokine elevation. Cytokine elevations may affect CYP450 enzyme activities. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ What are the key determinants of the effect of transient cytokine elevation on CYP450 activities? Is transient cytokine elevation likely to result in clinically meaningful DDIs? • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ A PBPK model was established to evaluate the effect of transient cytokine elevation on CYP450 activities. The effects of cytokines on CYP450 depend primarily on duration of cytokine elevation and, to a lesser extent, the magnitude of elevation. Cytokine levels observed in clinical studies can exceed the EC50 for CYP450 suppression. Transient cytokine elevation observed during blinatumomab treatment has a low DDI potential. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ This model has utility for DDI risk assessment and development of effective DDI evaluation strategies.
Blinatumomab is a bispecific T-cell engager (BiTE®) antibody construct that redirects T cells to CD19-expressing cells; its anti-CD3 moiety binds T cells and its anti-CD19 moiety binds B cells.1 Thus, blinatumomab transiently connects a patient's own cytotoxic T cells to CD19-positive cells, such as those found in B-cell malignancies, and induces T-cell-mediated killing of malignant cells.2 Blinatumomab has demonstrated clinical efficacy for treatment of B-cell leukemia and lymphoma, including adult and pediatric patients with relapsed/refractory acute lymphoblastic leukemia (r/r ALL), adults with minimal residual disease (MRD) ALL, and adults with non-Hodgkin lymphoma (NHL) subtypes.3–7
In preclinical experiments, blinatumomab-mediated T-cell activation resulted in peripheral T-cell expansion.8,9 Patients receiving blinatumomab exhibited transient release of inflammatory cytokines. Some patients also exhibited expansion of T cells.5 Subsequent serial lysis of multiple malignant cells by a single blinatumomab-activated T cell closely resembles a natural cytotoxic T-cell reaction.1,2 In clinical studies, blinatumomab has been administered under continuous intravenous infusion for 4 or 8 weeks per treatment cycle.4,6 We measured serum cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-2, IL-6, IL-8, IL-10, IL-12, IL-4, and interferon (IFN)-γ and observed transient cytokine elevation primarily with IL-6, IL-10, and IFN-γ.5,10 Elevation of cytokines generally took place on the first day of blinatumomab treatment and returned to baseline during the second day, while infusion was ongoing.5,10 Interpatient variability in cytokine elevations was large.4,5 Cytokine dynamic profiles were consistent across studies in patients with r/r ALL, MRD-positive ALL, and NHL, and the magnitude of cytokine elevation appeared to be associated with the initial blinatumomab dose levels, especially at doses >15 μg/m2/day or 28 μg/day.4,5,10
Because elevation of cytokines, especially IL-6, has been shown to suppress cytochrome P450 (CYP450) enzymes in humans,11–13 we evaluated the impact of transient cytokine elevation on CYP450 enzyme activities and potential implications for drug–drug interactions (DDIs). A physiologically based pharmacokinetic (PBPK) modeling approach using the Simcyp program was implemented.14 This approach has been successful in predicting the suppressive effect of the cytokine IL-6 on CYP3A4 substrates in patients with rheumatoid arthritis (RA), bone marrow transplantation, and surgical trauma.15 Our investigation focused on evaluation of 1) magnitude and duration of the effect of transient cytokine elevation on CYP450 enzyme activities, 2) potential of DDIs resulting from transient cytokine elevation during blinatumomab treatment, 3) key determinants of the magnitude of IL-6–mediated DDIs, and 4) utility of PBPK modeling for assessing IL-6–mediated suppression of CYP enzymes.
METHODS
Patients
IL-6 elevation profiles from Study MT103-104 in patients with NHL were used in this assessment. The doses studied ranged from 0.5–90 μg/m2/day4,16; the range was broader than that in other clinical trials (5–30 μg/m2/day or 9–28 μg/m2/day). Because of its short half-life, patients received blinatumomab via continuous intravenous infusion for 4 or 8 weeks in Study MT103-104.17 Demographic, safety, and efficacy data are reported elsewhere.16
Evaluation of the effect of blinatumomab and cytokines in human hepatocytes
Human hepatocytes from three donors were incubated for 24 or 48 hours with solvent (control group), blinatumomab 3,000 pg/mL, or a cocktail of cytokines (IL-2, IL-6, IL-10, IFN-γ, and TNF-α) corresponding to the low (125 pg/mL for all cytokines), middle (2,000 pg/mL for IL-6, IL-10, and IFN-γ; 500 pg/mL for IL-2 and TNF-α), or high (20,000 pg/mL for IL-6, IL-10, and IFN-γ; 1,000 pg/mL for IL-2 and TNF-α) concentrations of cytokines observed in Study MT103-104. Enzyme activity was measured by comparing the formation of a specified metabolite of CYP450 enzyme substrates in control and experimental groups. These metabolites include phenacetin/acetaminophen for CYP1A2, tolbutamide/4-methylhydroxytolbutamide for CYP2C9, bufuralol/1′-hydroxybufuralol for CYP2D6, S-mephenytoin/4′-hydroxymephenytoin for CYP2C19, and midazolam/1′-hydroxymidazolam for CYP3A4/5. The percentage of CYP450 enzyme suppression was calculated using the following equation:
PBPK modeling
CYP450 enzymes CYP3A4, CYP1A2, and CYP2C9 were selected for PBPK modeling based on in vitro results reported here. Among patients receiving blinatumomab, IL-6, IL-10, and IFN-γ are the most commonly elevated cytokines. Because of its demonstrated suppressive effects on CYP450 activity and expression, both in vitro and in vivo,15,18 IL-6 was selected for PBPK modeling.
The clinical profile of IL-6 was modeled assuming a zero-order input rate and first-order elimination rate using Phoenix WinNonlin v. 6.3.0 (Certara, St. Louis, MO). The resulting IL-6 kinetic parameters, total clearance (CL) and volume of distribution at steady state (Vd,ss), were used as input parameters for the simulation.
The Simcyp population-based simulator (v. 12.0; Simcyp, Sheffield, UK) was used for simulation. Simcyp models complex systems by using in vitro experimental data and demographic, physiological data from a specific patient population.14,19 It allows for evaluation of CYP450 enzymes in both the liver and the gut. The algorithm, physiological basis, and differential equations used by the Simcyp software have been previously described.14,19
Simulation parameters were based on data from clinical trials and in vitro studies in human hepatocytes (Table 2). Time–concentration IL-6 profiles from patients with NHL receiving blinatumomab (Study MT103-104; ClinicalTrials.gov, NCT00274742) were used. IL-6 parameters for CYP enzyme suppression such as Emin and EC50 were taken from in vitro data (median values) from Dickmann et al.18 Default values supplied by the Simcyp library were adopted for some parameters: the mean degradation constant (kdeg) values for CYP3A4, CYP1A2, and CYP2C9 in the liver were 0.0193, 0.0183, and 0.0067 h−1, respectively; the mean kdeg value in the intestine for both CYP3A4 and CYP2C9 was 0.03 h−1.20 CYP1A2 activity in the intestine was not considered because it is not expressed in that tissue.21
Table 2.
Parameter estimates of IL-6 for Simcyp simulation
| Parameter, unit | Value |
|---|---|
| Molecular weight, g/mol | 24,500 |
| Compound type | Neutral |
| Blood to plasma ratio | 1.0 |
| fu, plasma | 1.0 |
| Dose, mga | 0.018a; 1.04b |
| Systemic plasma clearance - CLiv, L/h | 0.6c; 3.2d |
| Volume of distribution - Vd,ss, L/kg | 0.12c,d |
| CYP3A4 Emine,f | 0.25 |
| CYP3A4 EC50f (μM) | 2.1 × 10−6 (51 pg/mL) |
| CYP1A2 Emine,f | 0.18 |
| CYP1A2 EC50f (μM) | 2.4 × 10−5 (587 pg/mL) |
| CYP2C9 Emine,f | 0.05 |
| CYP2C9 EC50f (μM) | 5.0 × 10−6 (121 pg/mL) |
CYP, cytochrome P450; EC50, half-maximal effective concentration; Emin, minimum CYP enzyme activity; fu, plasma, plasma fraction unbound; IL, interleukin; IV, intravenous; Vd,ss, volume of distribution at steady state.
To describe the mean IL-6 profile in patients, a hypothetical dose of 0.018 mg was assumed. IL-6 was administered as a constant IV infusion for 6 h, with the starting time = 0.
To describe the IL-6 profile in the patient with the highest IL-6 elevation (maximum observed (IL-6) = 59,231 pg/mL), a hypothetical dose of 1.04 mg was assumed. IL-6 was administered as a constant IV infusion for 4.15 h, with the starting time = 1.92 h.
Parameter used to model the mean serum IL-6 profile.
Parameters used to model the serum IL-6 profile from the patient with the highest elevation (may not reflect the physiologic volume of distribution or clearance of IL-6).
The CV% for CLiv and Vd,ss parameters in the population was assumed to be 50%.
Emin was the minimum CYP enzyme activity (the maximum suppression) expressed as a fraction of vehicle control.
It was assumed that the magnitude of IL-6 suppression for CYP3A4 and CYP2C9 in the intestine was the same as that in the liver.15,22 In addition, within the Simcyp simulator, it was assumed that IL-6 suppresses the rate of synthesis of CYP450 enzymes in the liver according to the following equation,15 in which [Enzyme]t is the amount of active CYP enzyme at any given time in the liver; [Enzyme]0 is the basal amount of CYP enzyme in the liver; [Enzyme]t = [Enzyme]0 at t = 0; Emin is the minimum CYP enzyme activity (i.e., the maximum suppression) expressed as a fraction of vehicle control; EC50 is the concentration that results in 50% of Emin (i.e., half of the maximum suppressive effect); [I]t is the perpetrator (IL-6) concentration at time t; and kdeg is the degradation rate of each respective CYP enzyme in the liver:
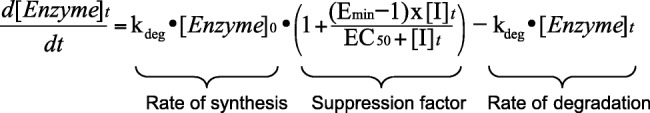 |
The simulated population size was 100 (10 trials with 10 virtual patients each). For each trial, virtual patients were between 18–50 years of age, with a 1:1 male:female ratio. The suppressive effect was calculated as a ratio of the area under the curve (AUC) or maximum concentration (Cmax) of the victim drug in the presence vs. absence of IL-6 elevation. The mean values for the trials and 95% confidence intervals (CIs) were calculated.
Tests of model predictability
Model predictability was assessed through comparison with the in vitro study results in human hepatocytes (presented here) and literature on in vivo drug interactions in RA. To model RA, IL-6 infusion was simulated for 24 days at steady-state concentrations of 50, 100, or 500 pg/mL and simvastatin was dosed on day 22. Results from the simulation were compared to in vivo results reported in Schmitt et al.23
Sensitivity analyses were conducted by changing the Emin and EC50 of IL-6 by threefold to determine the effect on the exposures of the selected CYP450 substrates.
RESULTS
Magnitude and duration of transient cytokine elevation
Dose-dependent increases in peak cytokine levels were demonstrated in studies of blinatumomab in patients with NHL and ALL.4,10 Doses administered to patients with ALL ranged from 5–30 μg/m2/day24; higher doses (up to 90 μg/m2/day) were administered in a dose-escalation study in NHL (Study MT103-104; ClinicalTrials.gov, NCT00274742).16
Peak cytokine levels increased with initial blinatumomab dose, reaching a plateau around the 60-μg/m2/day dose (Figure 1a). The cytokine profile of a representative patient is depicted in Figure 1b: cytokine elevation was transient, increasing rapidly after infusion started, peaking at ∼6 hours, and declining to baseline starting at 48 hours, despite ongoing infusion with blinatumomab. Along with cytokine elevation, there was a transient elevation of C-reactive protein (CRP; Figure 1c), an acute phase protein, which peaked within the first 2 days of treatment and declined thereafter; elevation of CRP was highly variable and showed a degree of dose dependency but appeared to reach a plateau at doses ≥30 μg/m2/day. Mean (SD) peak CRP levels were: 28 (26), 51 (74), 115 (115), 81 (37), and 91 (22) mg/L at doses of ≤5, 15, 30, 60, and 90 μg/m2/day, respectively.
Figure 1.
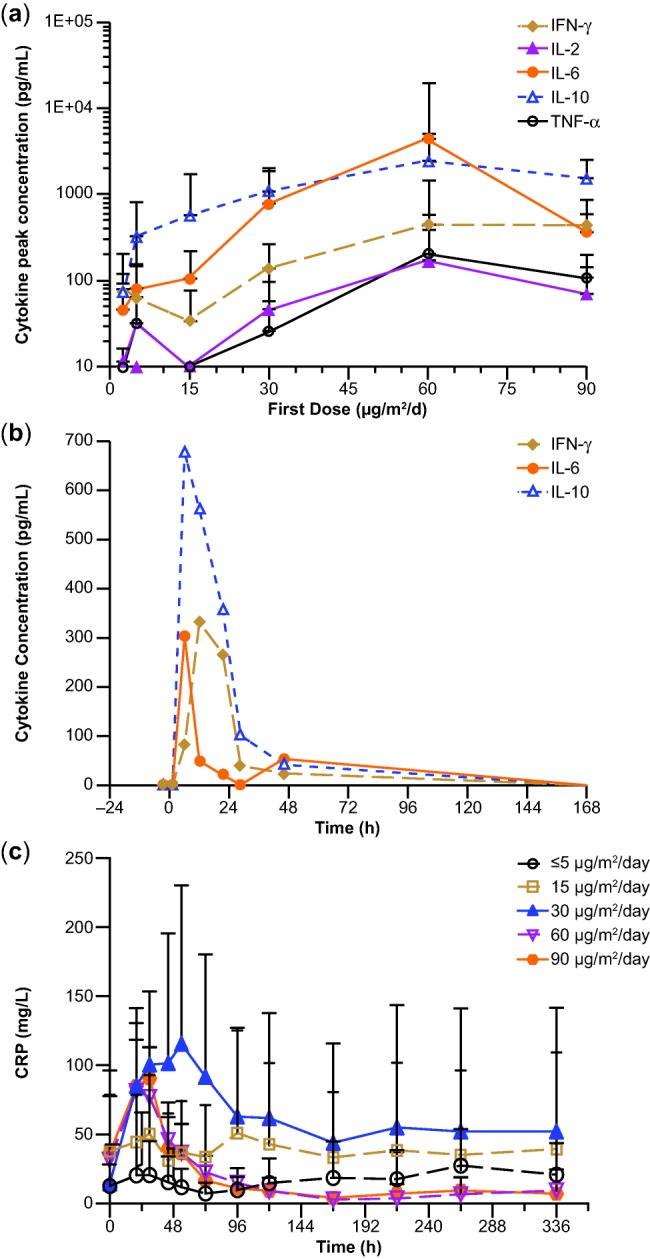
(a) Serum peak cytokine concentrations after initiation of blinatumomab, (b) cytokine concentrations over time in an individual patient after initiation of blinatumomab, and (c) CRP concentrations over time by blinatumomab dose (n = 4–13 for each dose group). The dose group of ≤5 μg/m2/day combined patients' doses at 0.5, 1.5, and 5 μg/m2/day. CRP, C-reactive protein; IFN, interferon; IL, interleukin; TNF-α, tumor necrosis factor alpha.
PBPK model
Selection of CYP450 enzymes
To determine the effect of blinatumomab and cytokines on suppression of CYP450 enzymes, an in vitro experiment was conducted with human hepatocytes. Blinatumomab concentration was tested at 3,000 pg/mL, corresponding to serum concentrations in patients receiving a 60-μg/m2/day dose. A cytokine cocktail was tested at "low-strength" (125 pg/mL: IL-2, IL-6, IL-10, IFN-γ, and TNF-α), "mid-strength" (500 pg/mL: IL-2 and TNF-α; 2,000 pg/mL: IL-6, IL-10, and IFN-γ), and "high-strength" (1,000 pg/mL: IL-2 and TNF-α; 20,000 pg/mL: IL-6, IL-10, and IFN-γ). The choice of cytokine strength and the components of the cytokine cocktail were based on cytokine data obtained from a clinical trial in NHL patients that tested a wide dose range of blinatumomab (0.5 to 90 μg/m2/day).4 The "low-strength" corresponds to the lower limit of quantification of 125 pg/mL in the cytokine assay,1 the "mid-strength" corresponds to peak cytokine levels in a majority of patients, and the "high-strength" corresponds to peak cytokine levels observed in the few patients with extremely high cytokine levels (Figure 1a). Consistent with US Food and Drug Administration guidance for drug interaction studies,25 hepatocytes from three donors were incubated for 24 or 48 hours (the approximate length of transient cytokine elevation observed in clinical studies).4,5 The potential effect of blinatumomab alone on CYP450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A) was tested separately from the cytokine cocktail in in vitro experiments.
The in vitro experiments showed that blinatumomab did not have any direct effect on CYP450 enzyme activity in hepatocytes. The cytokine cocktail showed a suppressive effect on CYP450 enzyme activities, and generally the suppression was greater with 48-hour incubation than with 24-hour incubation. The extent of suppression was similar with "mid-" and "high-strength" cytokine concentrations, which was higher than that with "low-strength" cytokine concentrations (Table 1), indicating that suppression of CYP450 enzymes was dependent on the cytokine concentrations but was maximized at "mid-strength" cytokine levels. This finding is consistent with a report that the EC50 values for IL-6 suppression of CYP450 activities ranged from 50–600 pg/mL, with the median EC50 value of 121 pg/mL, which is much lower than the "mid-strength" concentration (2,000 pg/mL) tested here.18
Table 1.
Suppression of CYP450 activities by blinatumomab or cytokine cocktails in human hepatocytes
| CYP450 enzyme suppression, %a | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4/5 | ||||||||||||||
| Test agents | Time | Hu 4197 | Hu 8123 | Hu 8125 | Hu 4197 | Hu 8123 | Hu 8125 | Hu 8125b | Hu 4197 | Hu 8123 | Hu 8125 | Hu 8125b | Hu 4197 | Hu 8123 | Hu 8125 | Hu 4197 | Hu 8123 | Hu 8125 |
| Blinatumomab | 24 h | 0 | −3 | 15 | 17 | −23 | −3 | −11 | 3 | −9 | −35 | −7 | −2 | −8 | −2 | −1 | 13 | −6 |
| Cytokine lowc | 56 | 8 | 53 | −3 | −46 | −44 | −47 | 10 | 5 | −1 | −29 | 12 | −3 | −11 | 21 | −18 | −17 | |
| Cytokine midd | 67 | 36 | 66 | 42 | −6 | −6 | −35 | 14 | 15 | −46 | 1 | 27 | 24 | 1 | 35 | 19 | −7 | |
| Cytokine highe | 67 | 47 | 69 | 47 | 10 | 14 | −6 | 16 | 16 | −21 | 9 | 29 | 24 | 10 | 29 | 3 | 10 | |
| Blinatumomab | 48 h | −5 | −52 | −16 | 7 | −31 | −1 | −17 | 6 | −6 | −63 | −19 | −8 | −3 | −13 | 0 | 5 | −3 |
| Cytokine lowc | 71 | −33 | 47 | 24 | −45 | −39 | −73 | 29 | 12 | −1 | −25 | 21 | 10 | −37 | 30 | 15 | −17 | |
| Cytokine midd | 92 | 76 | 58 | 66 | 38 | 16 | −56 | 47 | 49 | −56 | 20 | 43 | 57 | 4 | 60 | 46 | 29 | |
| Cytokine highe | 89 | 85 | 62 | 71 | 63 | 42 | −30 | 49 | 50 | −54 | 37 | 47 | 67 | 30 | 60 | 62 | 35 | |
CYP, cytochrome P450; IFN, interferon; IL, interleukin; TNF-α, tumor necrosis factor alpha.
Positive and negative numbers indicate the percentage of suppressed and increased enzyme activity, respectively.
Repeated experiment.
Low-strength cytokines: 125 pg/mL: IL-2, IL-6, IL-10, IFN-γ, and TNF-α.
Mid-strength cytokines: 500 pg/mL: IL-2 and TNF-α; 2000 pg/mL: IL-6, IL-10, and IFN-γ.
High-strength cytokines: 1,000 pg/mL: IL-2 and TNF-α; 20,000 pg/mL: IL-6, IL-10, and IFN-γ.
With the "high-strength" cytokines and 48 hours of incubation, >50% suppression of CYP1A2 activity was observed in cells from all donors; >50% suppression of CYP3A4/5 and CYP2C9 activities occurred in cells from two of the three donors; and negligible suppression was observed for CYP2C19 and CYP2D6 (Table 1). Therefore, CYP1A2, CYP3A4/5, and CYP2C9 were identified as the most sensitive to suppression of the cytokines tested and were selected for evaluation with PBPK modeling. Sensitive substrates of these enzymes were selected for use in the PBPK simulation, including simvastatin (40 mg) and midazolam (5 mg) for CYP3A4, theophylline (125 mg) and caffeine (150 mg) for CYP1A2, and (S)-warfarin (10 mg) for CYP2C9. The Simcyp built-in library profiles for these compounds were used without modification.
Cytokine selection
Of the three most elevated cytokines (IL-10, IL-6, and IFN-γ), IL-6 was selected as a perpetrator of DDI for PBPK modeling. IL-6 is known to have suppressive effects on CYP450 mRNA expression and enzyme activities in vitro and in vivo,15,18 whereas IL-10 was reported to have no clinically meaningful effects on CYP450 enzymes.26,27 Studies in human hepatocytes revealed no effect of IL-10 on CYP450 enzymes even at 5,000 pg/mL (Amgen Inc. Thousand Oaks, CA, data not shown). To date, results from in vitro studies reporting the suppression of CYP3A by IFN-γ are conflicting.28 In addition, no clinical data have been published on the effect of IFN-γ on CYP450 activities.12 The effect of other interferons tested, such as IFN-α, on CYP450s appears to be dose-dependent and more selective to CYP1A2.12
The IL-6 concentration–time profiles representing the mean and highest cytokine levels from the MT103-104 study were selected for PBPK evaluation of DDI.
Estimation of IL-6 parameters
To describe the changes in serum IL-6 levels in patients, IL-6 kinetic parameters (such as systemic plasma clearance and volume of distribution) were estimated by a one-compartment pharmacokinetic model following intravenous infusion with a zero-order input rate constant and a first-order elimination rate constant (Table 2). The mean IL-6 profile was modeled to cover all of the concentration data points above the suppression EC50 of IL-6 for CYP3A4 to maximally estimate IL-6–mediated DDI risk. The increase of mean IL-6 level peaked at 6 hours and returned to baseline by 48 hours (Figure 2a). A similar IL-6 dynamic profile was shown in the patient with the highest IL-6 elevation (Figure 2b); IL-6 peaked at 6 hours but returned to baseline by 30 hours. The estimated mean clearance and volume of distribution of IL-6 were 0.6 L/h and 0.12 L/kg, respectively, for the mean IL-6 profile (Table 2). The estimated IL-6 kinetic parameters and the median IL-6 kinetic parameters for CYP enzyme suppression reported in the literature18 were used as input parameters for PBPK modeling and simulation (Table 2).
Figure 2.
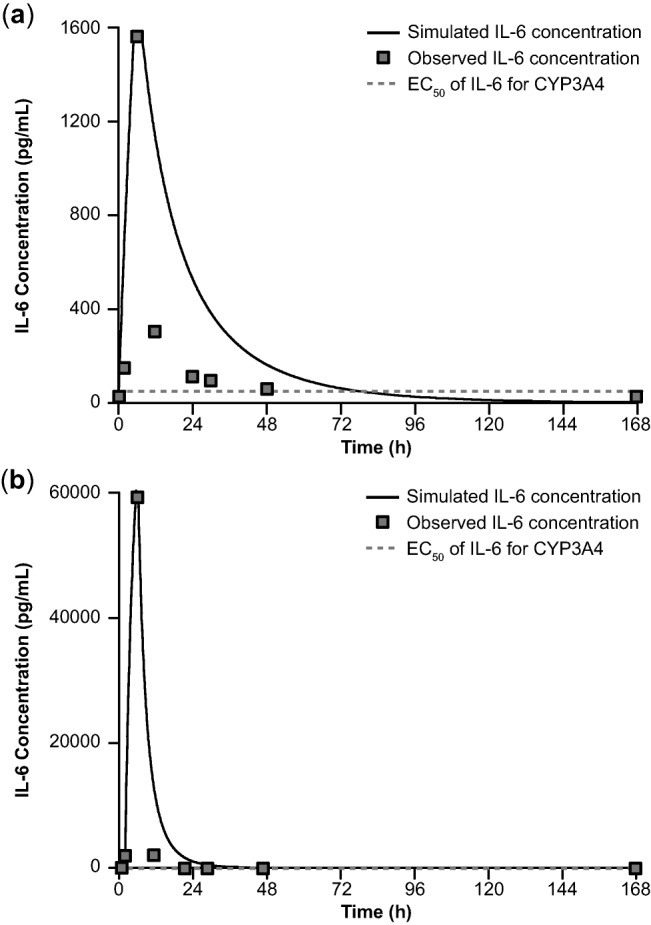
(a) Mean concentration-vs.-time profile of IL-6 following continuous intravenous infusion of blinatumomab and (b) IL-6 profile from a patient with the highest level of IL-6 elevation. CYP, cytochrome P450; EC50, half-maximal effective concentration; IL, interleukin.
PBPK model verification
The established PBPK model was verified before applying it to the evaluation of DDI resulting from blinatumomab-mediated cytokine elevations. Two separate evaluations were conducted.
Model validation via predicting in vitro observation
We assumed that IL-6 played a major role in suppression of CYP450 enzymes among the cytokines tested in the cocktail. Therefore, the established PBPK model for IL-6 should be able to predict the results from our in vitro experiment. Based on our simulation under a 48-hour incubation condition, the predicted mean effects of IL-6 at constant concentrations of 125 pg/mL, 2,000 pg/mL, and 20,000 pg/mL on suppression of intrinsic clearance of midazolam 1′-hydroxylation (a marker of CYP3A4 activity) in the liver were 25%, 39%, and 41%, respectively (Supplemental Table 1). The predicted percentages of suppression were comparable to the observed effect in human hepatocytes with the cytokine cocktail at low, mid, and high strengths after 48 hours of in vitro incubation (23%, 45%, and 52%, respectively). This indicates that the model is robust and can be used to replicate the in vitro results. The result also supported our assumption that IL-6 is the key cytokine contributing to the suppression of CYP450. Based on the result of model validation, it appears that the model has a potential for clinical DDI prediction for blinatumomab-mediated cytokine elevation.
Model validation via predicting clinical observation
To evaluate model predictability for clinical data, the established PBPK model was used to predict results from a clinical report. It was found in this study that tocilizumab treatment blocked IL-6 receptor signaling, which then decreased simvastatin concentrations in RA patients.23 Our model predicted that exposure to IL-6 treatment at a concentration range of 50–100 pg/mL for 24 days in RA patients would result in simvastatin AUC ratios between 2.0 and 2.6. This prediction is comparable to the observed AUC ratio of 2.4 (Supplemental Table 2), suggesting that the model predicted the clinical results of simvastatin in RA patients well; therefore, it should be useful in evaluating DDI risk associated with blinatumomab-mediated cytokine elevation.
PBPK model application for blinatumomab
Magnitude and duration of suppression of intrinsic clearance of CYP450 in the liver
The established PBPK model was used to project the effect of the mean IL-6 dynamic profile on CYP450 enzymes in patients treated with blinatumomab. The changes in hepatic intrinsic clearance of CYP3A4, CYP1A2, and CYP2C9 marker substrates over time were simulated with the model. The maximal suppression on intrinsic hepatic clearance of CYP3A4 was 28%, occurred at ∼48 hours, and lasted for ∼1 week (Figure 3a). Similarly, the maximal suppression on intrinsic hepatic clearance of CYP1A2 was 9%, occurred at ∼48 hours, and lasted for ∼1 week (Figure 3b). The maximal suppression on intrinsic hepatic clearance of CYP2C9 was 17%, occurred at ∼70 hours, and lasted for ∼9 days (Figure 3c).
Figure 3.

Predicted duration and magnitude of IL-6 suppression on hepatic CLint of (a) CYP3A4, as measured by CLint of simvastatin, (b) CYP1A2, as measured by CLint of theophylline N1-demethylation, and (c) CYP2C9, as measured by CLint of (S)-warfarin, based on the mean IL-6 profile in clinical trials of blinatumomab. (d) Predicted duration and magnitude of IL-6–mediated changes in simvastatin exposure. CLint, intrinsic clearance; CYP, cytochrome P450; IL, interleukin. Percentages shown are for maximal reductions observed.
Simulations were also performed with the highest IL-6 profile; the projected maximal suppression on hepatic intrinsic clearance of CYP3A4, CYP1A2, and CYP2C9 was 20%, 19%, and 14%, respectively. These results suggest that the 38-fold higher IL-6 concentration seen with the highest profile vs. the mean profile resulted in greater suppression of CYP1A2 (from 9% to 19%) but not of CYP3A4 and CYP2C9. This is likely due to a relatively high EC50 value of IL-6 suppression of CYP1A2 (Table 2).
Magnitude and duration of increased exposure to CYP450 substrates in vivo
Simulations were performed with the PBPK model to evaluate the magnitude of IL-6–induced increase of exposures to CYP3A4 substrates (simvastatin, midazolam), CYP1A2 substrates (theophylline, caffeine), and CYP2C9 substrate ((S)-warfarin) in virtual patients (the term "virtual patients" will be used hereafter to refer to both simulated "healthy subjects" and "patients" used in the model). The timing of administration of substrates was also evaluated, revealing that the maximal effect should not occur at the time of peak IL-6 concentration (6 hours) but around the time at which IL-6 showed maximal suppression of CYP450 hepatic intrinsic clearance (48 hours) (Figure 3d).
With administration of CYP450 substrates at 48 hours after the start of IL-6 infusion, the predicted AUC ratio of simvastatin, midazolam, theophylline, caffeine, and (S)-warfarin was 1.9-, 1.7-, 1.1-, 1.2-, and 1.2-fold, respectively (Table 3). In comparison, in a simulation dosing simvastatin at time of peak IL-6 concentration (6 hours), the predicted AUC ratio was only 1.1-fold (Figure 3d). Additional simulations were performed to dose simvastatin at 24, 30, and 55 hours after the start of IL-6 infusion; the predicted increase of AUC was less than that predicted for dosing at 48 hours.
Table 3.
Predicted maximum IL-6–mediated drug interaction on substrates of CYP3A4/1A2/2C9 based on the mean IL-6 profile*
| Substrate | CYP450 affected | Mean AUC ratio (95% CI) | Mean Cmax ratio (95% CI) |
|---|---|---|---|
| Simvastatin | 3A4 | 1.9 (1.8–2.0) | 1.7 (1.6–1.8) |
| Midazolam | 3A4 | 1.7 (1.6–1.8) | 1.2 (1.1–1.3) |
| Theophylline | 1A2 | 1.1 (1.0–1.1) | 1.0 (1.0–1.0) |
| Caffeine | 1A2 | 1.2 (1.1–1.3) | 1.0 (1.0–1.1) |
| (S)-Warfarin | 2C9 | 1.2 (1.0–1.4) | 1.0 (1.0–1.0) |
AUC, area under the concentration–time curve; CYP450, cytochrome P450; IL, interleukin.
The substrate was administered 48 h after the start of IL-6 intravenous infusion to maximize the potential for drug–drug interactions.
Because the highest observed IL-6 peak concentration was 38-fold greater than the mean peak concentration observed in Study MT103-104, simulations were performed to determine the suppressive effect on CYP450 with the highest observed IL-6 profile. Of note, duration of the highest IL-6 elevation was shorter than the mean IL-6 elevation (30 vs. 48 hours). The results showed the exposure ratio was <2 with the highest IL-6 profile. The predicted mean ratios of AUC and Cmax for simvastatin were 1.5 (95% CI, 1.4–1.6) and 1.4 (95% CI, 1.3–1.5), slightly lower than those with the mean IL-6 profile (1.9 (95% CI, 1.8–2.0) and 1.7 (95% CI, 1.6–1.8)). With the highest IL-6 profile, the predicted mean AUC ratios of theophylline (1.2), caffeine (1.2), and (S)-warfarin (1.1) were similar to those predicted based on the mean IL-6 profile (Table 3).
Key determinants of magnitude and duration of effects on CYP450
The PBPK model was used to evaluate the effect of duration of suppression on hepatic intrinsic clearance of CYP3A4. With constant IL-6 concentrations of 50, 100, and 500 pg/mL, which mimic conditions of chronic inflammation, the time to reach maximal suppression (i.e., steady state) on CYP3A4 activity could take >2 weeks. The magnitude of suppression depends on IL-6 concentration and duration of suppression (Figure 4). For instance, with a constant IL-6 concentration of 50 pg/mL over 3 weeks, the suppression on the hepatic intrinsic clearance of CYP3A4 is ∼17% by day 2 but 33% by week 3. This finding indicates that the duration of IL-6 elevation is a critical factor in determining the magnitude of CYP450 suppression. Given the transient nature of blinatumomab-induced cytokine elevation,5 its impact on exposures to CYP450 substrates is predicted to be limited.
Figure 4.
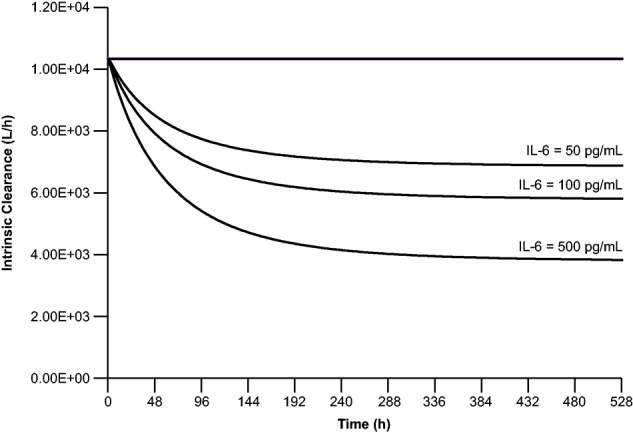
Predicted time course of IL-6 suppression on CLint of CYP3A4 at constant IL-6 concentrations (50, 100, and 500 pg/mL) for 3 weeks, as measured by CLint of simvastatin. CLint, intrinsic clearance; CYP, cytochrome P450; IL, interleukin.
Sensitivity analysis
To determine the impact of uncertainty of model parameters on simulation results, a sensitivity analysis was performed. The IL-6 kinetic parameters associated with CYP450 suppression, namely, maximum suppression (i.e., minimum CYP enzyme activity (Emin)) and EC50 for CYP3A4, were varied by threefold. No dramatic change in predicted suppressive effect on the CYP3A4 substrate simvastatin was found. In an extreme case (i.e., reducing EC50 (increasing the sensitivity of suppression) and reducing Emin (increasing the size of suppressive effect) by threefold simultaneously), the change in AUC ratio of simvastatin increased from 1.9-fold to 2.8-fold (Scenario 5, Supplemental Table 3).
Similarly, a threefold decrease in both EC50 and Emin of IL-6 did not change suppression of CYP1A2 or CYP2C9 substantially. The predicted mean AUC and Cmax ratios for CYP1A2 substrates were ≤1.4 and ≤1.1, respectively; for a substrate of CYP2C9, the predicted mean AUC and Cmax ratios were 1.3 and 1.0, respectively (data not shown).
DISCUSSION
PBPK modeling has been shown to predict DDIs between cytokines and small-molecule drugs in patients with inflammatory conditions successfully.15 In this analysis, we evaluated the application of PBPK modeling to prospectively predict the effect of transient cytokine elevation on suppression of CYP450 enzyme activities. The predictability of the PBPK model was verified by predicting transient CYP450 suppression from internal in vitro data and chronic CYP450 suppression observed in published clinical data. Model verification of the in vitro data indicated that, among the cytokines elevated in patients, IL-6 is the key contributor to suppression of CYP450 activities. PBPK modeling is a valuable approach for assessing risk of cytokine-mediated DDIs. Further, PBPK modeling has potential utility for developing strategies for clinical DDI assessment by evaluating the need to conduct clinical DDI studies and by guiding study design.
The verified PBPK model predicts the effect of transient elevation of IL-6 level on exposures of CYP3A4, CYP2C9, and CYP1A2 substrates to be less than twofold. Among the substrates tested, the CYP3A4 substrates (i.e., simvastatin and midazolam) appeared to be the most sensitive to the suppression of IL-6, partially because of the higher susceptibility of CYP3A4 to IL-6 suppression in vitro18 and the presence of dual first-pass metabolism of these substrates in both the liver and the gut.29 Because simvastatin and midazolam are sensitive substrates of CYP3A4, the effect on less-sensitive CYP3A4 substrates should be lower than the effect projected for simvastatin and midazolam. Owing to the negligible suppression observed in vitro, the effect of cytokine elevation on substrates of CYP2C19 and CYP2D6 was not evaluated by the PBPK approach. However, the effect is expected to be lower than that for CYP3A4 substrates.
PBPK prediction of the magnitude and time course of IL-6–mediated CYP3A4 suppression requires values for suppressor-dependent and intrinsic parameters. A key intrinsic factor is the turnover/half-life of the inhibited enzyme. This cannot be determined directly in vivo in humans. In simulations, we used the default Simcyp setting for hepatic and gut CYP3A4 half-lives (36 and 23 hours, respectively). This hepatic half-life value is consistent with estimates from clinical DDI data involving either time-dependent inhibition or induction time course of CYP3A4, whereas the half-life of gut CYP3A4 reflects the faster turnover of enterocytes relative to that of the enzyme.30,31
In this analysis, the predicted effect of the highest IL-6 profile on CYP450 enzymes was slightly lower than that from the observed mean IL-6 profile, indicating that duration of IL-6 elevation may be a more critical factor in determining the magnitude of CYP450 suppression than extent of IL-6 elevation, particularly when the elevated IL-6 level exceeds the EC50 for CYP450 suppression. This was supported by simulations of the time course of CYP3A4 suppression with constant IL-6 concentrations (Figure 4). Our sensitivity analysis also demonstrated that the predictions were not sensitive to the variation of key model parameters, EC50 and Emin, possibly due to the transient nature of IL-6 elevation15; this further increased our confidence in the prediction.
One consequence of increased IL-6 concentrations can be elevation of CRP levels.32 A transient increase of CRP was observed in parallel with increases in cytokines. An inverse correlation between CRP levels and CYP3A activity in patients with cancer has been reported.33,34 Large changes in circulating CRP may be a predictive marker of the clinical suppression and desuppression of CYPs.34 The duration of CRP elevation in patients receiving blinatumomab treatment lasted for 1–2 weeks, and the elevation peaked in the first 1–2 days after blinatumomab infusion start. The duration of CRP elevation may be indicative of the duration of CYP3A4 enzyme suppression, which is consistent with our model prediction.
The degree of cytokine elevation during blinatumomab treatment is highly variable from patient to patient. Hence, a dedicated clinical DDI study in patient populations with a meaningful sample size may not be practical. Using the PBPK model to assess the effects of elevated cytokine levels on suppression of CYP450 enzymes was a viable approach for risk assessment for blinatumomab, and potentially for other therapeutics or diseases that influence cytokine levels.35,36 For example, some patients treated with CD19-targeting chimeric antigen receptor (CAR) T-cell therapy had a 75-fold increase over pretreatment baseline levels in two of the seven measured cytokines.37 Similarly, in patients with psoriasis, mean systemic IL-6 concentrations were elevated 1.3–12.1-fold compared with matched controls.36
There have been reports that elevation of cytokines can modulate the levels of transporters and other proteins that play a role in the disposition of some drugs.38 Our model did not assess transporter-mediated DDI with drugs such as pravastatin and pitavastatin39 because of the absence of kinetic parameters for the influence of cytokines on transporters. Future research on the effects of cytokines on transporters is highly encouraged. Despite the limitation, this analysis provided a rational assessment of the influence of transient cytokine elevation on CYP450 enzymes and their substrates.
In summary, our investigation suggests that the magnitude of the suppressive effect of transient cytokine elevation on hepatic CYP450 enzyme activities is <30% for up to a week. In addition, the changes in exposures to substrates of CYP3A4, CYP1A2, and CYP2C9 are expected to be <twofold and the magnitude of CYP450 suppression is highly dependent on the duration of cytokine elevation. Since transient elevation of cytokines may suppress CYP450 enzyme activities, routine monitoring of exposure to concomitant CYP450 substrates with a narrow therapeutic index should be considered.
Acknowledgments
The authors thank Erica S. Chevalier-Larsen, PhD (Complete Healthcare Communications, Chadds Ford, PA), for assistance in the preparation of this article, which was funded by Amgen Inc.
Author Contributions
Y.X. and M.Z. wrote the manuscript. All authors participated in revising the manuscript for important intellectual content. Y.H. and A.W. designed the in vitro experiments. Y.X. developed the PBPK model and performed the simulation. Y.X., M.Z., Y.H., A.W., B.W., and Y-N.S. analyzed the data.
Conflict of Interest
Y.X., B.W., Y.-N.S. and M.Z. are employees of and shareholders in Amgen. A.W. is an employee of Amgen Research (Munich) and a shareholder in Amgen. Y.H. was an employee of Amgen Research (Munich) and a shareholder in Amgen at the time the research was conducted.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- Bargou R, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- Nagorsen D. Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp. Cell Res. 2011;317:1255–1260. doi: 10.1016/j.yexcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Gore L, et al. 2013. . Cytological and molecular remissions with blinatumomab treatment in second or later bone marrow relapse in pediatric acute lymphoblastic leukemia (ALL). American Society of Clinical Oncology Annual Meeting; May 31–June 4; Chicago, IL;
- Hijazi Y, et al. Blinatumomab exposure and pharmacodynamic response in patients with non-Hodgkin lymphoma (NHL) J. Clin. Oncol. 2013;31 , abstract 3051 ( ) [Google Scholar]
- Klinger M, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–6233. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- Topp MS, et al. Confirmatory open-label, single-arm, multicenter phase 2 study of the BiTE antibody blinatumomab in patients (pts) with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL) J. Clin. Oncol. 2014;32 , abstract 7005 ( ) [Google Scholar]
- Viardot A, et al. Open-label phase 2 study of the bispecific T-cell engager (BiTE) blinatumomab in patients with relapsed/refractory diffuse large B-cell lymphoma. Blood. 2013;122:1811. doi: 10.1182/blood-2015-06-651380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier T, et al. T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3-bispecific single-chain antibody construct. J. Immunol. 2003;170:4397–4402. doi: 10.4049/jimmunol.170.8.4397. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer. 2005;115:98–104. doi: 10.1002/ijc.20908. [DOI] [PubMed] [Google Scholar]
- Schub A, et al. 2013. . Immunopharmacodynamic responses following treatment with the bispecific T-cell engager antibody blinatumomab among patients with relapsed/refractory B-precursor acute lymphoblastic leukemia. 18th Congress of the European Hematology Association June 13–16; Stockholm, Sweden;
- Haas CE, Kaufman DC, Jones CE, Burstein AH. Reiss W. Cytochrome P450 3A4 activity after surgical stress. Crit. Care Med. 2003;31:1338–1346. doi: 10.1097/01.CCM.0000063040.24541.49. [DOI] [PubMed] [Google Scholar]
- Lee JI, Zhang L, Men AY, Kenna LA. Huang SM. CYP-mediated therapeutic protein-drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin. Pharmacokinet. 2010;49:295–310. doi: 10.2165/11319980-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Nakai K, et al. Decreased expression of cytochromes P450 1A2, 2E1, and 3A4 and drug transporters Na+-taurocholate-cotransporting polypeptide, organic cation transporter 1, and organic anion-transporting peptide-C correlates with the progression of liver fibrosis in chronic hepatitis C patients. Drug. Metab. Dispos. 2008;36:1786–1793. doi: 10.1124/dmd.107.020073. [DOI] [PubMed] [Google Scholar]
- Jamei M, et al. The Simcyp population-based ADME simulator. Expert Opin. Drug Metab. Toxicol. 2009;5:211–223. doi: 10.1517/17425250802691074. [DOI] [PubMed] [Google Scholar]
- Machavaram KK, et al. A physiologically based pharmacokinetic modeling approach to predict disease-drug interactions: suppression of CYP3A by IL-6. Clin. Pharmacol. Ther. 2013;94:260–268. doi: 10.1038/clpt.2013.79. [DOI] [PubMed] [Google Scholar]
- Viardot A, et al. Treatment of patients with hon-Hodgkin lymphoma (NHL) with CD19/CD3 bispecific antibody blinatumomab (MT103): double-step dose increase to continuous infusion of 60 μg/m2/d is tolerable and highly effective. Blood (ASH Annual Meeting Abstracts) 2010;116:2880. [Google Scholar]
- Baeuerle PA. Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69:4941–4944. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- Dickmann LJ, Patel SK, Rock DA, Wienkers LC. Slatter JG. Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab. Dispos. 2011;39:1415–1422. doi: 10.1124/dmd.111.038679. [DOI] [PubMed] [Google Scholar]
- Rowland Yeo K, Jamei M, Yang J, Tucker G. Rostami-Hodjegan A. Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut—the effect of diltiazem on the time-course of exposure to triazolam. Eur. J. Pharm. Sci. 2010;39:298–309. doi: 10.1016/j.ejps.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Simcyp. A Guide for IVIVE and PBPK/PD Modeling Using the Simcyp Population Based Simulator. 2012. 231-249 (Simcyp, Sheffield, UK,)
- Paine MF, et al. The human intestinal cytochrome P450 “pie.”. Drug Metab. Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno S, Kawase A, Tsuji A, Tanino T. Iwaki M. Decreased intestinal CYP3A and P-glycoprotein activities in rats with adjuvant arthritis. Drug Metab. Pharmacokinet. 2007;22:313–321. doi: 10.2133/dmpk.22.313. [DOI] [PubMed] [Google Scholar]
- Schmitt C, Kuhn B, Zhang X, Kivitz AJ. Grange S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin. Pharmacol. Ther. 2011;89:735–740. doi: 10.1038/clpt.2011.35. [DOI] [PubMed] [Google Scholar]
- Topp MS, et al. Effect of anti-CD19 BiTE blinatumomab on complete remission rate and overall survival in adult patients with relapsed/refractory B-precursor ALL. J. Clin. Oncol. 2012;30 , abstract 6500 ( ) [Google Scholar]
- U.S. Food and Drug Administration Center for Drug Evaluation and Research. Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations (Silver Springs, MD, 2012)
- Gorski JC, et al. In vivo effects of interleukin-10 on human cytochrome P450 activity. Clin. Pharmacol. Ther. 2000;67:32–43. doi: 10.1067/mcp.2000.103860. [DOI] [PubMed] [Google Scholar]
- Huhn RD, et al. Pharmacodynamics of subcutaneous recombinant human interleukin-10 in healthy volunteers. Clin. Pharmacol. Ther. 1997;62:171–180. doi: 10.1016/S0009-9236(97)90065-5. [DOI] [PubMed] [Google Scholar]
- Azam YJ, Machavaram KK. Rostami-Hodjegan A. The modulating effects of endogenous substances on drug metabolising enzymes and implications for inter-individual variability and quantitative prediction. Curr. Drug Metab. 2014;15:599–619. doi: 10.2174/1389200215666140926151642. [DOI] [PubMed] [Google Scholar]
- Thummel KE. Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu. Rev. Pharmacol. Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- Rowland Yeo K, Walsky RL, Jamei M, Rostami-Hodjegan A. Tucker GT. Prediction of time-dependent CYP3A4 drug-drug interactions by physiologically based pharmacokinetic modelling: impact of inactivation parameters and enzyme turnover. Eur. J. Pharm. Sci. 2011;43:160–173. doi: 10.1016/j.ejps.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhou Y, Hayashi M, Shou M. Skiles GL. Simulation of clinical drug-drug interactions from hepatocyte CYP3A4 induction data and its potential utility in trial designs. Drug Metab. Dispos. 2011;39:1139–1148. doi: 10.1124/dmd.111.038067. [DOI] [PubMed] [Google Scholar]
- Rhodes B, Furnrohr BG. Vyse TJ. C-reactive protein in rheumatology: biology and genetics. Nat. Rev. Rheumatol. 2011;7:282–289. doi: 10.1038/nrrheum.2011.37. [DOI] [PubMed] [Google Scholar]
- Rivory LP, Slaviero KA. Clarke SJ. Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br. J. Cancer. 2002;87:277–280. doi: 10.1038/sj.bjc.6600448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter JG, Wienkers LC, Dickmann LJ. Drug interactions of cytokines and anticytokine therapeutic proteins. In: Zhou H, Meibohm B, editors. Drug-Drug Interactions for Therapeutic Biologics. Hoboken, NJ: John Wiley and Sons; 2013. pp. 215–237. [Google Scholar]
- Harvey RD. Morgan ET. Cancer, inflammation, and therapy: effects on cytochrome p450-mediated drug metabolism and implications for novel immunotherapeutic agents. Clin. Pharmacol. Ther. 2014;96:449–457. doi: 10.1038/clpt.2014.143. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang YM. Ahn HY. Biological products for the treatment of psoriasis: therapeutic targets, pharmacodynamics and disease-drug-drug interaction implications. AAPS J. 2014;16:938–947. doi: 10.1208/s12248-014-9637-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila ML, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Vee M, Lecureur V, Stieger B. Fardel O. Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab. Dispos. 2009;37:685–693. doi: 10.1124/dmd.108.023630. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Sato H. Sugiyama Y. Evaluation of drug-drug interaction in the hepatobiliary and renal transport of drugs. Annu. Rev. Pharmacol. Toxicol. 2005;45:689–723. doi: 10.1146/annurev.pharmtox.44.101802.121444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information