

Abstract
Hydrogels with photocleavable units incorporated into the cross-links have provided researchers with the ability to control mechanical properties temporally and study the role of matrix signaling on stem cell function and fate. With a growing interest in dynamically tunable cell culture systems, methods to synthesize photolabile hydrogels from simple precursors would facilitate broader accessibility. Here, a step-growth photodegradable poly(ethylene glycol) (PEG) hydrogel system cross-linked through a strain promoted alkyne–azide cycloaddition (SPAAC) reaction and degraded through the cleavage of a nitrobenzyl ether moiety integrated into the cross-links is developed from commercially available precursors in three straightforward synthetic steps with high yields (>95%). The network evolution and degradation properties are characterized in response to one- and two-photon irradiation. The PEG hydrogel is employed to encapsulate embryonic stem cell-derived motor neurons (ESMNs), and in situ degradation is exploited to gain three-dimensional control over the extension of motor axons using two-photon infrared light. Finally, ESMNs and their in vivo synaptic partners, myotubes, are coencapsulated, and the formation of user-directed neural networks is demonstrated.
Introduction
Recent directions in the synthesis of scaffolds for cell culture and tissue regeneration have made tremendous progress in recapitulating aspects of the complex milieu of signals found in in vivo microenvironments. (1, 2) This direction represents a significant advancement for the field, but one often considers which features are most important and how much mimicry is required. In some applications, such as cartilage tissue engineering, early efforts found success when focused primarily on mechanical characteristics of the scaffolds, thereby recapitulating the basic tissue properties while simultaneously supporting chondrocyte function (e.g., survival and secretory properties). (3–5) However, other tissue engineering applications necessitated the development of scaffolds that introduced biochemical signals to promote or direct desired cell function, and this led to significant advances in bioconjugation methods to tether proteins and small molecules, conferring functionalization and signaling to embedded cells. Such approaches have been exploited to design materials that preferentially differentiate stem cells into different lineages, (6, 7) increase production of extracellular matrix (ECM) components, (8) direct cell migration, (9–11) and control extension of axons. (11, 12) However, in vivo signals are not presented in such a static manner, but rather, complex spatially and temporally regulated presentation of biological cues drive many fundamental processes. (13, 14) With this in mind, more recent efforts in scaffold design have exploited advances in polymer chemistry to demonstrate in situ and real time manipulation of cellular microenvironments, thus beginning to recreate the dynamic cellular milieu in vitro. Such approaches have relied on advances of bio-orthogonal reactions and cytocompatible photochemical reactions to render advanced cell culture niches. (15–23) Specifically, photopatterned protein–ligand interactions have enabled the spatially controlled presentation of bioactive signaling proteins to drive stem cell migration, (12, 24) orthogonal photocleavage reactions have been used to more efficiently differentiate cells by mimicking in vivo pathways, (15) and in situ photodegradation has been used to isolate the effect of cellular traction on stem cell differentiation. (25) For example, the Shoichet group developed a system exploiting strong protein–ligand interactions to immobilize gradients of sonic hedgehog protein to drive neural progenitor cell migration into an agarose gel. (12) The Lutolf group used a photoactivated enzymatic ligation to pattern gradients of VEGF to demonstrate control over mesenchymal stem cell migration. (24) Previous work in the Anseth group identified orthogonal photodegradation reactions that allowed the sequential release of bone morphogenic proteins, which enabled more efficient differentiation of human mesenchymal stem cells. (15)
While the above advances have demonstrated the power of controlled biological signaling to cells through regulation of material properties in space and time, complementary materials and simplified systems provide opportunities to expand the accessibility of the chemistry to a broader community of biomaterial researchers. (26, 27) Here, recent advances in molecular building blocks are exploited to synthesize a step-growth poly(ethylene glycol) (PEG) hydrogel cross-linked through copper-free click chemistry capable of undergoing degradation upon irradiation by either single- (334 and 365 nm) or two-photon light (740 nm). A SPAAC reaction between dibenzylcyclooctyne (DBCO) and an aliphatic azide is employed for cross-linking and a nitrobenzyl ether moiety is engineered into the cross-linker to enable degradation. The macromolecular monomer components used to synthesize the hydrogel are readily synthesized in one or three steps in gram quantities and at high yields (>95%) from commercially available precursors. The gel evolution and subsequent photodegradation properties are characterized by shear rheology and confocal microscopy and shown to behave in a predictable way based on step-growth network structure–property relationships (e.g., initial gel modulus) and fundamental photochemical reactions for the cleavage reaction. This predictable behavior and precise control of the initial hydrogel allows design of cell scaffolds with spatiotemporally tunable properties.
McKinnon et al. (28) previously synthesized an MMP-degradable PEG hydrogel system for the culture of embryonic stem cell-derived motor neurons (ESMNs) and identified chemical and physical signals that were critical to promoting long-term ESMN survival and axon extension. While this synthetic method allowed facile introduction of multiple biofunctional cues and subsequent control of critical aspects of the cellular microenvironment, the materials render the experimenter a passive observer as the cells dictate and remodel the surrounding environment. Perhaps not surprisingly, axon outgrowth was observed to be isotropic, which then motivates the question as to how one might direct or guide this outgrowth in a spatially regulated manner. Gaining control over the course of motor axon extension using an accessible photodegradable hydrogel platform could allow fundamental and quantitative studies of the effect of neurotrophins and growth factors on these processes. Such information may prove useful treating motor neuron injuries or treating neurodegenerative diseases, such amyotrophic lateral sclerosis, especially in the context of better understanding how to present cues that might guide neuromuscular junctions in vivo. (29, 30) Combining a dynamic, accessible hydrogel with embryonic stem cell-derived motor neurons provides a strong platform for further understanding motor axons extension on which to build future research.
Experimental Section
Macromer Synthesis
Eight-arm 40 kDa PEG-nitrobenzyl-azide (8-NBA) macromers were synthesized by first coupling a Fmoc-photolabile linker (16.8 equiv, Advanced ChemTech, RT1095) with 8-arm 40 kDa PEG-amine (1 equiv, JenKem, A8012) using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU, ChemPep, 120801) (16 equiv) as an activator and 4-methyl morpholine (32 equiv, Aldrich, M56557) as a base. The reaction was allowed to proceed overnight, after which the product was precipitated in ice cold ether, collected via centrifugation, and dissolved in 80% DMF, 20% piperidine to remove the Fmoc protecting group. After 4 h of deprotection, the product was again precipitated in ice cold ether, dissolved in DMF, and coupled to azido butanoic acid (16.8 equiv), which was synthesized as previously described, (31) again using HATU (16 equiv) as an activator and 4-methyl morpholine at a base (32 equiv). The final product was precipitated in diethyl ether for a third time, dissolved in a minimal volume of DI water, and dialyzed for 24 h, after which it was lyophilized and used for experimentation.
Four-arm 20 kDa PEG-dibenzylcyclooctyne (4-DBCO) macromers were synthesized by coupling 4-arm 20 kDa PEG-amine (1 equiv, JenKem, A7026) with DBCO-acid (8.8 equiv, Click Chemistry Tools, A101) using HATU (8 equiv) as an activator and 4-methyl morpholine (18 equiv) as a base. The product was precipitated in ice-cold diethyl ether, dissolved in DI water, dialyzed for 24 h, lyophilized, and finally used for experimentation (Scheme 1).
Scheme 1.

Facile, One, or Three Step Syntheses of Starting Materials for Copper-Free Click Chemistry Cross-Linked Photodegradable Hydrogels. aa8-NBA is synthesized through coupling commmericially available Fmoc-protected photolabile group to 8-arm 40 kDa PEG-amine, deprotecting the Fmoc group with piperidine, and coupling azido butanoic acid. 4-DBCO is synthesized by simply coupling DBCO-acid to 4-arm 20 kDa PEG-amine.
4-DBCO 1H NMR (DMSO-d6, 400 MHz)
δ = 7.73 (t, J = 4 Hz, 1H), 7.45 (m, 8H), 5.04 (d, J = 16 Hz, 1H), 3.62 (s, 2H), 3.51 (s, PEG), 3.11 (q, J = 4 Hz, 2H), 2.17 (m, 1H), 1.85 (t, J = 8 Hz, 2H), 1.74 (m, 1H), 1.20 (m, 4H).
8-NBA 1H NMR (DMSO-d6, 400 MHz)
δ = 8.69 (d, J = 8 Hz, 1H), δ= 7.96 (t, J = 4 Hz, 1H), 7.47 (s, 1H), 7.21 (s, 1H), 5.37 (m, 1H), 3.90 (s, 3H), 4.03 (m, 4H), 2.21 (m, 4H) 1.93 (m, 3H), 1.70 (m, 2H), 1.41 (d, J = 8 Hz, 3H).
Peptide Synthesis
N3-YIGSR was synthesized on a Protein Technologies Tribute Peptide Synthesizer using standard Fmoc chemistry and Rink Amide MBHA resin. The final azide reside was incorporated onto the N-terminus by including azido-butanoic acid in the final cartridge. Peptide cleavage solution was prepared by dissolving 250 mg of dithiothreitol (DTT) and 250 mg of phenol in a solution of 95% trifluoroacetic acid (TFA), 2.5% triisopropylsilane (TIPS), and 2.5% deionized water. Synthesized peptides were cleaved in the solution for 2 h. Cleaved peptides were precipitated in cold diethyl ether, recovered via centrifugation, desiccated overnight, and then purified by reverse-phase HPLC (Waters Delta Prep 4000) purification on a C18 column using a linear acetonitrile:water gradient. The collected fractions of purified peptides were identified by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry. Expected mass, 706 Da; measured mass, 706 Da.
Gel Formation
The PEG macromers were dissolved in PBS to form 20 wt % (50 mM functional groups) stock solutions for the 4-DBCO and 8-NBA. Solutions were combined stoichiometrically at 6.7 wt % (8.3 mM functional groups) to form hydrogels in situ on a temperature-controlled Peltier plate set to 25 °C.
Rheology
Hydrogels were formed in situ by pipetting 12 μL of monomer solutions (2 μL 4-DBCO (20 wt %, 50 mM functional groups), 2 μL of 8-NBA (20 wt %, 50 mM functional groups), and 8 μL of PBS) between the bottom quartz plate and a flat tool 8 mm in diameter on a shear rheometer (TA DH-R3). This yielded a stoichiometrically balanced 6.7 wt % (8.3 mM functional group concentration) gel. The gap was closed to 100 μm, and the experiments were commenced as quickly as possible, typically in less than 1 min. Frequency and strain sweeps were performed to ensure measurements were made in the linear region. Experiments designed to monitor the hydrogel evolution and its subsequent photodegradation were performed at 1% strain and 1 rad/s, and frequency sweeps were performed at 1% strain. The light source used for the photodegradation experiments was an EXFO Actacure with a 320–390 nm filter in between the mercury lamp and the rheometer, allowing the 365 and 334 nm bands to erode the material. Light intensities between 102 and 204 mW/cm2 were used, and based on initial functional group concentrations 40% of the light was attenuated through the hydrogel.
ES Cell Culture
ES cells were differentiated into spinal motor neurons as previously described. (32, 33) Briefly, Hb9::GFP mouse embryonic stem cells were plated into ES cell medium (ES DMEM, ES FBS, glutamine, nonessential amino acids, nucleosides, 2-mercaptoethanol, LIF (Life Technologies)) at approximately 5 × 105 cells per gelatinized T25 flask. After 24 h the media was replaced, and on day 2 of culture ES cells were trypsinized and placed in suspension culture in motor neuron media (Advanced-DMEM/F12, Neurobasal, and Knockout Serum Replacement (Life Technologies)) at 5 × 105 cells per untreated 10 cm tissue culture dish. In suspension culture, the cells aggregated into embryoid bodies (EBs). Two days after initial seeding, the EBs were split 1:4 and induced into motor neurons with 1 μM retinoic acid (RA) (Sigma) and smoothened agonist (SAG) (Millipore). After 3 days of exposure to RA and SAG, the EBs displayed strong expression of the Hb9::GFP transgene.
Cell Encapsulation
Gels were prepared with a total volume of 30 μL from stock solutions of 4-DBCO (20 wt %, 50 mM functional groups), 8-NBA (20 wt %, 50 mM functional groups), N3-YIGSR (20 mM), and N3-Alexa 594 (0.1 mM). 4-DBCO was prereacted for 5 min with peptides and dye to minimize cellular uptake of small molecules. Dozens of EBs were then gently mixed into the solution and 8-NBA was added. The solution was then quickly, but carefully, transferred into a mold 5 mm in diameter and 1.5 mm tall and allowed to polymerize for 5 min before transferring into motor neuron media.
Confocal Microscopy Imaging and Gel Degradation
Gels were placed between a glass slide and a coverslip separated by a rubber gasket and were imaged using a NA 0.3 10× water immersion objective. Hydrogel features were eroded using a two-photon laser tuned to 740 nm emitting a total averaged power of 300 mW dwelling 1.58 μs per pixel. The pixel size was adjusted to match the theoretical x–y resolution of the 0.3 NA lens. A 488 nm laser was used to excite eGFP, calcein AM, and ethidium homodimer-1. A 594 nm laser was used to excite the Alexa 594-labeled alpha-bungarotoxin.
Axon Growth Videomicroscopy and Tracking
Dynamic axon growth in photodegraded channels was observed using a Nikon TE 2000-E microscope with a Nikon environmental chamber, an external heater (InVivo Scientific), and a CO2 regulator (InVivo Scientific). Hydrogels were degraded as described previously, and imaged within a 24-well culture insert plate (BD Falcon, Fisher). The gels were held stationary by the addition of a modified transwell insert that lacked a bottom filter to allow for clear visualization of the axon growth. Immediately following photodegradation, images were taken at 5 min intervals for 36 h using Metamorph software for automated stage control, image collecting, and postexperiment axon tracking. Specifically, the axon growth cone was tracked from the initial distinction from the embryoid body until reaching the end of the channel. Axonal growth speed was then calculated from positional information over time.
C2C12 Culture Conditions
C2C12 mouse myoblasts were plated on tissue culture polystyrene (TCPS) and cultured in myoblast growth medium (high glucose DMEM supplemented with 20% fetal bovine serum (FBS) and penicillin and streptomycin (P/S)). When the cells reached confluency, the media was changed to differentiation medium (high glucose DMEM with 2% FBS and P/S) to induce myotube formation. After 7–10 days in differentiation medium, myoblast fusion and myotube alignment were observed.
Coculture
Differentiated C2C12 myotubes were mechanically removed from TCPS, triturated until the clusters were similar in size to EBs, and gently rinsed twice to remove single cells. Encapsulation was performed as before, except myotubes and EBs were encapsulated together in the same gel. The medium used for coculture was motor neuron media supplemented with 10 ng/mL glial cell-derived neurotrophic factor (GDNF).
Bungarotoxin Staining
Once axons had reached the muscle cells and begun to innervate the cell mass, alpha-bungarotoxin (α-BTX) conjugated with Alexa Fluor 594 was introduced to the culture media at 10 ng/mL and allowed to swell into the gel for 3 h on a shaker. The media was then replaced with PBS to allow excess α-BTX to diffuse out for 3 h, changing the PBS each hour.
Results and Discussion
Four-arm 20 kDa PEG macromers were functionalized with dibenzylcyclooctyne (DBCO) moieties and 8-arm 40 kDa PEG macromers were functionalized with nitrobenzyl azide (NBA) moieties and dissolved into phosphate buffered saline (PBS) at 20 wt % (50 mM functional groups) to form stock solutions (Scheme 1). As first reported by the Bertozzi group, (34) DBCO reacts with a variety of azides quickly and specifically via a strain promoted azide–alkyne cycloaddition (SPAAC) (Figure 1a). Mixing macromer solutions of 8-NBA and 4-DCBO under physiological conditions results in the formation of a hydrogel, abbreviated by 8-NBA:4-DBCO, with an equilibrium shear elastic modulus of 3600 ± 200 Pa. Furthermore, evolution of the network is rapid with a mean time of 240 ± 40 s to achieve 90% of the final modulus (Figure 1b). While the gelation kinetics and final modulus are acceptable for the experiments discussed herein, both of these parameters can be easily tailored to the application at hand by varying the stoichiometry, molecular weight, or functionality of the initial PEG macromers. The results can be predicted using fundamental theories describing step-growth gelation. (23, 35, 36)
Figure 1.
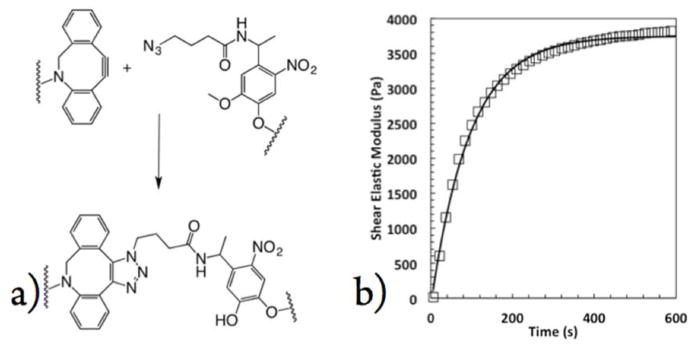
(a) Chemical structures showing the formation of the triazole bond linking the PEG macromers driving the formation of the hydrogel. (b) Evolution, left, of synthetically accessible click hydrogel (open squares) along with exponential fit (line). Gels reached 90% of their equilibrium moduli of 3600 ± 200 Pa after 240 ± 40 s.
While SPAAC coupling between DBCO and NBA drives gel formation, the presence of NBA in the final network structure can serve as a photolabile linker, allowing externally directed degradation of the gel by exposure to UV single-photon or NIR two-photon irradiation. Upon absorption of a photon, the NBA cleaves between the nitrogen and the tertiary carbon through an intramolecular reaction into an amide-terminate fragment and an aldehyde-terminated fragment (Figure 2a). An important difference between this material system and previously published studies (37, 38) is that the nitrobenzyl ether group is linked to the PEG via an amide bond instead of an ester bond. The rationale for choosing the amide-linker is that the small molecule Fmoc-protected photolabile linker is commercially available, which is then readily coupled to PEG-NH2 through standard peptide coupling conditions. This Fmoc-protected photolabile linker allows the synthesis of photodegradable macromers in 1–2 steps with >95% yield. However, the photocleavage properties of amide-linked NBA have not been previously studied and we hypothesized that its degradation properties may differ significantly from the ester-linked nitrobenzyl due to the dramatically different stabilities of ester and amide bonds.
Figure 2.
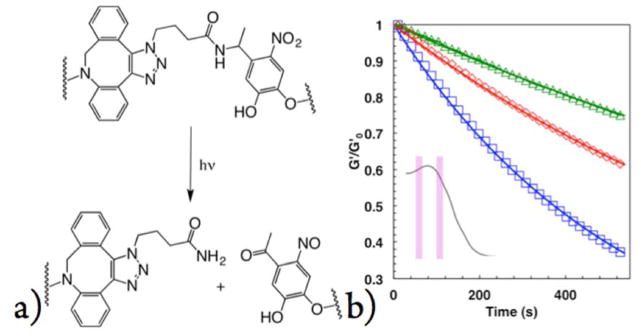
(a) Chemical structures showing the degradation of the bond cross-linking the hydrogel network. Cleavage occurs between the tertiary carbon and the amide nitrogen in a radical-mediated mechanism. Degradation of the same hydrogels under 204 mW/cm2 (blue), 154 mW/cm2 (red), and 102 mW/cm2 (green) UV irradiation (mercury arc lamp with 320 nm–390 nm bandpass filter) along with first-order fits. The absorbance spectrum of the nitrobenzyl cleavable unit is shown in the inset along with the bands used for degradation (334 and 365 nm).
Consistent with previously published degradation studies, hydrogels were formed in situ on a rheometer and exposed to varying intensities of UV light derived from a mercury arc lamp coupled to a 320–390 nm bandpass filter. Hydrogel degradation was observed to be much slower than that of similar ester-linked nitrobenzyl hydrogels. Because hydrogel degradation is a first-order process, plots of decaying shear elastic modulus were fit to eq 1,
| (1) |
where G(t) is the shear elastic modulus as a function of time, G0 is the initial shear elastic modulus, t is the time, Φ is the quantum yield, ε is the absorbance, λ is the wavelength, I0 is the incident light intensity, NA is Avogadro’s number, h is Planck’s constant, and c is the speed of light. Hydrogels were exposed to 204, 154, and 102 mW/cm2 UV irradiation for 1200 s, and, as expected, rates of degradation increased with increasing light intensity. Interestingly, immediately upon exposure to light, the hydrogel modulus noticeably increased, a phenomenon previously observed by Kasko and Griffin. (37) However, we eliminated this effect from our rheological traces and fit degradation to a first-order exponential decay model (Figure 2b).
Upon fitting and analyzing the data, we observed much slower rates of degradation compared to previously published reported values for the ester-linked nitrobenzyl.(37) Apparent rates of degradation varied from 1.6 ± 0.2 × 10−3 s−1 under 204 mW/cm2 irradiation to 0.5 ± 0.1 under 102 mW/cm2. If photodegradation occurs in a thin film and follows a first-order process, the apparent kinetic constant of cleavage normalized to the light intensity (kapp/I0 × 104) for the amide linked NBA is simply 6.3 ± 1.4 cm2 mW−1 s−1. Note that this value is 5 times slower than the kinetic of cleavage of the slowest nitrobenzyl group previously reported and 130 times slower than the fastest. (37)
We then sought to characterize the degradation properties of this material in response to two-photon light. Two-photon light has been used widely in the biomedical sciences due to its limited dispersion and biocompatibility. However, intense two-photon light has been shown to disrupt mitochondrial function, (39, 40) damage the cytoskeleton, (40) and cleave axons. (41) Furthermore, some potential existed that the slow single-photon degradation characteristics would preclude two-photon degradation, due to the intensity of light required cavitating the hydrogel before degrading cross-links, an effect that was previously observed at high pulse energies. (42) However, the two-photon response of an organic molecule is not necessarily correlated to its single photon response, a phenomenon that is abundantly clear when designing genetically encoded fluorescent proteins. (43)
In using two-photon degradation to introduce topographical features in 3D, we first wanted to ensure that the light dose required for degradation of the material would be within a range tolerated by encapsulated cells and not cause material cavitation before erosion of desired features. As ESMN embryoid bodies are generally between 100 and 500 μm in diameter, a 0.3 NA 10× water objective was selected to erode mesoscale features relevant to the length scales of axon extension. Since the focal volumes of lenses used for focusing two-photon light are well understood (calculations in the Supporting Information), we estimated that the 10× water immersion lens with 740 nm light would have a focal volume of 2.5 μm3, and at the maximum laser power of 279 mW only 0.25 nJ/μm3 is delivered per pulse. This is several orders of magnitude lower than doses known to damage cells in the literature. (39, 40) For example, axotomy has been reported at 400 nJ/μm3, (41) mitochondrial damage at 77 nJ/μm3, (40) and the lowest threshold for cellular damage reported as 58 nJ/μm3. (39)
Next, we sought to estimate the minimum time of irradiation to erode gel features as a function of the gel connectivity and exposure parameters. Specifically, the time for reverse gelation, tc, was estimated as a function of the percolation threshold (pc), the average power of irradiation (Pavg), the two-photon cross section of the nitrobenzyl unit (δuϕu), the period of the laser pulses (T, 12.5 ns), the duration of the laser pulses (τp, 140 fs), the wavelength (λ, 740 nm), the focal area (ωxy, 532 nm), and the focal volume (VF, 2.5 μm3) (eq 2). h and c are Planck’s constant and the speed of light.
| (2) |
Equation 2 defines a curve combining photodegradation kinetics, two-photon physics, and statistical models of reverse gelation to define what combination of light intensity and exposure time will result in total material erosion. (42) Thus, the extent of photodegradation can be controlled by varying light exposure, connectivity of the hydrogel, objective lens, and quantum yield of photodegradable cross-linker. Given ca. 25% overlap in pixel size and the 8 μm z-resolution of the 0.3 NA 10× objective, the light exposure is effectively increased by a factor of 8 when eroding planes are separated by 1 μm, which is a typical spacing used in these studies. With these assumptions and equations, we calculated expected critical exposure times for different laser powers using previously published quantum efficiencies of a similar nitrobenzyl moiety (Figure 3). (44) Using these plots, we calculated the theoretical minimum power and exposure time required for complete gel degradation.
Figure 3.
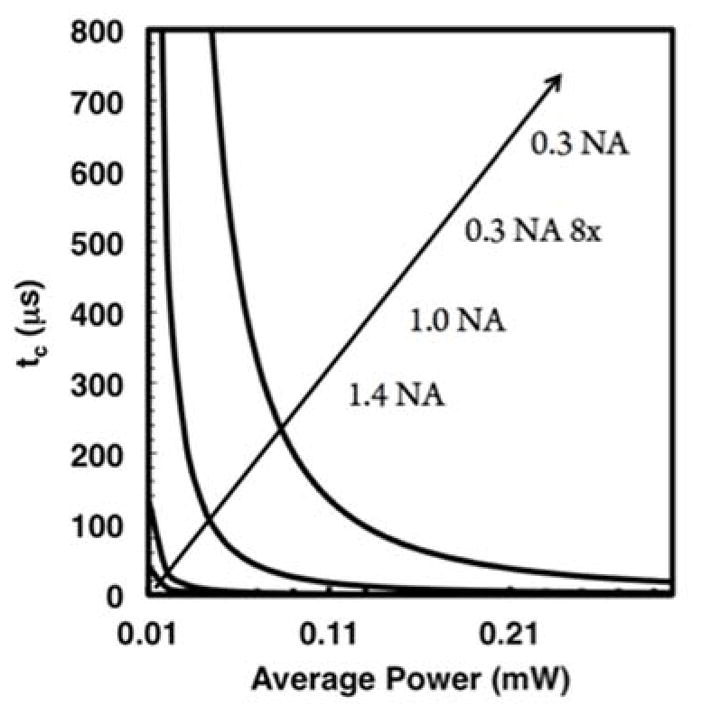
Theoretical plots of critical exposure time versus power with curves representing lenses of several common numerical apertures. The critical exposure time decreases dramatically for a given power as the numerical aperture increases. The numerical aperture of the lenses are shown offset from the curves with the 0.3 NA 8× curve representing the effective light dose a 1 μm plane receives when scanned by a 0.3 NA objective lens, given the 8 μm z-resolution.
To test the validity of these estimates and their relevance to the experimental samples, wild type ESMN embryoid bodies were encapsulated in gel formulations identical to those characterized on the rheometer (6.3 wt %, 8.3 mM functional groups) aside from the addition of 0.7 mM YIGSR adhesive peptide and 3 μm Alexa-594 to enable hydrogel visualization. The cell-laden gels were placed in motor neuron media and stained for markers of viability at 72 h (Figure 4). Note, ESMNs lacking the Hb9::GFP transgene were used to avoid convolution with the viability staining and imaging. Since primary motor neurons are highly sensitive to oxidative damage, (45) it is important to note that the SPAAC cross-linking allows gelation in the presence of ESMNs and leads to high levels of cell survival (>95%). These results are very similar to our previously reported thiol–ene hydrogel system, (28) but in this case, we added an azide-terminated YIGSR peptide as an adhesive cue and also incorporated the photolabile linker to create topographical features for guidance of axon extension in 3D.
Figure 4.
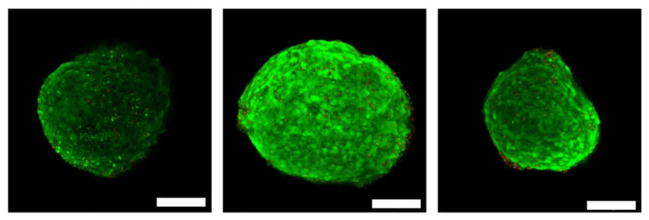
Images of ESMN embryoid bodies encapsulated in 8-NBA:4-DBCO hydrogel 72 h after encapsulation. 6.3 wt % (8.3 mM functional groups) SPAAC cross-linked PEG gels were functionalized with 0.7 mM YIGSR peptide and 3 μm Alexa-594 to allow hydrogel visualization. Cells were stained with calcein (green) and ethidium homodimer (red) to indicate live and dead cells, respectively. Cell viability is excellent in all cases, although difficult to quantify based on the tight packing of cells into embryoid bodies. Scale bar is 100 μm.
Next, ESMN embryoid bodies were encapsulated in the same hydrogel formulations, and ten 10 × 10 × 300 μm channels were eroded in proximity to the embryoid bodies, scanning each pixel 3.16 μs and using laser powers varying from 10 to 100%, corresponding to 15–110 mW/μm3. Theoretically, the critical threshold for reverse gelation should be crossed at 40% laser power (50 mW/μm3), if the quantum yield for the amide-linked nitrobenzyl cross-linker is similar to that of the ester-linked version. (44) Experimentally, axon extension was observed in channels created at only 20%, 28 mW/μm3 laser power, indicating efficient two-photon degradation (Figure 5).
Figure 5.
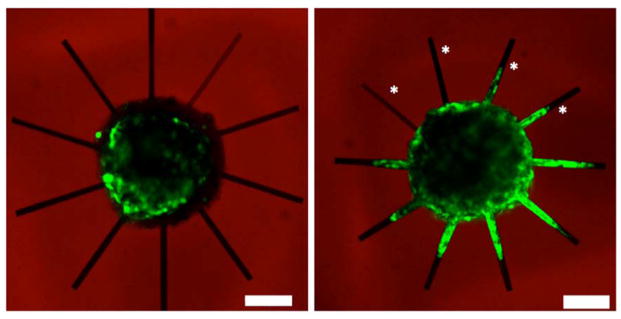
ESMN embryoid body encapsulated in the 6.3 wt % 8-NBA:4-DBCO hydrogel immediately after encapsulation and erosion of channels (left) and 48 h after. Axons extend into channels that have been exposed to sufficient light to erode the material. Channels represent varying erosion where the exposure time was held constant at 3.16 μs and the power was varied from 15 to 110 mW/μm3. These powers correspond to a tc from 815 to 0.90 μs, based on a δϕ value of 0.20 GM. Channels marked with an asterisks were exposed to light under conditions that do not theoretically lead to complete gel erosion, which is theoretically crossed at <63 mW/μm3 and the ray at 3 o’clock. In all of these conditions, axon growth is not observed. Because axons extend through channels exposed to 39 mW/μm3, it is likely the quantum efficiency, δϕ, of the nitrobenzyl linker under study is actually higher than the previously reported value of a similar compound.(44) Scale bar is 100 μm.
After experimentally confirming exposure conditions necessary to erode gel features and allow axon extension, the effects of channel dimensions on this extension were investigated. In contrast to literature reported of cell migration as a function of channel dimensions, (46, 47) the speed and extent of axon outgrowth were found to be relatively independent of channel width, except when the channel dimensions (2 by 2 μm) approached those of extending axons (1 μm) where hindered outgrowth was observed (Figure 6). Example axon tracks are shown in Supporting Information Figure 3.
Figure 6.
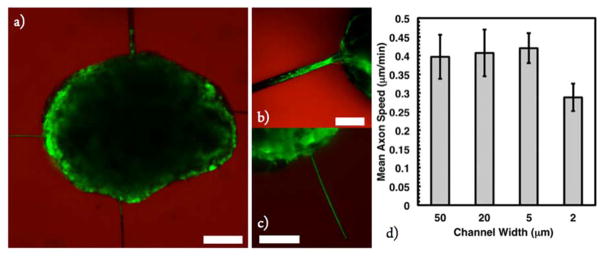
Square channels of 50 μm × 50 μm, 20 μm × 20 μm, 5 μm × 5 μm, and 2 μm × 2 μm cross-sectional area were eroded proximal to encapsulated ESMN embryoid bodies in 6.3 wt % 8-NBA:4-DBCO hydrogels. Axon growth rates were then monitored using a real-time microscope. Axon extension rates were found to be independent of channel size, until the size of the channel cross-sectional area approached 2 μm, which is close to the size of the axon, and progress is physically impeded. (a) ESMN embryoid body extending axons through channels of interest. (b) Motor axons extending through a 20 μm × 20 μm channel. (c) Motor axons extending through a 2 μm × 2 μm channel. Scale bar is 100 μm for (a) and 50 μm for (b) and (c). Examples tracks are shown in Supporting Information Figure 3.
After identifying conditions for channel erosion and physiologically relevant channel dimensions that promote axon extension, we next sought to conduct experiments to explore aspects of motor axon path-finding and decision-making. In vivo, motor axons must follow a complex array of signal gradients to reach their final synaptic destination. Numerous proteins implicated in this process have been identified, but few quantitative methods exist to explore their role in vitro or to study aspects of competing signals. Thus, to develop an in vitro model of this, we engineered a patterned hydrogel system to present extending axons with pathway options, specifically physical channels of varying orientation to the initial axon extension (Figure 7). In the absence of externally presented biochemical stimuli, motor axons prefer to extend in a persistent manner through channels that are in the direction of the extension (i.e., straight or at ±45° to the this line). From this baseline characterization, experiments are currently underway to present gradients of attractive cues through unfavorable forks at 135° angles or repulsive cues through favorable forks. Through such experiments, we seek to quantification the relative role of biophysical versus biochemical signals on axon guidance with the goal of providing fundamental insight into competing and synergistic mechanisms of axon guidance.
Figure 7.
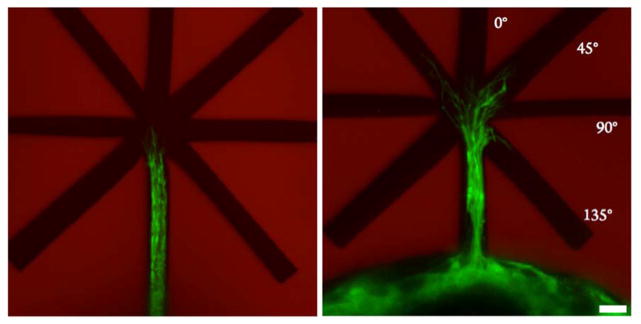
Fork-shaped patterns were eroded into the 6.3 wt % 8-NBA:4-DBCO using 740 nm two-photon light providing extending motor axons with decisions regarding their direction of extension. The image on the left shows axons hours before reaching the fork and the image on the left shows axons shortly after selecting a direction. We observed that the vast majority of axons entered the 0° or ±45° forks, indicating persistence in axon outgrowth when no external stimulus is applied. Scale bar is 10 μm.
The rationale of studying and manipulating axon guidance is to maintain functionality of neuromuscular junctions after traumatic injury or chronic disease. Toward the first step of a model in vitro system of physiological interest, we aimed to demonstrate the ability of photodegradable and patternable hydrogel systems to allow user-directed axon outgrowth -and formation of a simplified neural network. ESMNs have been shown to spontaneously form functional synapses with C2C12 myotubes in vitro, (48, 49) so we sought to demonstrate matrix control over motor axon extension and assemble a neural circuit in this hydrogel. Specifically, C2C12 myoblasts were differentiated into myotubes using established literature techniques. (50, 51) Briefly, myoblasts were allowed to become confluent and then switched to a low-serum media, which drives fusion and myotube formation. The resulting myotubes were then mechanically removed from culture plates through gentle scraping, forming myotube bundles that were roughly the same size as the ESMN embryoid bodies. These myotube bundles were then coencapsulated with ESMN embryoid bodies at roughly one dozen aggregates per hydrogel. The gels were then imaged to identify spatial location of the two cell types, and then eroded via two-photon process to create channels connecting the two cell types. Images captured motor axon extension toward their synaptic targets within 2 days, as well as observation of synaptic markers between the two via staining with alpha-bungarotoxin (Figure 8). Specially, significant axon branching and interrogation of the myotube surface were observed, and evidence of the formation of functional junctions was characterized by staining for acetylcholine receptor clustering using alpha-bungarotoxin.
Figure 8.

User-directed in vitro assembly of neural circuits. ESMN embryoid bodies expressing eGFP (green) were cultured with myotube bundles (gray, marked with asterisks) and connected using a 740 nm two-photon laser to erode 10 μm × 10 μm channels between the two cell types. Multiple channels were eroded to increase the chances of axons extending from the embryoid body to the myotube aggregate. Within 2 days, motor axons extended up to millimeters to reach the myotubes. Bungarotoxin stain (red) suggests the formation of functional synapses. Asterisks are added to mark the location of myotube bundles. Scale bar is 100 μm.
Conclusions
The synthesis and characterization of a photodegradable hydrogel formed through bioorthogonal click chemistries is reported. Gelation characteristics were characterized via rheological methods, and found to be appropriate primary ESMN cultures and to yield a wide range of properties for biomaterial applications. Cell-laden gels can be modified in real time at user defined time points by single and multiphoton degradation. While single photon degradation was significantly slower than previously reported systems two-photon degradation occurred at cytocompatible rates, namely, conditions below the threshold exposure that can lead to possible cell damage or material cavitation. This hydrogel was then functionalized with laminin-derived peptide to enable the culture of ESMN embryoid bodies. These cells were shown to remain nearly 100% viable in the material and two-photon degradation was employed to gain control over their axon extension. Speed and degree of axon extension was observed to be independent of eroded-channel width and axons were observed to generally extend in a persistent manner. Finally, ESMNs and C2C12 myotubes were cocultured in the gel and channels were eroded connecting aggregates of both cell types. Motor axons extended from the embryoid bodies to the myotube aggregates and branched over their surface. Strong, punctate alpha-bungarotoxin staining indicated the formation of functional neuromuscular junctions. These results suggest that dynamic materials can be used to direct the formation of neural networks in vitro and should have broad implications in the biomaterials, neuroscience, and neural engineering communities.
Supplementary Material
Acknowledgments
Funding from the National Science Foundation (CBET 1236662) and the Howard Hughes Medical Institute is acknowledged. The authors acknowledge Dr. Malar Azagarsamy for helpful discussions.
Abbreviations
- ECM
extracellular matrix
- PEG
poly(ethylene glycol)
- ESMN
embryonic stem cell derived motor neuron
- ALS
amyotrophic lateral sclerosis
- DBCO
dibenzyl cyclooctyne
- NBA
nitrobenzyl azide
- SPAAC
strain promoted azide–alkyne cycloaddition
- PBS
phosphate buffered saline
- HATU
(1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate)
- FBS
fetal bovine serum
- P/S
penicillin and streptomycin
- GDNF
glial cell-derived neurotrophic factor
- α-BTX
alpha-bungarotoxin
Footnotes
Author Contributions
D.D.M. and T.E.B. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- 1.Azagarsamy MA, Anseth KS. ACS Macro Lett. 2012;2(1):5–9. doi: 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirschner CM, Anseth KS. Acta Mater. 2013;61:931–944. doi: 10.1016/j.actamat.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryant SJ, Anseth KS. J Biomed Mater Res. 2001;59:63–72. doi: 10.1002/jbm.1217. [DOI] [PubMed] [Google Scholar]
- 4.Bryant SJ, Anseth KS. J Biomed Mater Res. 2002;64A:70–79. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 5.Bryant SJ, Durand KL, Anseth KS. J Biomed Mater Res. 2003;67A:1430–1436. doi: 10.1002/jbm.a.20003. [DOI] [PubMed] [Google Scholar]
- 6.Keselowsky BG, Collard DM, García A. J Proc Natl Acad Sci USA. 2005;102(17):5953–5957. doi: 10.1073/pnas.0407356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benoit DSW, Schwartz MP, Durney AR, Anseth KS. Nat Mater. 2008;7:816–823. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCall JD, Luoma JE, Anseth KS. Drug Delivery Transl Res. 2012;2:305–312. doi: 10.1007/s13346-012-0090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLong SA, Moon JJ, West JL. Biomaterials. 2005;26:3227–3234. doi: 10.1016/j.biomaterials.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 10.Guarnieri D, De Capua A, Ventre M, Borzacchiello A, Pedone C, Marasco D, Ruvo M, Netti PA. Acta Biomater. 2010;6:2532–2539. doi: 10.1016/j.actbio.2009.12.050. [DOI] [PubMed] [Google Scholar]
- 11.Luo Y, Shoichet MS. Nat Mater. 2004;3:249–253. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
- 12.Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Nat Mater. 2011:799–806. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 13.Ashe HL, Briscoe J. Development. 2006;133:385–394. doi: 10.1242/dev.02238. [DOI] [PubMed] [Google Scholar]
- 14.Weng G, Bhalla US, Iyengar R. Science. 1999;284:92–96. doi: 10.1126/science.284.5411.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azagarsamy MA, Anseth KS. Angew Chem, Int Ed. 2013;52(51):13803–13807. doi: 10.1002/anie.201308174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azagarsamy MA, Alge DL, Radhakrishnan SJ, Tibbitt MW, Anseth KS. Biomacromolecules. 2012;13:2219–2224. doi: 10.1021/bm300646q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azagarsamy MA, McKinnon DD, Alge DL, Anseth KS. ACS Macro Lett. 2014;3:515–519. doi: 10.1021/mz500230p. [DOI] [PubMed] [Google Scholar]
- 18.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Adv Mater. 2013;26:865–872. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Chem Mater. 2014;26(7):2382–2387. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grover GN, Lam J, Nguyen TH, Segura T, Maynard HD. Biomacromolecules. 2012;13:3013–3017. doi: 10.1021/bm301346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koehler KC, Anseth KS, Bowman CN. Biomacromolecules. 2013;14(2):538–547. doi: 10.1021/bm301789d. [DOI] [PubMed] [Google Scholar]
- 22.Rickett TA, Amoozgar Z, Tuchek CA, Park J, Yeo Y, Shi R. Biomacromolecules. 2011;12:57–65. doi: 10.1021/bm101004r. [DOI] [PubMed] [Google Scholar]
- 23.Fan Y, Deng C, Cheng R, Meng F, Zhong Z. Biomacromolecules. 2013;14:2814–2821. doi: 10.1021/bm400637s. [DOI] [PubMed] [Google Scholar]
- 24.Mosiewicz KA, Kolb L, van der Vlies AJ, Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, Lutolf MP. Nat Mater. 2013;12:1072–1078. doi: 10.1038/nmat3766. [DOI] [PubMed] [Google Scholar]
- 25.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Nat Mater. 2013;12:458–465. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kloxin AM, Tibbitt MW, Anseth KS. Nat Protoc. 2010;5:1867–1887. doi: 10.1038/nprot.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeForest CA, Polizzotti BD, Anseth KS. Nat Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKinnon DD, Kloxin AM, Anseth KS. Biomater Sci. 2013;1:460–469. doi: 10.1039/c3bm00166k. [DOI] [PubMed] [Google Scholar]
- 29.Brooks BR. Neurology. 1996;47:S71–S82. doi: 10.1212/wnl.47.4_suppl_2.71s. [DOI] [PubMed] [Google Scholar]
- 30.Jessell TM, Sürmeli G, Kelly JS. Neuron. 2011;72:419–424. doi: 10.1016/j.neuron.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 31.DeForest CA, Anseth KS. Nat Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wichterle H, Lieberam I, Porter J, Jessell TM. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 33.Wichterle H, Peljto M. Curr Protoc Stem Cell Biol. 2008 doi: 10.1002/9780470151808.sc01h01s5. [DOI] [PubMed] [Google Scholar]
- 34.Jewett J, Sletten E, Bertozzi CR. J Am Chem Soc. 2010;132:3688–3390. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeForest CA, Sims EA, Anseth KS. Chem Mater. 2010;22:4783–4790. doi: 10.1021/cm101391y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, Filion TM, Prifti F, Song J. Chem—Asian J. 2011;6:2730–2737. doi: 10.1002/asia.201100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griffin DR, Kasko AM. J Am Chem Soc. 2012;134:13103–13107. doi: 10.1021/ja305280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe W, Arakawa N, Matsunaga S, Higashi T. Opt Express. 2004;12:4203–4213. doi: 10.1364/opex.12.004203. [DOI] [PubMed] [Google Scholar]
- 40.Shen N, Datta D, Schaffer CB. MCB. 2005;2:17–25. [PubMed] [Google Scholar]
- 41.Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y, Ben-Yakar A. Nature. 2004;432:822–822. doi: 10.1038/432822a. [DOI] [PubMed] [Google Scholar]
- 42.Tibbitt MW, Kloxin AM, Dyamenahalli KU, Anseth KS. Soft Matter. 2010;6:5100–5108. doi: 10.1039/C0SM00174K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drobizhev M, Makarov NS, Tillo SE, Hughes TE, Rebane A. Nat Meth. 2011;8:393–399. doi: 10.1038/nmeth.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aujard I, Benbrahim C, Gouget M, Ruel O, Baudin J-B, Neveu P, Jullien L. Chem—Eur J. 2006;12:6865–6879. doi: 10.1002/chem.200501393. [DOI] [PubMed] [Google Scholar]
- 45.Uttara B, Singh A, Zamboni P, Mahajan R. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang TQ, Qu X, Liu J, Chen S. Biomed Microdevices. 2013;16:127–132. doi: 10.1007/s10544-013-9812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan W, Desai TA. Tissue Eng. 2003;9:255–267. doi: 10.1089/107632703764664729. [DOI] [PubMed] [Google Scholar]
- 48.Miles GB, Yohn DC, Wichterle H, Jessell TM, Rafuse VF, Brownstone RMJ. Neurosci. 2004;24:7848–7858. doi: 10.1523/JNEUROSCI.1972-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Umbach JA, Adams KL, Gundersen CB, Novitch BG. PLoS One. 2012;7:e36049. doi: 10.1371/journal.pone.0036049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yaffe D. Proc Nat Acad Sci USA. 1968;61:477–483. doi: 10.1073/pnas.61.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blau HM, Pavlath GK, Hardeman EC, Chiu CP. Science. 1985;230:758–766. doi: 10.1126/science.2414846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.