

Abstract
Objective:
To characterize the clinical features and MRI abnormalities of leucine-rich glioma-inactivated 1 (LGI1)-autoantibody (Ab) faciobrachial dystonic seizures (FBDS).
Methods:
Forty-eight patients with LGI1-Ab encephalopathy were retrospectively identified by searching our clinical and serologic database from January 1, 2002, to June 1, 2015. Of these, 26 met inclusion criteria for this case series: LGI1-Ab seropositivity and FBDS. In a separate analysis of all 48 patients initially identified, the MRIs of patients with (n = 26) and without (n = 22) FBDS were compared by 2 neuroradiologists blinded to the clinical details.
Results:
The median age of the 26 included patients was 62.5 years (range 37–78); 65% were men. FBDS involved arm (26), face (22), and leg (12). Ten were previously diagnosed as psychogenic. Ictal EEGs were normal in 20 of 23 assessed. Basal ganglia T1 and T2 signal abnormalities were detected in 11 patients (42%), with excellent agreement between neuroradiologists (κ scores of 0.86 and 0.93, respectively), and included T1 hyperintensity alone (2), T2 hyperintensity alone (1), or both (8). The T1 hyperintensities persisted longer than the T2 hyperintensities (median 11 weeks vs 1 week, p = 0.02). Improvement with immunotherapy (18/18) was more frequent than with antiepileptic medications (10/24). A separate analysis of all 48 patients initially identified with LGI1-Ab encephalopathy showed that basal ganglia MRI abnormalities were present in 11 of 26 with FBDS but not present in those without FBDS (0/22) (p < 0.001). In contrast, mesial temporal MRI abnormalities were less common among those with FBDS (42%) than those without (91%) (p < 0.001).
Conclusions:
Basal ganglia T1 hyperintensity is a clinically useful MRI biomarker of LGI1-Ab FBDS and suggests a basal ganglia localization.
Faciobrachial dystonic seizures (FBDS) are a recently recognized immunotherapy-responsive disorder first described in 2008.1 They are characterized by unilateral, short-lived dystonic posturing of the upper limb and face accompanied by autoantibodies (Abs) to leucine-rich glioma-inactivated 1 (LGI1), a component of the voltage-gated potassium channel complex (VGKC-complex).2,3 Although they have characteristics suggesting seizures, ictal scalp EEG is usually normal.2,3 The unusual presentation and normal EEGs have led to them frequently being mislabeled as functional or psychiatric in origin.2 MRI abnormalities accompanying FBDS are not well characterized but may assist in making the diagnosis and confirming organicity. We describe 26 patients with FBDS, 10 of whom had a novel finding of unilateral basal ganglia (BG) T1 hyperintensity.
METHODS
Through a search of our clinical and serologic database from January 1, 2002, to June 1, 2015, we retrospectively identified 89 patients with VGKC-complex-Ab (>0.02 nmol/L) encephalitis (including those with seizures, amnesia/limbic encephalitis, and dystonia/dyskinesia). Of these, 48 patients were seropositive for LGI1-Ab (median VGKC-complex titer 0.51 nmol/L, range 0.07–5.96), 6 were seropositive for contactin-associated protein-2 (CASPR2)-Ab (median VGKC-complex titer 0.21 nmol/L, range 0.11–0.36), and the remaining 35 were negative for both LGI1- and CASPR2-Ab (median VGKC-complex titer 0.28 nmol/L, range 0.05–3.5). Twenty-six met our inclusion criteria for this case series: LGI1-Ab seropositivity and FBDS (involving the arm, face, or both).2 Four patients with FBDS with VGKC-complex-Ab positivity were negative for both LGI1 and CASPR2 and thus excluded. Twenty-eight of the 48 LGI1-Ab-positive patients identified have been included in prior studies.4–8
Neuroimaging.
To identify which patients had BG MRI abnormalities, 2 neuroradiologists (A.L.K., R.E.W.) blinded to the clinical details independently reviewed all available MRIs of the 48 LGI1-Ab-positive patients. In cases of disagreement, a consensus was reached for the purpose of analysis. To determine whether BG MRI abnormalities were exclusive to FBDS, we compared findings in patients with (n = 26) and without FBDS (n = 22). All available 18F-fluorodeoxyglucose (FDG)-PET images were reviewed by a nuclear imaging specialist (V.L.) blinded to the clinical details.
VGKC-complex-Ab and LGI1-Ab evaluation.
Our current practice in the Mayo Clinic Neuroimmunology Laboratory is to screen for Abs specific for any component of the solubilized VGKC-complex using a radioimmunoassay (RIA) that precipitates the VGKC ligated with radioiodinated α-dendrotoxin, as described previously.5 This RIA is most sensitive and cost-effective as a screening test for VGKC-complex autoimmunity. Sera testing positive by RIA (>0.02 nmol/L) are then tested for LGI1-Abs by cell-based immunofluorescence assay (Euroimmun, Lübeck, Germany). This assay uses HEK293 cells transfected with plasmid encoding human LGI1.5,9,10 The dilution of serum is 1:10 and the result is reported as positive or negative based on visual comparison of transfected and nontransfected cells under a microscope. All assays are run using both positive and negative control sera; the specific LGI1-Ab titers are not measured.
Statistical methodology.
Agreement between the 2 neuroradiologists was measured using κ values (κ values <0.4 represent poor agreement, 0.4–0.75 represent fair to good agreement, and >0.75 represent excellent agreement). Comparisons were performed using Wilcoxon rank-sum test or Fisher exact test, as appropriate, using JMP 8.0 software (SAS Institute, Cary, NC).
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Mayo Clinic Institutional Review Board and all patients consented to the use of their medical records for research purposes. The patient in the video (see Neurology.org/nn) consented to publication of a video of his FBDS episode.
RESULTS
Clinical characteristics.
The clinical features of the 26 patients included are summarized in table 1. Fifteen patients reported FBDS during sleep and 10 described unexplained falls during the episodes. Fifteen patients (58%) presented with exclusively unilateral FBDS, and the remaining 11 patients (42%) presented with (or later developed) bilateral FBDS. These episodes occurred for a median of 5.5 months (range 1–19 months), and 5 patients continued to have FBDS at last follow-up. Twenty-one patients (81%) had memory difficulties (67% were preceded by FBDS) suggestive of coexisting limbic encephalitis; 1 patient had prominent behavioral difficulties in addition to memory troubles, mimicking frontotemporal dementia. The median Kokmen Short Test of Mental Status score (when available) was 30 of 38 (range 7–36; ≤29 is consistent with dementia).11 Ten patients (38%) were initially diagnosed with psychiatric or functional neurologic disorders and 5 (19%) were suspected of having Creutzfeldt-Jakob disease. Two patients had neoplasms detected (both prostate adenocarcinoma) during their evaluation and were suspected of having a paraneoplastic LGI1-Ab syndrome. Twelve patients (46%) had a personal history of one or more autoimmune disorders (hypothyroidism [6], rheumatoid arthritis [2], chronic inflammatory polyneuropathy [2], diabetes [2], vitiligo [2], psoriasis [1]). Nine patients had a family history of an autoimmune disorder in a first-degree relative (diabetes [5], hypothyroidism [2], Graves disease [1], pernicious anemia [1]).
Table 1.
Summary of the demographics and clinical and laboratory characteristics of LGI1-autoantibody faciobrachial dystonic seizures (n = 26)
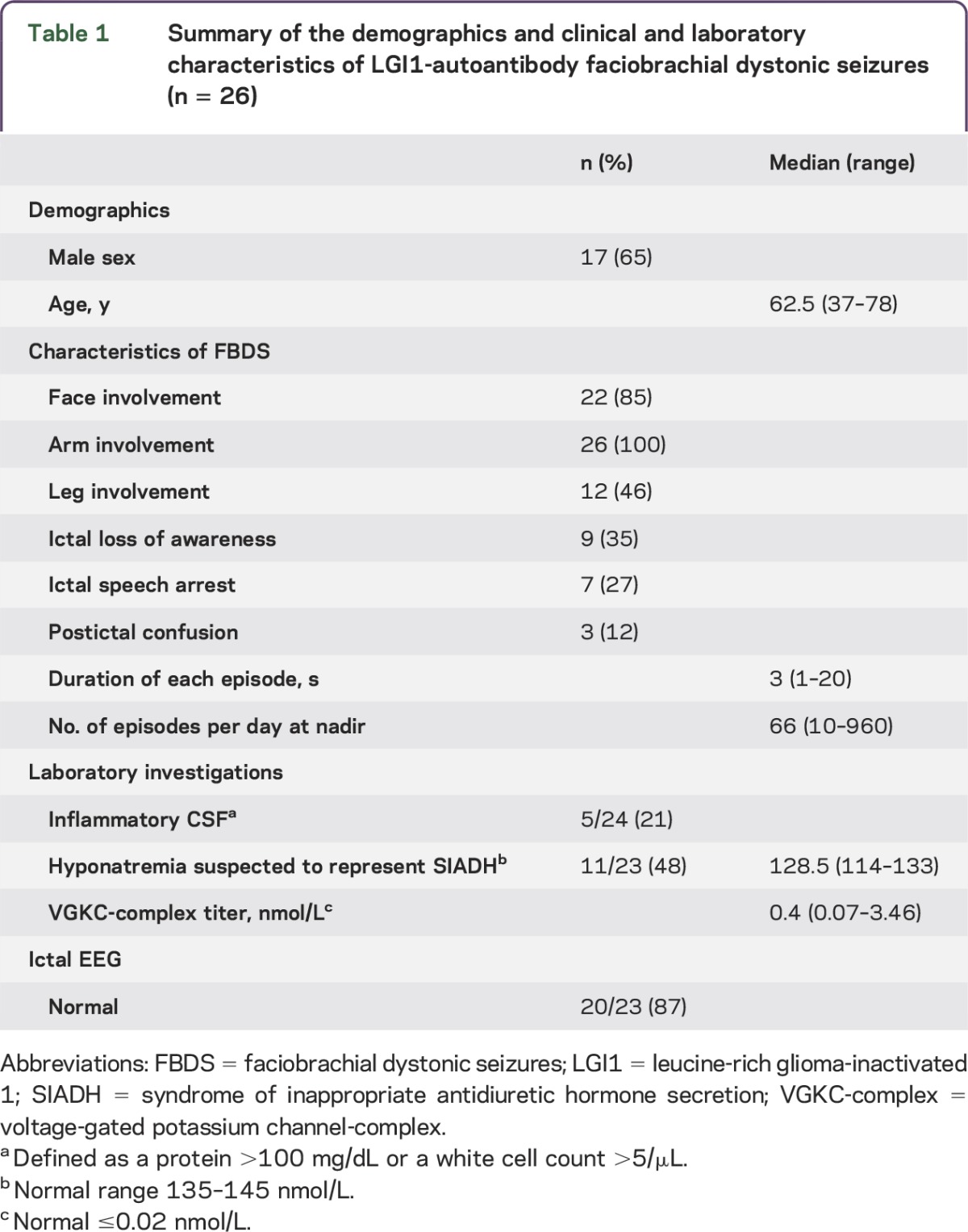
Laboratory investigations.
The details are summarized in tables 1 and 2. Eight patients were seropositive for one or more other neural Abs, including the following: glutamic acid decarboxylase 65 antibody (3 patients; 0.04, 0.06, and 2.11 nmol/L [normal ≤0.02 nmol/L]); ganglionic acetylcholine receptor Ab (2 patients; 0.04 and 0.45 nmol/L [normal ≤0.02 nmol/L]); muscle acetylcholine receptor binding Ab (2 patients; 0.06 and 0.10 nmol/L [normal ≤0.02 nmol/L]); N-type calcium channel Ab (1 patient; 0.05 nmol/L [normal ≤0.03 nmol/L]); and striated muscle Ab (1 patient; 1:30,720 [normal <1:60]).
Table 2.
Characteristics of 11 LGI1-autoantibody-positive patients with faciobrachial dystonic seizures and basal ganglia MRI abnormalities

Neuroimaging features.
The neuroimaging features and other clinical characteristics of the 11 patients with BG MRI abnormalities are summarized in table 2. Representative images are shown in figures 1 and 2. T1 hyperintensities either occurred concurrently with (62%) or were preceded by (38%) short-lived BG T2 hyperintensity during the episodes. The imaging abnormalities were generally contralateral to the FBDS (figure 2). No patients had gadolinium-enhancing lesions; 1 patient had bilateral T1 hyperintensities at the time of bilateral FBDS. T1 hyperintensities persisted for longer than T2-hyperintense lesions (median of 11 weeks [range 0–68] vs median of 1 week [range 0–5], p = 0.02). FBDS resolved prior to BG signal abnormalities in 3 patients, and BG T2 hyperintensity resolution and clinical resolution occurred concurrently in 1 patient. This could not be assessed in the other 7 patients because of variable MRI timing, ongoing FBDS at last follow-up, or ongoing BG signal abnormalities at last follow-up. Two patients developed caudate atrophy. In 5 of the 11 patients (45%) with BG signal change, T2 signal hyperintensities within the mesial temporal lobes coexisted. Brain FDG-PET showed focal hypermetabolism in the BG of 4 of 14 patients (29%) and focal BG hypometabolism in 2 of 14 (14%) (table 2). Other FDG-PET findings included diffuse hypometabolism (3), mesial temporal hypermetabolism (3), and focal bifrontal hypometabolism (2).
Figure 1. MRI head in faciobrachial dystonic seizures.

(A) Axial fluid-attenuated inversion recovery image from patient 3 in table 2 shows T2-weighted hyperintensity within the right caudate nucleus (A.a, arrow), and T1-weighted images prior to gadolinium administration reveal T1 hyperintensity in the caudate and upper putamen on axial images (A.b, arrow). Restricted diffusion is notable on diffusion-weighted images (A.c, arrow) and corresponding apparent diffusion coefficient images (A.d, arrow). (B) Axial T1-weighted images prior to gadolinium administration from patient 2 in table 2 reveal an initial T1 hyperintensity in the left putamen on axial (B.a, arrow) and sagittal images (B.b, arrow). (C) Coronal T1-weighted images prior to gadolinium administration from patient 5 in table 2 reveal T1 hyperintensity in the left globus pallidus and putamen (arrow).
Figure 2. Evolution of MRI abnormalities in faciobrachial dystonic seizures.

Axial fluid- attenuated inversion recovery (FLAIR)-weighted images from patient 4 in table 2 reveal left putamen and globus pallidus T2 hyperintensity (A.a, arrow) with T1 hyperintensity prior to gadolinium administration in the left putamen (A.b, arrow). The T2 hyperintensity resolved on follow-up FLAIR image (B.a), but T1 hyperintensity persisted (B.b, arrow); both resolved on further follow-up images (C.a, C.b). During a clinical relapse, a right-sided FLAIR T2 hyperintensity appeared (D.a, arrow) with concurrent T1 hyperintensity on pregadolinium images (D.b, arrow). The T2 hyperintensity subsequently resolved (E.a) prior to the T1 hyperintensity (E.b, arrow). FBDSz = faciobrachial dystonic seizures.
EEG findings.
Ictal EEG was normal in 20 of 23 patients (87%) (tables 1 and 2). Three patients had an ictal EEG correlate to the FBDS: 2 had left temporal rhythmic discharge (figure 3) and 1 had a right temporal rhythmic discharge. Interictal EEG showed coexisting epileptiform activity (spikes, sharp waves, subclinical seizures, or clinical seizures with a different semiology from FBDS) in 12 of 26 patients (46%).
Figure 3. EEG demonstrating ictal seizure discharges in a patient with faciobrachial dystonic seizures.

The EEG of a 56-year-old woman with faciobrachial dystonic seizures is shown. The EEG was unchanged at the onset of her dystonic seizures, manifested by bilateral upper extremity (bil UE) extension followed by left hand dystonia lasting 12 seconds. The EEG eventually showed rhythmic seizure activity over the left temporal region (arrows), which persisted beyond termination of the left hand dystonia.
Treatments, resolution, recurrence, and follow-up.
Acute immunotherapies used (alone or in combination) included IV methylprednisolone (16), oral prednisone (4), IV immunoglobulin (IVIg) (4), and plasma exchange (3). Antiepileptic medications used (median of 2 medications per person [range 1–5]) included levetiracetam (15), phenytoin (12), valproic acid (8), clonazepam (5), carbamazepine (4), oxcarbazepine (3), topiramate (3), lacosamide (3), gabapentin (3), clorazepate (2), midazolam (1), lamotrigine (1), and phenobarbital (1). Improvement (defined as decreased frequency of seizures reported by the patient or caregiver) with immunotherapy (18/18; complete resolution in 14, >50% reduction in 3, and <50% reduction in 1) was more frequent than with antiepileptic medications (10/24; complete resolution in 4 [3 from antiepileptics alone: levetiracetam and carbamazepine (1), clonazepam (1), and oxcarbazepine (1)], >50% reduction in 4, and <50% reduction in 2). In 2 patients without improvement from acute IV immunotherapy, resolution occurred with prolonged oral prednisone. In the 2 patients treated with IVIg alone, a sustained response was not seen, and both required subsequent corticosteroid therapy. Two received treatment (brachytherapy and hormonal treatment) for coexisting prostate cancer in addition to immunotherapy. Two patients resolved spontaneously while on no medication 6 and 15 months after onset. Eight of 9 patients (89%) had reductions in VGKC-complex titer after immunotherapy. The median duration from first symptom to last follow-up was 24.5 months (range 1–90). Seven patients (27%) had a recurrence (return of symptoms after initial resolution). Four of 5 patients with ongoing FBDS at last follow-up had never received immunotherapy; 1 had FBDS until death 10 months after onset.
A separate analysis of MRI abnormalities in all 48 patients initially identified with LGI1-Ab encephalopathy was conducted. Agreement between neuroradiologists was excellent for each signal abnormality: BG T1 hyperintensity (κ = 0.86), BG T2 hyperintensity (κ = 0.93), and mesial temporal T2 hyperintensity (κ = 0.82). BG T1 or T2 signal abnormalities were found in 11 of 26 patients (42%) with FBDS but were not found in any of the 22 patients without FBDS (p < 0.001). Mesial temporal lobe MRI abnormalities were less common in those with FBDS (11 of 26 [42%]) than those without FBDS (20 of 22 [91%]) (p < 0.01).
DISCUSSION
Because FBDS are frequently misdiagnosed as psychogenic or indeterminate, a brain MRI that reveals BG T1 or T2 hyperintensities helps alert the clinician to suspect an LGI1-Ab encephalitis.
The nomenclature for these dystonic spasms (FBDS) implies an epileptic focus, and evidence in favor of this presumption includes the characteristic semiology, the paroxysmal nature, and the abrupt onset and offset in all cases. These episodes may also resemble myoclonic or tonic seizures, which occur in LGI1 knockout mice.12,13 Other features, although infrequently present, may also suggest seizure, including the following: aura, interictal speech arrest or loss of awareness, postictal confusion, and response to antiepileptic drugs in some.2 Normal ictal scalp EEG recordings in the majority of patients with FBDS in this study may reflect a deep seizure origin or the fact that scalp EEG often cannot detect simple partial seizures.14 The ictal scalp EEG may show electrodecremental events that are typical of tonic motor seizures.12 The phenomenologic similarity with paroxysmal movement disorders suggests that LGI1-Ab testing should be considered in adults evaluated for paroxysmal dyskinesia/dystonia (without a family history).15 Similar to prior reports,2,3 in this study, FBDS occurred in older individuals (median age 62.5 years), had a male predominance, usually lasted just a few seconds, and typically occurred 50–100 times per day at nadir. Also in agreement with prior studies,2,3 an accompanying hyponatremia/syndrome of inappropriate antidiuretic hormone secretion was a useful diagnostic clue and occurred in approximately half the patients, but an inflammatory CSF was found in a minority.
Patients with VGKC-complex (including LGI1-Ab) encephalopathy frequently have MRI T2 hyperintensities in the mesial temporal lobe,2,7 which is a highly epileptogenic region. However, our data suggest that this may not be the substrate for FBDS, because mesial temporal T2 hyperintensities were less frequent among those with FBDS than those without FBDS. These abnormalities presumably reflect the common, coexisting limbic encephalitis but cast doubt on the temporal lobe as the origin of FBDS. In contrast, BG MRI abnormalities were exclusive to LGI1-Ab encephalopathy patients with FBDS and were typically contralateral to episodes (e.g., figure 2), suggesting a BG origin, which is consistent with the conclusions of prior publications on FBDS.2,3,16 This is further supported by the characteristic semiology and the few previously reported cases of T2 hyperintensities,2,3,17,18 FDG-PET hypermetabolism,2,3 and T1 hyperintensity in the BG (only 2 other cases to our knowledge)19,20; furthermore, LGI1 mutant mice develop dystonia.13 Our study demonstrated that BG T1 hyperintensity can be reliably detected by neuroradiologists. The higher frequency (38.5%) of BG T1 hyperintensity in FBDS than that reported in prior series2,3 may reflect the availability of multiple MRIs in each patient in our series, the subtle nature, and the fact that only limited pregadolinium T1-weighted sequences may be available (e.g., only sagittal) and may be reviewed in less detail when compared to other sequences. Reports of gadolinium enhancement in FBDS also exist,15,21 but this was not found in our study. Future studies should carefully evaluate for pregadolinium T1 hyperintensity, as this can be mistakenly labeled as gadolinium enhancement.
There are few causes of BG T1 hyperintensities and most result in bilateral abnormalities,22 potentially allowing unilateral BG T1 hyperintensity be a useful diagnostic biomarker of FBDS (compared to the shorter-lasting T2 hyperintensities, which have a broader differential diagnosis). However, this retrospective study resulted in variable MRI timing, making it difficult to determine the exact duration of BG T1 and T2 signal abnormalities. Other causes of BG T1 hyperintensity include manganese accumulation (liver failure, hyperalimentation), Creutzfeldt-Jakob disease, chorea from nonketotic hyperglycemia or lupus, HIV infection, multiple system atrophy, calcification (Fahr disease), hereditary disorders of metal deposition (Wilson disease), medications (cyclosporine), Cockayne syndrome, hypoxic/ischemic injury, intracranial hemorrhage, and multiple MRI scans with gadolinium (possibly due to gadolinium deposition in the BG).22,23 The cause of T1 hyperintensity in FBDS is uncertain. The absence of calcification on CT and T2 hyperintensity excluded calcification and melanin, respectively22; persistent T1 hyperintensity after FBDS resolution suggested that it was not from ongoing seizures. Delayed ischemic T1 hyperintensity (akin to cortical laminar necrosis) described with incomplete infarction evolves similarly: arising 7–10 days after injury and persisting for months to years.24 The restricted diffusion in some FBDS fits that hypothesis. The ischemia could be from perivascular inflammation or immune-mediated basal ganglia swelling and damage. The histologic underpinning of this T1 hyperintensity may be the paramagnetic free radical manganese superoxide dismutase in injured mitochondria, although iron deposition, swollen astrocytes (gemistocytes), lipid-laden macrophages, and protein denaturation are other potential etiologies (many of which have been described in VGKC-complex-Ab pathology studies).24,25
We excluded VGKC-complex-Ab-seropositive patients who were negative for both LGI1-Ab and CASPR2-Ab because the clinical significance in this situation is less well characterized. Finally, we noted that FBDS responded more frequently and more completely to immunotherapy than to antiepileptic drugs, similar to prior reports,2,3 suggesting that immunotherapy should be first-line treatment for FBDS, either alone or in combination with antiepileptic medications (resolution rarely occurred with antiepileptics alone). In this study, IV corticosteroid therapy (either alone or in combination with other immunotherapies) was the mainstay of immunotherapy, and in 2 patients treated with IVIg alone, IV steroids were eventually required for resolution. More-prolonged oral prednisone (for months) was helpful in rare patients not responding to acute IV immunotherapies. Recognition of the hallmark BG T1 hyperintensity may allow earlier diagnosis, more rapid initiation of immunotherapy, and potentially better outcomes.
Supplementary Material
GLOSSARY
- Ab
autoantibody
- BG
basal ganglia
- CASPR2
contactin-associated protein-2
- FBDS
faciobrachial dystonic seizures
- FDG
18F-fluorodeoxyglucose
- IVIg
IV immunoglobulin
- LGI1
leucine-rich glioma-inactivated 1
- RIA
radioimmunoassay
- VGKC-complex
voltage-gated potassium channel complex
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Eoin P. Flanagan: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data, statistical analysis, study supervision. Amy L. Kotsenas: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data. Jeffrey W. Britton: drafting/revising the manuscript, analysis or interpretation of data, analysis of EEGs and clinical manifestations. Andrew McKeon: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Robert E. Watson: analysis or interpretation of data, acquisition of data. Christopher J. Klein: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. Bradley F. Boeve: analysis or interpretation of data. Val Lowe: drafting/revising the manuscript, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data. J.E. Ahlskog: analysis or interpretation of data. Cheolsu Shin: drafting/revising the manuscript, acquisition of data, study supervision. Christopher J. Boes: drafting/revising the manuscript, analysis or interpretation of data. Brian A. Crum: drafting/revising the manuscript, acquisition of data. Ruple S. Laughlin: drafting/revising the manuscript, acquisition of data. Sean J. Pittock: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
E.P. Flanagan and A.L. Kotsenas report no disclosures. J.W. Britton received research support from GW Pharmaceuticals and Mr. and Mrs. David Hawk Charity. A. McKeon received research support from Guthy Jackson Charitable Foundation. R.E. Watson reports no disclosures. C.J. Klein is on the editorial board for Journal of the Peripheral Nervous System and received research support from National Institute of Neurological Disorders and Stroke. B.F. Boeve is on the scientific advisory board for Tau Consortium, has consulted for Isis Pharmaceuticals, and received research support form GE Healthcare, FORUM Pharmaceuticals, and NIH. V. Lowe is on the scientific advisory board for Bayer Schering and Piramal Imaging and received research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, NIA, NCI, MN Partnership for Biotechnology and Medical Genomics, Elsie and Marvin Dekelboum Family Foundation, and Leukemia & Lymphoma Society. J.E. Ahlskog received the American Parkinson's Disease Association Fred Springer Award, received speaker honoraria for CME activities, is on the editorial board for Parkinsonism & Related Disorders and Clinical Neuropharmacology, and receives royalties from Oxford University Press and Humana Press. C. Shin reports no disclosures. C.J. Boes is a contributing editor for Headache Currents and was the book review editor for Neurology. B.A. Crum and R.S. Laughlin report no disclosures. S.J. Pittock has a financial interest in patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker; has consulted for Alexion Pharmaceuticals, Medimmune, and Chugai Pharma; and received research support from Alexion Pharmaceuticals, NIH, and Guthy Jackson Charitable Foundation. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Irani SR, Buckley C, Vincent A, et al. Immunotherapy-responsive seizure-like episodes with potassium channel antibodies. Neurology 2008;71:1647–1648. [DOI] [PubMed] [Google Scholar]
- 2.Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900. [DOI] [PubMed] [Google Scholar]
- 3.Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013;136:3151–3162. [DOI] [PubMed] [Google Scholar]
- 4.Quek AM, Britton JW, McKeon A, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 2012;69:582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein CJ, Lennon VA, Aston PA, et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol 2013;70:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008;70:1883–1890. [DOI] [PubMed] [Google Scholar]
- 7.Kotsenas AL, Watson RE, Pittock SJ, et al. MRI findings in autoimmune voltage-gated potassium channel complex encephalitis with seizures: one potential etiology for mesial temporal sclerosis. AJNR Am J Neuroradiol 2014;35:84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toledano M, Britton JW, McKeon A, et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology 2014;82:1578–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kokmen E, Naessens JM, Offord KP. A short test of mental status: description and preliminary results. Mayo Clin Proc 1987;62:281–288. [DOI] [PubMed] [Google Scholar]
- 12.Andrade DM, Tai P, Dalmau J, Wennberg R. Tonic seizures: a diagnostic clue of anti-LGI1 encephalitis? Neurology 2011;76:1355–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chabrol E, Navarro V, Provenzano G, et al. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain 2010;133:2749–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devinsky O, Sato S, Kufta CV, et al. Electroencephalographic studies of simple partial seizures with subdural electrode recordings. Neurology 1989;39:527–533. [DOI] [PubMed] [Google Scholar]
- 15.Aradillas E, Schwartzman RJ. Kinesigenic dyskinesia in a case of voltage-gated potassium channel-complex protein antibody encephalitis. Arch Neurol 2011;68:529–532. [DOI] [PubMed] [Google Scholar]
- 16.Striano P. Faciobrachial dystonic attacks: seizures or movement disorder? Ann Neurol 2011;70:179–180; author reply 180. [DOI] [PubMed] [Google Scholar]
- 17.Duncan M, Cholfin J, Restrepo L. Clinical reasoning: a 72-year-old man with rapid cognitive decline and unilateral muscle jerks. Neurology 2014;82:e194–e197. [DOI] [PubMed] [Google Scholar]
- 18.Yoo JY, Hirsch LJ. Limbic encephalitis associated with anti-voltage-gated potassium channel complex antibodies mimicking Creutzfeldt-Jakob disease. JAMA Neurol 2014;71:79–82. [DOI] [PubMed] [Google Scholar]
- 19.Boesebeck F, Schwarz O, Dohmen B, et al. Faciobrachial dystonic seizures arise from cortico-subcortical abnormal brain areas. J Neurol 2013;260:1684–1686. [DOI] [PubMed] [Google Scholar]
- 20.Jaychandran R, Patel MJ, Netravathi M, Saini J, Satishchandra P, Pal PK. Unusual clinical and imaging characteristics in a patient with voltage-gated potassium channel antibody-associated encephalitis. Neurol India 2012;60:533–534. [DOI] [PubMed] [Google Scholar]
- 21.Plantone D, Renna R, Grossi D, Plantone F, Iorio R. Teaching NeuroImages: Basal ganglia involvement in facio-brachial dystonic seizures associated with LGI1 antibodies. Neurology 2013;80:e183–e184. [DOI] [PubMed] [Google Scholar]
- 22.Ginat DT, Meyers SP. Intracranial lesions with high signal intensity on T1-weighted MR images: differential diagnosis. Radiographics 2012;32:499–516. [DOI] [PubMed] [Google Scholar]
- 23.Kanda T, Ishii K, Kawaguchi H, Kitajima K, Takenaka D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology 2014;270:834–841. [DOI] [PubMed] [Google Scholar]
- 24.Fujioka M, Taoka T, Hiramatsu KI, Sakaguchi S, Sakaki T. Delayed ischemic hyperintensity on T1-weighted MRI in the caudoputamen and cerebral cortex of humans after spectacular shrinking deficit. Stroke 1999;30:1038–1042. [DOI] [PubMed] [Google Scholar]
- 25.Bien CG, Vincent A, Barnett MH, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012;135:1622–1638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.