

Abstract
Although the early antidepressant trials which included severely ill and hospitalized patients showed substantial drug-placebo differences, these robust differences have not held up in the trials of the past couple of decades, whether sponsored by pharmaceutical companies or non-profit agencies. This narrowing of the drug-placebo difference has been attributed to a number of changes in the conduct of clinical trials. First, the advent of DSM-III and the broadening of the definition of major depression have led to the inclusion of mildly to moderately ill patients into antidepressant trials. These patients may experience a smaller magnitude of antidepressant-placebo differences. Second, drug development regulators, such as the U.S. Food and Drug Administration and the European Medicines Agency, have had a significant, albeit underappreciated, role in determining how modern antidepressant clinical trials are designed and conducted. Their concerns about possible false positive results have led to trial designs that are poor, difficult to conduct, and complicated to analyze. Attempts at better design and patient selection for antidepressant trials have not yielded the expected results. As of now, antidepressant clinical trials have an effect size of 0.30, which, although similar to the effects of treatments for many other chronic illnesses, such as hypertension, asthma and diabetes, is less than impressive.
Keywords: Major depression, antidepressants, placebo, clinical trials, expectation bias, drug development regulators
Twenty years ago we believed that antidepressants worked in 70% of depressed patients and placebo in 30% of them, as stated in the U.S. Department of Health and Human Services report on treatment of major depression (1). This notion, however, has undergone a major revision in the past two decades.
Kuhn’s original report describing the therapeutic effects of imipramine was based on clinical vignettes (2). As is the case with most disorders, it was evident even in this first report that not all depressed patients responded to the new drug. Kuhn pointed out that patients with endogenous or vital depression were most likely to respond.
A considerable body of research subsequently explored which depressed patients responded to select antidepressants such as imipramine and phenelzine compared to electroconvulsive therapy (ECT) (e.g.,(3)). As part of this development, the need arose to quantify the depressive syndrome, and pioneers like M. Hamilton designed depression rating scales (4).
In the U.S., Klerman and Cole produced a detailed review of trials evaluating the effectiveness of imipramine (5). Consistent with Kuhn’s findings, they reported that hospitalized depressed patients with a melancholic pattern of symptoms were most likely to respond to the drug. Much of the wisdom about the magnitude of antidepressant and placebo response was based on these early clinical trials of tricyclic antidepressants, and these data carried well into the early 1990s (6).
However, in the 1970s and 1980s, several important changes were occurring in psychiatry. Most significant was the advent of DSM-III. Using an atheoretical approach, this diagnostic system minimized differences between subtypes of depression and conceptualized a broad syndrome called major depressive disorder, characterized by a single or recurrent bouts named major depressive episodes. Such a “uniform” diagnosis now included millions of patients and became an attractive target for the pharmaceutical industry. Thus, the American Psychiatric Association, sponsor of the DSM-III, unintentionally expanded the market for antidepressants.
Not surprisingly, a plethora of new drugs were developed, and almost all of the trials for the new compounds, such as selective serotonin reuptake inhibitors (SSRIs) and serotonin and norepinephrine reuptake inhibitors (SNRIs), included depressed patients meeting the DSM-III generic criteria for “major depressive episode”. Some attempts were made to recruit in antidepressant trials the more classical “endogenous” or “melancholic” subtypes of patients. However, these attempts were often half-hearted and criteria were not always strictly followed.
So, when we accessed the public domain data from the U.S. Food and Drug Administration (FDA) archives for the antidepressants approved between 1985 and 1997 (7), it quickly became apparent that many of the assumptions about the relative potency of antidepressants compared to placebo were not based on data from the contemporary trials but from an earlier era. Specifically, it became evident that the magnitude of symptom reduction was about 40% with antidepressants and about 30% with placebo.
The U.S. FDA public domain reports used symptom reduction as a measure of improvement and did not include therapeutic response rates. Even with this caveat, however, it was evident that the conventional wisdom of 70% response with antidepressants was at best an overestimate.
Not surprisingly, Walsh et al (8) also noted that the magnitude of symptom reduction with placebo had been increasing in the past three decades, based on an analysis of published antidepressant clinical reports. This publication prompted considerable attention and speculation from a number of investigators.
The effectiveness of modern antidepressants was not only questioned by placebo-controlled clinical trials, but also by trials based on a clinical practice model that did not include placebo. An experiment about antidepressant effectiveness started by J. Rush in Texas became a large scale national effort, supported by the U.S. National Institute of Mental Health, known as the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) project (9). This project showed that antidepressants such as citalopram led to a therapeutic response in only about 4 out of 10 depressed outpatients.
These challenges to the assumptions about the effectiveness of antidepressants brought about close scrutiny of the clinical trial data provided by the pharmaceutical companies, since the development, manufacturing and marketing of antidepressants is obviously a commercial venture. Specifically, criticism was raised by both academics and the general public as to the integrity of antidepressant clinical data generated by the industry (e.g., 10,11).
As a reaction, JAMA Network editors refused to accept data analyses completed by pharmaceutical companies (12). Instead, they insisted that they would only consider industry papers for publication if the original clinical trial data were independently reviewed by academic, non-industry statisticians.
THE IMPACT OF EXPECTATION BIAS
Given such an acrimonious situation with potential conflicts, we compared depression clinical trial data from non-pharmaceutical industry sources to antidepressant clinical data from the FDA Freedom of Information Act (FOIA) sources. In this analysis, we evaluated the magnitude of symptom reduction with all of the acknowledged depression treatments as well as their active or passive controls, including placebo (13).
This rather complex set of data, illustrated in Figure 1, contains several significant findings. On the left side of the figure (striped bars, black bars, grey bar) are data from non-pharmaceutical company sources, and on the right side (checkered bars) are data based on FDA reports. The magnitude of symptom reduction with placebo pill is higher in the non-pharmaceutical industry depression trials (grey bar) compared to pivotal registration trials (checkered bar).
Figure 1.
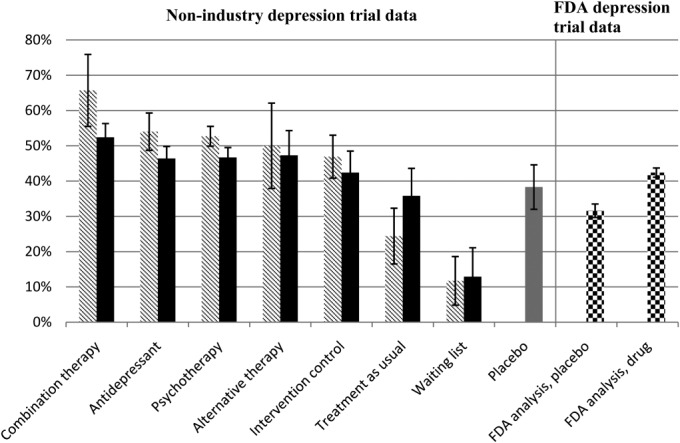
Mean percentage symptom reduction in unblinded and blinded treatment arms from published depression trials compared to data from pivotal registration depression trials as reported by the U.S. Food and Drug Administration (FDA) (adapted from 13). Striped bars represent unblinded trial arms; black bars represent blinded trial arms; the grey bar represents placebo control arms from published non-registration trials; checkered bars represent data from pivotal registration trials. The mean percentage symptom reduction was weighted by the number of assigned patients. Error bars represent 95% confidence intervals
Of even more interest is the pattern of response among the non-pharmaceutical industry double- or triple-blinded depression trials. The striped bars indicate the magnitude of depressive symptom reduction in trials where the investigators and their staff were aware of the design and expectations of the study. The black bars indicate the magnitude of symptom reduction when the investigators and raters were “blinded” to the design and execution of the study.
Clearly, investigator and rater bias influences the magnitude of symptom reduction with all treatments, whether they are approved treatments, active controls, passive controls, sham treatments, treatment as usual, waiting list, or placebo. For example, the magnitude of symptom reduction where the design of the trials was known to the investigators and raters (striped bars) followed the pattern of accepted expectations. The combined pharmacotherapy and psychotherapy had the best outcome, followed by antidepressants alone, known forms of psychotherapy (e.g., cognitive behavioral therapy), alternative therapies such as acupuncture or exercise, intervention controls (e.g., sham acupuncture, control psychotherapies such as educational sessions), and placebo. Not unexpectedly, “treatment as usual” fared worse than placebo, and waiting list had the smallest improvement.
On the other hand, the pattern was quite different if the investigators and their staff were blinded to the design and execution of the trials. Under these circumstances, the symptom reduction with each treatment was of smaller magnitude and the differences among the various treatments and controls were also smaller. The depressed patients assigned to all the treatments (active or control) – antidepressants, psychotherapy, acupuncture, exercise, sham acupuncture, sham psychotherapy and “treatment as usual” – experienced a symptom reduction that was comparable to that observed with placebo. In other words, when the level of blinding was high and it was difficult for the investigators, their staff and depressed patients to guess treatment assignment, the differences between these treatments, controls and placebo became quite small.
For two of the treatment paradigms, it is difficult to blind both clinicians and patients completely. One of these paradigms is combined pharmacotherapy and psychotherapy and the other is the waiting list. Not surprisingly, the magnitude of symptom reduction compared to placebo was significantly different using these two paradigms. The combined pharmacotherapy and psychotherapy showed a superior treatment response compared to placebo, and the waiting list an inferior treatment response than placebo. Simply put, clinicians and depressed patients continued to fulfill expectations of each treatment based on prior assumptions.
The effect of expectation bias is well illustrated by other experimental data. Sinyor et al (14) reported that, if there was no placebo control in an antidepressant trial comparing two antidepressants, the magnitude of symptom reduction was 65.7%. If the trial included two antidepressant treatments and one placebo arm (33% placebo exposure risk), the magnitude of symptom reduction with the antidepressants was 57.7%, while that with placebo was 44.6%. If the antidepressant trial included one antidepressant arm and one placebo arm (50% placebo exposure risk), the magnitude of symptom reduction with antidepressant was 51.7% and that with placebo was 34.3%.
In short, the apparent therapeutic effect of antidepressants is related to the risk of exposure to placebo, when this is known to clinicians and depressed patients from the consent form. If you lower the risk of exposure to placebo, then the apparent therapeutic effect with the antidepressants and placebo is greater.
These data from antidepressant clinical trials are applicable to clinical practice. First and foremost, it is critical to note that patients with mild to moderate depression are prone to non-specific therapeutic effects. The comments made by Brown (15) regarding the experience of patients assigned to placebo are pertinent. He states: “The capsule they receive is pharmacologically inert, but hardly inert with respect to its symbolic value and its power as a conditioned stimulus. In addition, placebo-treated patients receive all the components of the treatment situation common to any treatment, i.e., a thorough evaluation; an explanation for distress; an expert healer: a plausible treatment; a healer’s commitment, enthusiasm, and positive regard; an opportunity to verbalize their distress”.
Indeed, Frank has argued that these elements of the treatment situation are the active ingredients of all the psychotherapies (16). Since antidepressant clinical trials involve extensive evaluations, long visits, many experts and “new and exotic treatments”, it is not surprising that, under such conditions, the differences between active treatments and inactive treatments including sham acupuncture and placebo are, at best, small.
Although considerable attention has been paid to the magnitude of placebo response in depression and the small antidepressant-placebo differences, this phenomenon is not unique to depressive disorders. Illnesses that are chronic, have a fluctuating course and are associated with subjective distress are prone to placebo response. The following are some disorders that show the same pattern as depression.
Among patients with irritable bowel syndrome, treatment response occurs in 56% of cases, whereas the response rate to placebo is 46% (17). Thirty-six percent of patients with ulcerative colitis experience a therapeutic response with 5-aminosalicylic acid, whereas the response rate among those assigned to placebo is 20% (18).
A therapeutic response to one of six different anti-hypertensive agents was observed in 58% of patients with hypertension, while the response rate with placebo was 30% (19). The magnitude of change in one-second forced expiratory volume was 7% with bronchodilators compared to 4% with placebo (20).
In patients with osteoarthritis, the frequency of therapeutic response measured after arthroscopic lavage and debridement is lower than with sham procedures (21). Parkinson’s disease patients are also prone to placebo response: the reduction of symptoms with selegiline is 12%, while that with placebo is 10% (22).
Lastly, non-pharmacological somatic treatments for depression such as ECT and vagal nerve stimulation (VNS), under controlled clinical trial conditions, also show the same pattern. For example, sham ECT can result in 30% of severely depressed patients experiencing a therapeutic effect (23). Similarly, the implant of an “inactive” VNS pacemaker results in a 10% treatment response, while the response rate to “active” VNS is 15% among patients with chronic and treatment resistant depression (24).
These data clearly suggest that a high magnitude of placebo response is not unique to depressed patients, but inherent in an experimental paradigm. Thirty years ago, Quitkin et al (25) noted that the placebo response has an early onset and a fluctuating course, and it was assumed that depressed patients who respond to placebo relapse quickly back into depression. However, there is now evidence that, once patients respond to placebo, they remain well for a considerable period of time.
In a select sample of nine antidepressant trials, depressed patients who responded to either the investigational antidepressant or placebo during the double-blind trial continued on the same treatment assignment for six months or longer (26). Seventy-nine percent (333/420) of the depressed patients assigned to placebo did not relapse, compared to 93% (1074/1154) of the depressed patients assigned to antidepressants. In other words, four out of five depressed patients who improved with placebo remained well without relapse for six months or longer.
Mayberg et al (27) noted that clinical improvement with either fluoxetine or placebo was associated with cerebral glucose metabolism increases in depressed patients. Such a potential biological basis for placebo response is further supported by similar studies in pain and Parkinson’s disease (28).
In summary, depressed patients are prone to non-specific treatment effects, in particular when receiving placebo. Expectations by both patients and clinicians play a significant role in the magnitude of treatment effects in depression clinical trials. Once set, placebo response tends to persist and there are sufficient data to suggest that this is associated to changes in brain glucose metabolism.
THE IMPACT OF REGULATORY DECISIONS
The decisions of the FDA have strongly influenced what has happened to the design and execution of antidepressant trials in the past three decades. It is important to note that some of these decisions were taken by the regulators based on their assessment of prevailing wisdom and knowledge. Their ultimate intent was to reassure themselves and the public that pharmaceutical companies must demonstrate that their antidepressant is consistently superior to placebo before the drug is approved for marketing. This is part of the public health mandate being enforced by the FDA.
Although many of these regulatory decisions have a major impact on the design, execution and interpretation of antidepressant clinical trials, this fact is not well understood. As an illustration, although the concept of therapeutic response is easy to grasp, the FDA staff has never accepted this as a valid method. Actually, the counting of the number of depressed patients who responded versus those who did not was abandoned by the FDA after the approval of the antidepressant amitriptyline (29).
The rationale is as follows. A single measure may focus on factors that are not related to the specific disorder. As an example, opiates may produce a sense of well-being and “be therapeutic” globally for patients with malignancies, but they have little or no effect on the disease itself. Thus, the documentation of the impact of a drug on a disorder such as depression, as defined by the prevailing wisdom (in this instance, that of the DSM-III), requires a syndromal improvement, rather than a global feeling of well-being.
Hence, the FDA has considered rating scales such as the Hamilton Depression Rating Scale (HAM-D) (4) or the Montgomery-Asberg Depression Rating Scale (MADRS) (30) as surrogate markers to indicate a syndromal improvement for clinical depression. Interestingly, the FDA has accepted that the total score on these scales (that leads to a single number) is a valid method to assess improvement.
Not surprisingly, the variability produced in an antidepressant clinical trial is inherently influenced by this key decision. Specifically, the Clinical Global Impression (CGI) score can only be between 1 and 7, a rather narrow range, while the maximum total score for the HAM-D can be as high as 54 and as low as 0, and the maximum total score for the MADRS can be as high as 60 and as low as 0. This potential scatter has been seen by the FDA staff as an advantage in reducing the odds of a false positive result. However, not surprisingly, the use of CGI almost always leads to a better antidepressant-placebo separation.
Besides using that conservative outcome method, FDA also adopts very stringent criteria for data analyses. The FDA staff has considered the last observation carried forward (LOCF) method of analysis as the optimal one. In this model, if a depressed patient quits participating in a trial, the last known total HAM-D or MADRS score is replicated for the rest of the measurement points. Since the onset of response to placebo is early (31) and response to antidepressants occurs later, this acts to minimize antidepressant-placebo differences.
The FDA has recently accepted the concept of mixed-effect model repeated measure (MMRM) analysis, which consists of substituting missing data with a computational statistical model based on the overall pattern of the outcome measures. Although this method may be better than the LOCF (not proven yet), it is still mired in statistical concepts and not easily translated for interpretation, and certainly is not designed to favor outcome with antidepressants.
To complicate matters further, the European Medicines Agency (EMA) uses alternate models in evaluating new antidepressants. For example, it requires a relapse prevention model, in which depressed patients are treated with the new antidepressant and only those who respond to it are randomized into an experimental paradigm. In a double-blind manner, a segment of the responders continue to be assigned to the new antidepressant and another segment to placebo. Depressed patients are followed for approximately six months and the numbers of patients relapsing into another depressive episode testify to the effectiveness of the new antidepressant compared to placebo. As a rule, the differences between the two groups are larger than in the acute, parallel design models. For example, Geddes et al (32) showed that the relapse rate using this model was 41% for depressed patients assigned to placebo compared to 18% for those assigned to antidepressants.
The FDA does not accept such models to approve a new antidepressant and thus pharmaceutical companies are forced to come up with multiple models that are not complementary and leave both the clinician and the researcher confused, making it difficult to transfer common sense ideas into clinical practice. Such a conundrum can be used in a masterly way by marketers or be cynically dismissed as a marketing ploy.
This major hobbling of antidepressant clinical trials and the fact that the results of these trials need to be interpreted with caution is neither appreciated nor heeded by researchers or clinicians (10), nor by the media, which need sensational stories (11).
As these data about the weaknesses of the double-blind placebo-controlled antidepressant trials were gathered, several attempts have been made to address this situation. As noticed in a recent review (33), the best way to show antidepressant-placebo differences is to reduce the number of investigative sites, say to ten to twelve. This fact is currently ignored, as most multi-center pharmaceutical industry antidepressant trials include an average of 60 sites, some studies going to 120 sites worldwide.
Another major factor, as we emphasized, is the placebo risk exposure. Simply put, a two treatment option (with a placebo risk of 50%) has the best chance of keeping placebo response to a minimum. However, most contemporary antidepressant clinical trials have a minimum of three treatment arms, with a considerable number having four or more. This approach is significantly influenced by regulatory agencies such as the FDA. Specifically, the FDA requires that trials attempt to show a dose-response relationship for new antidepressants. In other words, it requests the use of doses of the new antidepressant that may not be effective, so that the minimum effective dose can be identified. For example, a dose of 10 mg of fluoxetine has to be consistently shown not to be superior to placebo, so that FDA staff can consider the next higher dose being the possibly lowest effective one. Thus, several studies are conducted in futility that simply make the results of antidepressant trials look worse than they are. In this context, it is important to note that no clear dose-response relationship has been established to date for most of new antidepressants.
Another regulatory burden, although not universally required by the FDA, is the use of an active control – i.e., an approved antidepressant such as fluoxetine – to show what is technically termed “assay sensitivity”. Such a paradigm is not only likely to increase the magnitude of placebo response, as the placebo exposure risk goes down, but also leads to many trials showing that the active comparator is not superior to placebo, adding more confusion.
The original concept (5) that more severely depressed patients respond better to antidepressants, whereas less severely depressed patients tend to respond to placebo, has held true in recent antidepressant trials (34,35). However, the implementation of this principle has not yielded any better results. Attempts at including patients who have a higher score on rating scales such as the HAM-D prospectively and prior to randomization has simply led to a greater magnitude of placebo response, although the factors behind such a phenomenon remain elusive (36).
Indeed, among the seven antidepressant trials where the severity of depression at baseline using HAM-D-17 was set at a score of 14 or higher, the magnitude of symptom reduction with placebo was 28.2%, while among the ten antidepressant trials where the threshold was set at 20, the magnitude of symptom reduction with placebo was 35.7%. Among the twenty antidepressant trials where the requested severity of depression at baseline using HAM-D-21 was 18 or higher, the magnitude of symptom reduction with placebo was 27.1%, while among the fourteen antidepressant trials where the threshold was set at 20, the magnitude of symptom reduction with placebo was 34.2%. These data raise the concern that forcing a higher pre-randomization severity may simply not work.
In this context, we have observed that using the longer version of HAM-D (21 items) results in a 60% increase in the antidepressant-placebo difference. Such a pattern has persisted over the past twenty-five years. It is possible that this version of HAM-D captures improvement in a larger group of depressive symptoms. Indeed, HAM-D-21 symptoms include diurnal variation in mood, paranoia and sense of hopelessness, reflecting additional dimensions of depression that may be more sensitive to antidepressant effects.
An alternative explanation of the high rate of placebo response when more severe patients are included may be that the investigative site staff rate patients as being more depressed than they actually are for commercial gain (38). However, attempts at having the patients evaluated by clinicians who do not stand to gain commercially by inflating the scores of rating scales, via videos or audiotapes, have not yielded the expected decrease in the magnitude of placebo response. Actually, they have produced an increase in that response and lower antidepressant-placebo differences (39,40).
In summary, given the constraints enforced by the regulators and the inability to control for multiple factors that may influence antidepressant clinical trial outcomes, it is more realistic to set up low expectations. In this context, a recent report by Gibertini et al (41) provides a useful model. These investigators analyzed the data from 81 monoaminergic antidepressant trials conducted in the past three decades, submitted to the FDA for the approval of fifteen antidepressants. They found an effect size of 0.30, which is considered modest.
This finding translates into the design of a prospective antidepressant trial as follows. This trial should compare a known effective dose of the test antidepressant to placebo (50% placebo risk), a two treatment option. Each treatment arm should consist of a minimum of 120 depressed patients and should be implemented at a maximum of twelve investigative sites. A specific successful example of the application of these principles has been the relatively quick and easy approval of vilazodone, based on two out of two positive trials (42).
In this context, it is important to note that clinical trials of medications for other common disorders, such as hypertension (43), asthma (44) and diabetes (45), have produced similar effect sizes, although attracting much less attention and criticism.
CONCLUSIONS
Patients with a major depressive episode as defined by DSM-III, DSM-IV and DSM-5 are significantly prone to non-specific treatment effects. This applies to both industry and non-industry clinical trials. It is important to note that the high magnitude of response to placebo is not unique to depression, but common to other chronic illness associated with subjective distress. Increasing the blinding of clinicians conducting the antidepressant clinical trial may not result in a decrease in the magnitude of placebo response nor an increase in the magnitude of antidepressant response. In fact, it is likely to do just the opposite.
Drug development regulators such as the FDA and the EMA have a significant, albeit underappreciated, role in how modern antidepressant clinical trials are designed and conducted. Because of their concern about possible false positive results, these regulators require trials that may not have the best design and conduct. Interpretation of data from such trials is difficult and confusing.
Although there are known factors that may influence the outcome of antidepressant trials, taking these factors into account is not easy and is not routinely done. Attempts by researchers to select patients independently from site clinicians by video- or audiotaping have not yielded promising results.
The effect size of current antidepressant trials that include patients with major depressive episode is approximately 0.30 (modest), and this fact needs to be heeded for future antidepressant trials.
References
- 1.U.S. Department of Health and Human Services. Clinical Practice Guideline No. 5. Washington: U.S. Government Printing Office; 1993. Depression in primary care: treatment of major depression. Vol. 2. [Google Scholar]
- 2.Kuhn R. The treatment of depressive states with G 22355 (imipramine hydrochloride) Am J Psychiatry. 1958;115:459–64. doi: 10.1176/ajp.115.5.459. [DOI] [PubMed] [Google Scholar]
- 3.Clinical Psychiatry Committee. Clinical trial of the treatment of depressive illness: report to the Medical Research Council. BMJ. 1965;1:881–6. doi: 10.1136/bmj.1.5439.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klerman GL, Cole JO. Clinical pharmacology of imipramine and related antidepressant compounds. Pharmacol Rev. 1965;17:101–41. [PubMed] [Google Scholar]
- 6.Brown WA, Khan A. Which depressed patients should receive antidepressants? CNS Drugs. 1994;1:341–7. [Google Scholar]
- 7.Khan A, Warner H, Brown WA. Symptom reduction and suicide risk in patients treated with placebo in antidepressant clinical trials: an analysis of the FDA database. Arch Gen Psychiatry. 2000;57:311–7. doi: 10.1001/archpsyc.57.4.311. [DOI] [PubMed] [Google Scholar]
- 8.Walsh BT, Seidman SN, Sysko R, et al. Placebo response in studies of depression: variable, substantial, and growing. JAMA. 2002;287:1840–7. doi: 10.1001/jama.287.14.1840. [DOI] [PubMed] [Google Scholar]
- 9.Rush AJ, Fava M, Wisniewski SR. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials. 2004;25:119–42. doi: 10.1016/s0197-2456(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 10.Blier P. Do antidepressants really work? J Psychiatry Neurosci. 2008;33:89–90. [PMC free article] [PubMed] [Google Scholar]
- 11.Begley S. Why antidepressants are no better than placebos. Newsweek. 2010 January 29, [Google Scholar]
- 12.DeAngelis CD, Fontanarosa PB. Impugning the integrity of medical science: the adverse effects of industry influence. JAMA. 2008;299:1833–5. doi: 10.1001/jama.299.15.1833. [DOI] [PubMed] [Google Scholar]
- 13.Khan A, Faucett J, Lichtenberg P, et al. A systematic review of comparative efficacy of treatments and controls for depression. PLoS One. 2012;7:e41778. doi: 10.1371/journal.pone.0041778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinyor M, Levitt AJ, Cheung AH, et al. Does inclusion of a placebo arm influence active antidepressant treatment in randomized controlled trials? Results from pooled and meta-analyses. J Clin Psychiatry. 2010;71:270–9. doi: 10.4088/JCP.08r04516blu. [DOI] [PubMed] [Google Scholar]
- 15.Brown WA. The placebo effect in clinical practice. Oxford: Oxford University Press; 2013. [Google Scholar]
- 16.Frank JD, Frank JB. Persuasion and healing: a comparative study of psychotherapy. Baltimore: Johns Hopkins University Press; 1991. 3rd ed. [Google Scholar]
- 17.Patel SM, Statson WB, Legedza A, et al. The placebo effect in irritable bowel syndrome trials: a meta-analysis. Neurogastroenterol Motil. 2005;17:332–40. doi: 10.1111/j.1365-2982.2005.00650.x. [DOI] [PubMed] [Google Scholar]
- 18.Sutherland LR, May GR, Shaffer EA. Sulfasalazine revisited: a meta-analysis of 5-aminosalicylic acid in the treatment of ulcerative colitis. Ann Intern Med. 1993;118:540–9. doi: 10.7326/0003-4819-118-7-199304010-00009. [DOI] [PubMed] [Google Scholar]
- 19.Preston RA, Materson BJ, Reda DJ, et al. Placebo-associated blood pressure response and adverse effects in the treatment of hypertension. Arch Int Med. 2000;160:1449–54. doi: 10.1001/archinte.160.10.1449. [DOI] [PubMed] [Google Scholar]
- 20.Joyce DP. The placebo effect in asthma drug therapy trials: a meta-analysis. J Asthma. 2000;37:303–18. doi: 10.3109/02770900009055454. [DOI] [PubMed] [Google Scholar]
- 21.Mosely JB, O’Malley K, Petersen NJ, et al. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2002;347:81–8. doi: 10.1056/NEJMoa013259. [DOI] [PubMed] [Google Scholar]
- 22.Ondo WG, Sethi KD, Kricorian G. Selegiline orally disintegrating tablets in patients with Parkinson disease and “wearing off” symptoms. Clin Neuropharmacol. 2007;30:295–300. doi: 10.1097/WNF.0b013e3180616570. [DOI] [PubMed] [Google Scholar]
- 23.Pagnin D, de Quiroz V, Pini S, et al. Efficacy of ECT in depression. J ECT. 2004;20:13–20. doi: 10.1097/00124509-200403000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Rush AJ, Marangell LB, Sackeim HA. Vagus nerve stimulation for treatment resistant depression: a randomized, controlled acute phase trial. Biol Psychiatry. 2005;58:347–54. doi: 10.1016/j.biopsych.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 25.Quitkin FM, Rabkin JG, Ross D, et al. Identification of true drug response to antidepressants: use of pattern analysis. Arch Gen Psychiatry. 1984;41:782–6. doi: 10.1001/archpsyc.1984.01790190056007. [DOI] [PubMed] [Google Scholar]
- 26.Khan A, Redding N, Brown WA. The persistence of the placebo response in antidepressant clinical trials. J Psychiatr Res. 2008;42:791–6. doi: 10.1016/j.jpsychires.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Mayberg HS, Silva AJ, Brannan SK, et al. The functional neuroanatomy of the placebo effect. Am J Psychiatry. 2002;159:728–37. doi: 10.1176/appi.ajp.159.5.728. [DOI] [PubMed] [Google Scholar]
- 28.Benedetti F, Mayberg HS, Wager TD, et al. Neurobiological mechanisms of the placebo effect. J Neurosci. 2005;25:10390–402. doi: 10.1523/JNEUROSCI.3458-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burt CG, Gordon WF, Holt NF, et al. Amitriptyline in depressive states: a controlled trial. Br J Psychiatry. 1962;108:711–30. doi: 10.1192/bjp.108.456.711. [DOI] [PubMed] [Google Scholar]
- 30.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 31.Khan A, Cohen S, Dager S. Onset of response in relation to outcome in depressed outpatients with placebo and imipramine. J Affect Disord. 1989;17:33–8. doi: 10.1016/0165-0327(89)90021-9. [DOI] [PubMed] [Google Scholar]
- 32.Geddes JR, Carney SM, Davies C. Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet. 2003;361:653–61. doi: 10.1016/S0140-6736(03)12599-8. [DOI] [PubMed] [Google Scholar]
- 33.Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170:723–33. doi: 10.1176/appi.ajp.2012.12040474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan A, Leventhal RM, Khan S, et al. Severity of depression and response to antidepressants and placebo: an analysis of the FDA database. J Clin Psychopharmacol. 2002;22:40–5. doi: 10.1097/00004714-200202000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Khan A, Brodhead AE, Kolts RL, et al. Severity of depressive symptoms and response to antidepressants and placebo in antidepressant clinical trials. J Psychiatr Res. 2005;39:145–50. doi: 10.1016/j.jpsychires.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Khan A, Schwartz K, Kolts RL, et al. Relationship between depression severity entry criteria and antidepressant clinical trial outcomes. Biol Psychiatry. 2007;62:65–71. doi: 10.1016/j.biopsych.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 37.Khan A, Bhat A, Kolts R, et al. Why has the antidepressant-placebo difference in antidepressant clinical trials diminished over the past three decades. CNS Neurosci Ther. 2010;16:217–26. doi: 10.1111/j.1755-5949.2010.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mundt JF, Greist JH, Jefferson JW, et al. Is it easier to find what you are looking for if you think you know what it looks like? J Clin Psychopharmacol. 2007;27:121–5. doi: 10.1097/JCP.0b013e3180387820. [DOI] [PubMed] [Google Scholar]
- 39.Khan A, Faucett J, Brown W. Magnitude of placebo response and response variance in antidepressant clinical trials using structured, taped and appraised rater interviews compared to traditional rating interviews. J Psychiatr Res. 2014;51:88–92. doi: 10.1016/j.jpsychires.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Khan A, Faucett J, Brown W. Magnitude of change with antidepressants and placebo in antidepressant clinical trials using structured, taped and appraised rater interviews compared to traditional semi-structured interviews. Psychopharmacology. 2014;231:4301–7. doi: 10.1007/s00213-014-3584-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibertini M, Nations KR, Whitaker JA. Obtained effect size as a function of sample size in approved antidepressants: a real-world illustration in support of better trial design. Int Clin Psychopharmacol. 2012;27:100–6. doi: 10.1097/YIC.0b013e32834f504f. [DOI] [PubMed] [Google Scholar]
- 42.Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Investig Drugs. 2009;18:12–1. doi: 10.1517/13543780903286396. [DOI] [PubMed] [Google Scholar]
- 43.Patel HC, Hayward C, Ozdemir BA, et al. Magnitude of blood pressure reduction in the placebo arms of modern hypertension trials. Hypertension. 2015;65:401–6. doi: 10.1161/HYPERTENSIONAHA.114.04640. [DOI] [PubMed] [Google Scholar]
- 44.Schilling JP, Khan SRF, Khan A. Outcome measures and serious asthma exacerbation in clinical trials of asthma controller medications. Ann Allergy Asthma Immunol. 2012;108:448–53. doi: 10.1016/j.anai.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 45.Shojania KG, Sumant RR, McDonald KM, et al. Effects of quality improvement strategies for type II diabetes on glycemic control: a meta-regression analysis. JAMA. 2006;296:427–40. doi: 10.1001/jama.296.4.427. [DOI] [PubMed] [Google Scholar]