

Abstract
While it has long been established that the chemokine receptor CCR9 and its ligand CCL25 are essential for the movement of leukocytes into the small intestine and the development of small-intestinal inflammation, the role of this chemokine-receptor pair in colonic inflammation is not clear. Toward this end, we compared colonic CCL25 protein levels in healthy individuals to those in patients with ulcerative colitis. In addition, we determined the effect of CCR9 pharmacological inhibition in the mdr1a −/− mouse model of ulcerative colitis. Colon samples from patients with ulcerative colitis had significantly higher levels of CCL25 protein compared to healthy controls, a finding mirrored in the mdr1a −/− mice. In the mdr1a −/− mice, CCR9 antagonists significantly decreased the extent of wasting and colonic remodeling and reduced the levels of inflammatory cytokines in the colon. These findings indicate that the CCR9:CCL25 pair plays a causative role in ulcerative colitis and suggest that CCR9 antagonists will provide a therapeutic benefit in patients with colonic inflammation.
1. Introduction
The chemokine system is an intricate network of related cytokines and their cognate receptors, which together play a key role in the directed migration of leukocytes to their site of action [1, 2]. Expression of both the chemokine receptor CCR9 and the integrin α 4 β 7 is required for efficient lymphocyte homing to the intestine; these proteins form the so-called gut-homing phenotype of intestinal lymphocytes [3, 4]. CCR9 is expressed on the majority of CD4+ (67%) and CD8+ (54%) T cells [5] as well as B cells [4] and plasmacytoid dendritic cells [6, 7] isolated from the small intestine. In contrast, 2–4% of circulating T cells in healthy individuals express CCR9 [5]. The only known ligand for CCR9, CCL25/TECK, is abundantly produced in the small intestine [8], where its expression is localized to the epithelium [5, 9, 10]. The requirement for CCL25 in the recruitment of lymphocytes to the intestine has been highlighted in CCL25-deficient mice, which display a lack of antigen-specific T cell recruitment into the lamina propria and epithelium [11]. Indeed, targeting the CCR9/CCL25 axis has been shown to be of benefit in the TNF-ΔARE [12] and SAMP-1/YIT [13] models of ileitis. These observations are also consistent with those made in an adoptive transfer model in which pharmacological inhibition of CCR9 resulted in a significant reduction in the numbers of T cells recruited in the small intestine, without inhibiting cell trafficking to other organs [14]. Indeed, in the PROTECT-1 clinical trial, pharmacological inhibition of CCR9 resulted in a significant reduction in disease severity in Crohn's disease subjects, relative to those on placebo [15].
While it is clear that CCL25 is produced in the small intestine and regulates migration of circulating CCR9+ leukocytes to this tissue under both homeostatic and inflammatory conditions, the role of CCR9 and CCL25 in leukocyte migration to the colon is controversial. Although CCR9+ cells have been found in normal human colon [5], until recently CCL25 had not [5, 9, 16]. In colon from inflammatory bowel disease (IBD) patients with active colonic inflammation, CCL25 mRNA was detected by one group [17] but not by two others [16, 18]. CCL25 mRNA was recently detected in mouse colon and was found to be upregulated in the DSS model of acute colitis [19]. An early report indicated that the increased frequency of circulating CCR9+ T cells observed in Crohn's disease patients was not observed if the inflammation was limited solely to the colon [18]. However, two recent reports have noted changes in circulating CCR9+ cells in individuals with purely colonic inflammation. Mann et al. reported that the percentages of circulating CCR9+ γδ T cells are increased in ulcerative colitis patients compared to healthy controls [20]. Linton et al. reported that CCR9 levels are higher on circulating T cells and HLA-DRhi monocytes in IBD patients with active colonic inflammation compared to healthy controls [17]. These changes are reminiscent of those noted in patients with Crohn's disease [18] and are implicated in the pathogenesis of primary sclerosing cholangitis [21], an inflammatory liver disease with a high degree of association with ulcerative colitis [22, 23]. Importantly, the CCR9 antagonist Vercirnon/CCX282 reduced colonic inflammation in Crohn's disease patients with colonic involvement [15].
In this study, we investigated the role of CCR9 and CCL25 in colonic inflammation. First we demonstrate that CCL25 is expressed in healthy human colon and is localized to the epithelium. Then we show that the levels of colonic CCL25 protein are elevated, compared to healthy controls, both in patients with ulcerative colitis and in the mdr1a −/− mouse model of ulcerative colitis. Finally we show that two CCR9 small-molecule antagonists, both of which have been tested in humans [15], inhibit colonic inflammation in mdr1a −/− mice.
2. Materials and Methods
The selective small molecule CCR9 antagonists, CCX025 and CCX282-B, have been previously described [12, 24–26].
2.1. Northern Blotting
CCL25 and CCR9 DNA fragments were cloned from human thymus cDNA (Biochain, Hayward, CA) using primers 5′-ATGAACCTGTGGCTCCTG-3′ and 5′-TAACAGGCAGGAATGACTC-3′ (CCL25) and 5′-ATGACACCCACAGACTTC-3′ and 5′-GAGGGAGAGTGCTCCTG-3′ (CCR9). The fragments were isolated by agarose gel electrophoresis, labeled with 32P-cytidine 5′-triphosphate (GE Healthcare, Piscataway, NJ) using the NEBlot kit (New England Biolabs, Ipswich, MA) and purified on Sephadex G-50 columns (GE Healthcare, Piscataway, NJ). Normal tissue mRNAs were purchased from Ambion (Austin, TX), Biochain (Hayward, CA), Origene (Rockville, MD), and Cybrdi (Frederick, MD). RNAs were subjected to agarose gel electrophoresis (2 µg/lane) and blotted to nylon membranes using the Northernmax kit (Ambion, Austin, TX). The membranes were prehybridized with Ultrahyb (Ambion, Austin, TX) at 42°C for 1 hr, and the radiolabeled probes were boiled and added (~5 × 106 cpm/blot). After overnight hybridization at 42°C, the probes were removed and the blots were rinsed successively with 2x SSC 0.1% SDS and 0.1x SSC 0.1% SDS at 42°C. Blots were exposed to Hyperfilm MP (GE Healthcare, Piscataway, NJ) which were developed with a Mini-Med 90 (AFP, Elmsford, NY).
2.2. Immunofluorescence and Immunohistochemistry
Normal small intestine and colon frozen sections were purchased from Biochain (Hayward, CA). Sections were fixed for 5 min in cold acetone, air-dried, rinsed for 5 min in TBS, and immersed for 30 min in TBS containing 5% goat serum, 1% BSA, 0.1% triton X-100, and 0.05% Tween-20. Sections were exposed to anti-CCL25/TECK or its IgG2b isotype Ab (both from R&D Systems, Minneapolis, MN) at 2.5 µg/mL for 1 hr. Sections were rinsed for 3 × 2 min with TBST and exposed to Alexa Fluor 488-conjugated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA) at 4 µg/mL for 30 min. Sections were rinsed with PBS for 5 min and coverslips were mounted with Vectashield with DAPI (Vector Labs, Burlingame CA). For immunohistochemistry (IHC), the sections were treated as described above but were additionally pretreated to remove endogenous alkaline phosphatase (Dako, Carpinteria, CA) and biotin (Vector Labs, Burlingame, CA). CCL25 mAb was detected with biotinylated goat anti-mouse IgG (Jackson Immunoresearch, West Grove PA) at 1 : 1000 for 30 min. After rinsing with TBST, the sections were exposed to avidin-conjugated alkaline phosphatase (Dako, Carpinteria, CA) for 20 min. The sections were rinsed in TBST, stained with fuchsin+ reagent (Dako, Carpinteria, CA), rinsed in water for 2 min, counterstained with Mayer's hematoxylin (Sigma, St. Louis, MO) for 2 min, and rinsed in tap water for 3 min. Coverslips were mounted with Vectamount (Vector Labs, Burlingame, CA).
2.3. Tissue Cytokine Levels
Human terminal ileum and colon biopsies were obtained at baseline from Crohn's disease patients with a Crohn's Disease Activity Index (CDAI) ≥ 250 (PROTECT-1 trial) [15]. Since this was a multinational, multicenter clinical trial, central ethics committees reviewed the protocol and informed consent form and provided written approval prior to study initiation. All patients who agreed to undergo colonoscopic biopsies provided written informed consent prior to the biopsy procedure. Colon biopsies from healthy individuals were obtained from ILSbio (Chestertown, MD). Samples were homogenized on ice and homogenates were analyzed for CCL25 by ELISA (R&D Systems, Minneapolis, MN). Murine intestine tissue was harvested, cleaned, and snap-frozen. Homogenates were analyzed for CCL25, IL-1β, IL-6, IL-10, and IFNγ by ELISA (R&D Systems, Minneapolis, MN). Cytokine levels were normalized to total protein in the sample as determined by Bradford Reagent (Bio-Rad, Hercules, CA).
2.4. mdr1a −/− Mouse Model of Ulcerative Colitis
All animal experiments and procedures were approved by the ChemoCentryx Institutional Animal Care and Use Committee under protocol number CCX-154-2002. Female mdr1a −/− and wild-type FVB mice were purchased from Taconic (Germantown, NY). CCX025, formulated in 1% hydroxypropyl methylcellulose, was dosed at 100 mg/kg s.c. once daily. CCX282-B, formulated in 5% Cremophor, was dosed at 50 mg/kg c.c. twice daily. During the course of the study, body weights and the incidence of diarrhea were recorded on a weekly basis. Any animals that exhibited weight loss of greater than 20% of their peak body weight were euthanized. Final body weights for any euthanized or dead animals were carried forward for data analysis. Diarrhea was scored on a 0–5 scale; when animals reached a score of ≥3, their diarrhea was constant and irreversible and was thus considered as established diarrhea.
2.5. Histological Analysis
Histological analysis of H&E stained samples was conducted in a blinded fashion by a board-certified veterinary pathologist. Extent of leukocyte infiltration, reported as inflammation score, was defined as follows: 0: normal; 1: minimal—diffuse minimal with no separation of glands; 2: mild—diffuse mild with no separation of glands; 3: moderate—diffuse moderate with mild multifocal separation of glands; 4: marked—diffuse marked with multifocal solid areas of inflammation devoid of glands; 5: severe—diffuse severe with large areas of mucosa devoid of glands. The parameters reflecting gland loss or surface erosion were scored individually based upon the percentage of total mucosa area affected: 0: none; 1: 1–10%; 2: 11–25%; 3: 26–50%; 4: 51–75%; 5: 76–100%.
2.6. Statistical Analysis
Data are presented as mean ± SEM and were analyzed using Graphpad Prism 4.03 (Graphpad Software, La Jolla, CA). Statistical analysis was performed using unpaired Student's t-tests versus relevant control or Log rank test, as appropriate.
3. Results
3.1. CCL25 Is Expressed in Normal Human Colon
Normal small intestine and colon mRNA samples from different vendors were evaluated for CCL25 transcripts by Northern blot analysis. CCL25 mRNA was detected in all small intestine samples and in the majority (5 of 6) of colon samples (Figure 1). Immunohistochemical and immunofluorescence methods were utilized to determine whether CCL25 protein is expressed in normal colon. By each method, CCL25 protein was detected on epithelial cells in both the small bowel and colon (Figure 2).
Figure 1.

CCL25 mRNA is expressed in normal human colon. Normal intestinal mRNA samples purchased from multiple vendors were evaluated for CCL25 mRNA content by Northern Blotting. CCL25 mRNA was detected in 6 of 6 small intestine samples and 5 of 6 healthy colon samples.
Figure 2.
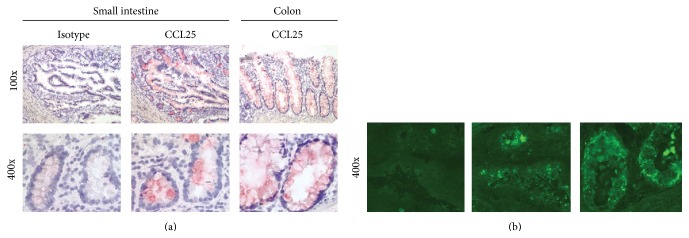
CCL25 protein is expressed in healthy human colon. Frozen sections of healthy human colon and small intestine were stained with CCL25 and isotype control antibodies. CCL25 protein was detected in epithelium in both colon and small bowel, as determined by IHC ((a), red color) and immunofluorescence ((b), green color). Magnification 100x (top row of (a)), 400x (bottom row of (a) and (b)).
3.2. Colonic Inflammation Is Associated with Increased CCL25 Levels
Levels of CCL25 protein were measured in colon tissue from healthy individuals and Crohn's disease and ulcerative colitis patients, as well as Crohn's disease terminal ileum. CCL25 levels in colon biopsies from individuals with ulcerative colitis were significantly higher than CCL25 levels in normal colon (Figure 3(a)). CCL25 levels were also significantly higher in colon biopsies from individuals with active Crohn's disease (CDAI ≥ 250) (Figure 3(a)). Interestingly, CCL25 levels in the terminal ileum of Crohn's disease patients were not higher than the levels of CCL25 in the colon of these patients or ulcerative colitis patients (Figure 3(a)). As a specificity control, we found no CCL25 in psoriatic plaques (data not shown).
Figure 3.

CCL25 protein levels are significantly elevated during colonic inflammation. CCL25 protein levels in homogenates of colon and terminal ileum were determined by ELISA and normalized to the levels of total protein. (a) CCL25 protein levels were significantly elevated in colon biopsies from individuals with active Crohn's disease or ulcerative colitis, relative to healthy controls. In Crohn's disease, the levels of CCL25 in the colon were equivalent to the levels in the terminal ileum. n = 6–8 individuals. (b) CCL25 levels were increased in presymptomatic mdr1a −/− mice and further increased in mdr1a −/− mice with severe disease, compared to wild-type mice. n = 6–8 mice. (c) The frequency of CCR9+CD4+ T cells among PBMC is significantly increased in mdr1a −/− mice compared to age-matched wild-type mice; n = 5 mice per time point. ∗ P < 0.05; ∗∗ P < 0.01.
As mdr1a −/− mice spontaneously develop a form of colitis that exhibits many of the hallmarks of human ulcerative colitis, such as marked lymphocyte infiltration into the lamina propria, elevation of inflammatory cytokines, and ulceration [27, 28], we chose this model to examine the therapeutic benefit of pharmacological CCR9 inhibition in the context of colonic inflammation. This model is an attractive one, since it recapitulates the therapeutic activity of IL-12/23 p40 inhibition and the disease-exacerbating activity of IL-17 inhibition observed in humans [29]. In 16-week-old mdr1a −/− mice, which exhibited no overt symptoms (diarrhea, weight loss) of colitis, colonic CCL25 levels were significantly higher than in age-matched wild-type mice (Figure 3(b)). Colonic CCL25 levels in mice with severe colitis, defined by persistent, severe diarrhea and wasting, increased more than 60-fold (P < 0.01), compared to presymptomatic mdr1a −/− mice, and were more than 350-fold higher than in wild-type mice (P < 0.05; Figure 3(b)). CCL25 levels in the colon of mdr1a −/− mice with severe disease were similar to the levels of CCL25 in the small intestine of mdr1a −/− mice (Figure 3(b)). As a specificity control, we found no CCL25 in normal or inflamed brains from C57BL/6 mice with experimental autoimmune encephalitis (data not shown).
As a surrogate measure of intestinal inflammation we noted changes in the frequency of CCR9+ cells in the circulation of mdr1a −/− mice prior to the onset of clinical symptoms. The frequency of CCR9+CD4+ T cells in PBMC of wild-type mice remained ~1% over time (Figure 3(c)), whereas this frequency increased over time in mdr1a −/− mice. At 11 weeks of age, the frequency of CCR9+CD4+ T cells in mdr1a −/− mice was ~1.5% of PBMC, but this frequency was ~4.5% at 17 weeks of age (Figure 3(c)). No significant differences were noted between wild-type and mdr1a −/− mice with respect to the frequency of the CCR9+CD8+ peripheral T cell population (data not shown).
3.3. CCR9 Inhibition Confers a Significant Therapeutic Benefit in the mdr1a −/− Mouse Model of Ulcerative Colitis
The role of CCR9 in the initiation and progression of colitis was tested by pharmacological CCR9 inhibition with two small molecule antagonists, CCX282-B and CCX025. Both compounds are potent and selective antagonists of human and mouse CCR9 [12, 24]. CCX025 inhibits CCR9-mediated chemotaxis of human Molt-4 cells (a T cell line) with an IC50 value of 23 nM in buffer and 78 nM in 100% human serum (Supplementary Figures S1A-B in Supplementary Material available online at http://dx.doi.org/10.1155/2015/628340). In addition, CCX025 inhibits CCR9-mediated chemotaxis of mouse thymocytes with an IC50 value of 12 nM in buffer (Supplementary Figure S1C). The IC50 values of CCX282-B have already been described [12]. For in vivo studies, the dose of CCX025 was selected based upon its ability to block T cell trafficking to the intestine (Supplementary Figure S1D), similar to the CCR9 antagonist CCX8037 [14]. The dose of CCX282-B was selected based on its ability to reduce ileitis severity in the TNF-ΔARE model [12].
mdr1a −/− mice were randomized into two groups and treated daily with either CCX025 or vehicle control, beginning at 11 weeks of age. Body weights and diarrhea incidence were recorded on a weekly basis. At 21 weeks of age, vehicle-treated mdr1a −/− mice began to exhibit weight loss consistent with the onset of colitis (Figure 4(a)). In contrast, CCX025-treated animals continued to gain weight at a rate similar to that of healthy wild-type mice, an effect that was maintained throughout the course of the study (Figure 4(a)). Because colitis-associated wasting is progressive, mdr1a −/− mice were euthanized if their body weight loss exceeded 20%. Over the course of the study, 32% of vehicle-treated mdr1a −/− mice had to be euthanized; in contrast, only 4% of CCX025-treated mice had to be euthanized (P < 0.05; Figure 4(b)). CCX025 also significantly reduced the incidence of established, severe diarrhea exhibited in this model (Figure 4(c)). At the end of the study, CCR9 expression on circulating T cells was assessed. Vehicle-treated mdr1a −/− mice exhibited a significant elevation in the frequency of CCR9+CD4+ T cells relative to wild-type mice; in contrast, CCX025-treated mdr1a −/− mice had normal levels of circulating CCR9+CD4+ T cells (Figure 4(d)).
Figure 4.

Pharmacological CCR9 inhibition results in a significant reduction in severity of colitis symptoms in mdr1 −/− mice. (a) CCX025 treatment, starting at 11 weeks of age, resulted in a significant inhibition of the growth retardation associated with disease. Mice treated with CCX025 (n = 25) gained weight in an identical fashion to wild-type controls (n = 5); vehicle-treated mdr1a −/− mice (n = 25) failed to gain weight after week 22. (b) CCX025 treatment resulted in a significant reduction in mortality compared to vehicle in mdr1a −/− mice. (c) CCX025 treatment resulted in a significant reduction in the percentage of mdr1a −/− mice that developed established diarrhea (score of ≥3 out of 5). (d) CCX025 treatment resulted in a significant inhibition of the disease-induced increase in CCR9+CD4+ PBMC frequencies in mdr1a −/− mice, measured at 17 weeks of age. (e) CCX282-B treatment (n = 34), starting at 10 weeks of age, resulted in a significant inhibition of the growth retardation associated with disease, compared to controls (n = 29). (f) Therapeutic treatment with CCX282-B (n = 17 per treatment group) resulted in a significant reduction in the weight loss associated with active disease. Animals were randomized at 16 weeks of age to either CCX282-B or vehicle treatment. All panels: ∗ P < 0.05; ∗∗ P < 0.01.
CCX282-B was also tested in the mdr1a −/− model. Like CCX025, CCX282-B blocked the colitis-associated weight loss inherent in the model (Figure 4(e)). In order to test CCR9 inhibition in a nonprophylactic regimen, that is, starting treatment during the onset of colitis, 16-week-old mdr1a −/− mice were randomized to receive either CCX282-B or vehicle. Even when used in this regimen, CCX282-B abrogated growth arrest (Figure 4(f)). CCX282-B also caused a marked reduction in the incidence of established diarrhea (4% incidence with CCX282-B versus 24% with vehicle; data not shown).
We analyzed other important parameters of disease in CCX025-treated mdr1a −/− mice at the end of the study. First, colonic inflammation in the mdr1a −/− model is mediated in part by the proinflammatory cytokines IFN-γ, IL-1β, and IL-6. The levels of each of these cytokines were significantly reduced in the colons of mdr1a −/− mice treated with CCX025, relative to vehicle-treated mice (Figure 5(a)). In addition, CCX025 treatment was associated with a significant increase in the colonic levels of the anti-inflammatory cytokine IL-10, relative to vehicle-treated mice (Figure 5(a)). Second, mdr1a −/− mice exhibit structural changes in the colon, which become both shorter in length and heavier as a result of tissue remodeling [27, 28]. CCX025-treated mice exhibited a significant reduction in the colon weight : length ratio, relative to vehicle-treated mice (Figure 5(b)). Third, mdr1a −/− mice exhibit inflammation, ulceration, and epithelial erosion of the colon [27, 28]. CCX025 treatment resulted in a significant reduction in both the inflammation and ulceration scores in proximal colon of mdr1a −/− mice (Figure 5(c)). In the more severely affected distal colon, CCX025 treatment significantly reduced the inflammation, ulceration, and epithelial erosion scores (Figure 5(d)).
Figure 5.

Pharmacological CCR9 inhibition results in a significant improvement in colonic inflammation in mdr1a −/− mice. (a) CCX025 treatment resulted in significantly lower levels of the proinflammatory cytokines IFN-γ, IL-1β, and IL-6 in the colon; at the same time CCX025 treatment resulted in a significant elevation in the levels of the anti-inflammatory cytokine IL-10 in the colon (n = 16–19). (b) CCX025 treatment, starting at 11 weeks of age, resulted in a significant reduction in the weight-to-length ratio of the colon in mdr1a −/− mice, compared to vehicle-treated mice (n = 25). CCX025 treatment resulted in significantly reduced histological damage scores in the proximal colon (c) and the distal colon (d). All panels: ∗ P < 0.05; ∗∗ P < 0.01.
4. Discussion
In this study we show that the CCR9 ligand CCL25 is expressed by normal colon epithelium; that levels of colonic CCL25 increase in both Crohn's disease and ulcerative colitis; and that pharmacological inhibitors of CCR9 prevent the development of disease in an experimental model of ulcerative colitis. These results suggest that CCL25-mediated recruitment of CCR9+ leukocytes into the colon plays a causative role in the inflammation that occurs in chronic colonic diseases such as ulcerative colitis, similar to the case with ileal inflammation in Crohn's disease.
Several groups have failed to detect CCL25 in normal human colon [5, 9, 16], even though CCR9+ T cells are abundant there [5]. We used three methods to assess CCL25 expression in normal colon. Using Northern blotting on multiple samples from multiple vendors, we found that CCL25 mRNA is indeed expressed in human colon. Using ELISA, we found that CCL25 protein is expressed in both human and mouse colon. Finally, using IHC, we found that CCL25 protein is expressed by the colonic epithelium, as is the case in small intestine [5, 9, 18]. We surmise that the earlier studies' inability to detect CCL25 in normal colon was due either to insufficient amounts of epithelium in their samples or to the possibility that CCL25 is expressed unevenly over the length of the colon, as has been previously described for the small intestine [30].
Even though CCL25 protein levels in normal human and mouse colon were lower than in the small intestine, substantial elevations were observed in both species in connection with colonic inflammation. Colon biopsies obtained from patients with active Crohn's disease (CDAI ≥ 250) had significantly higher levels of CCL25 protein than healthy colon samples. The increase in colonic CCL25 levels was not restricted to Crohn's disease, since colon biopsies from ulcerative colitis patients also contained significantly elevated levels of CCL25 protein. In the mdr1a −/− mouse model of ulcerative colitis, increased colonic CCL25 protein levels were noted before any clinical manifestations of disease and correlated temporally with increases in the frequency of circulating CCR9+CD4+ T cells, which we used as a surrogate for intestinal inflammation. Colonic CCL25 protein levels continued to increase dramatically as mice developed severe colitis, and the highest levels of colonic CCL25 protein were found in those mice with the most severe disease. An increase in CD4+ cells in the colonic lamina propria lymphocyte and intraepithelial lymphocyte populations of mdr1a −/− mice with active colitis has been described [27, 28]; it may well be that such an increase results from the concurrent upregulation of CCL25 in the colon coupled with an increase in circulating CCR9+CD4+ T cells, both documented in our studies.
Interference with the CCR9/CCL25 axis has been shown to improve disease symptoms and tissue inflammation in mouse models of ileitis such as the SAMP-1/Yit model [13] and the TNF-ΔARE model [12]. The mdr1a −/− mouse model shares many features with human ulcerative colitis, such as lymphocyte infiltration into the lamina propria and epithelial destruction and ulceration, and thus is a robust model in which the efficacy of new therapeutic agents is to be tested [27, 28]. Treatment of mdr1a −/− mice with the CCR9 antagonist CCX025 resulted in complete protection from the development of colitis. All aspects of the disease were ameliorated, including weight loss, diarrhea, inflammation, colon shortening, and mortality. These effects were also noted with the CCR9 antagonist CCX282-B, indicating that the CCX025-mediated changes were due to CCR9 inhibition and not the result of an off-target effect. While our results are consistent with reports that describe inflammation throughout the entire colon [27], we observed, histologically, a higher degree of inflammation in the distal colon compared to the proximal colon.
CCR9 inhibition resulted in improvements in the degree of ulceration and epithelial erosion in the colon. It is reasonable to assume a causal relationship between decreased lymphocyte infiltration, histological improvements, and improved clinical symptoms. The importance of mucosal healing to the clinical manifestation of ulcerative colitis has been described [31]. Inhibition of CCR9 also resulted in marked changes in the cytokine profiles observed in colonic tissue from mdr1a −/− mice. Significant decreases were seen in the levels of IFN-γ, IL-1β, and IL-6, all of which are elevated in human lamina propria mononuclear cells isolated from ulcerative colitis and Crohn's disease patients [32]. The reduced levels of IFN-γ are noteworthy, as CCR9+ cells have been shown to be a significant source of this cytokine in Crohn's disease [33]. The mice treated with CCX025 also had increased colonic levels of the anti-inflammatory cytokine IL-10, in concordance with the fact that IL-10−/− mice have a predisposition to develop colonic inflammation [34]. Finally, CCR9 inhibition resulted in normalization of the frequency of circulating of CCR9+CD4+ PBMC.
The results presented herein clearly demonstrate that pharmacological inhibition of CCR9 provides a therapeutic benefit in the mdr1a −/− mouse colitis model. However, there are reports in the literature that conclude that CCR9−/− mice fare worse in the DSS-induced acute colitis model [19] and are unable to develop tolerance to oral antigens [35]. Differences between CCR9 pharmacological inhibition and genetic deletion have also been reported in the TNF-ΔARE ileitis model: pharmacological inhibition of CCR9 [12] was beneficial, whereas CCR9 deletion either had no effect [36] or worsened disease [37]. We recently conducted a head-to-head comparison between the effects of CCR9 pharmacological inhibition and genetic deletion on oral tolerance in mice and found that CCR9 deletion does not predict the results obtained when using a pharmacological approach [38]. The inability of the CCR9−/− mouse to predict the outcome of pharmacological inhibition could be explained by differences in the intestinal composition of immune cells (such as γδ T cells [39], plasmacytoid DC [7], and IgA+ plasma cells [40]) reported in CCR9−/− mice relative to wild-type mice.
One limitation of our study is that we have not proven the mechanism of action of the CCR9 antagonists in the mdr1a −/− model. We surmise the mechanism to be the inhibition of CCR9+ T cell trafficking to the colon, based on short-term trafficking models in which CCR9 antagonists blocked antigen-mediated T cell trafficking to the small intestine of OT-1 mice [14] (and Figure S1D). Another limitation lies in the fact that activities occurring in mouse models of disease do not always recapitulate in human patients. However, given the lack of safe, orally administered treatments for ulcerative colitis, the increase in colonic CCL25 levels (Figure 3) and circulating CCR9+ leukocytes [17, 20] during disease, and the remarkable activities of CCR9 antagonists in the mdr1a −/− model of ulcerative colitis (Figures 4 and 5), exploring CCR9 antagonism as a therapy for ulcerative colitis seems appropriate. CCX282 and CCX025 have been shown to be safe and well-tolerated in healthy subjects in Phase I clinical trials, and trials in patients with colonic inflammatory disorders such as ulcerative colitis are indicated.
Supplementary Material
CCX025 is a potent inhibitor of T-cell migration. Increasing concentrations of CCX025 inhibit the chemotaxis of Molt-4 T-cells which constitutively express human CCR9, to human CCL25 in an in vitro migration assay. CCX025 was potent when the assay was run in buffer (a) or in 100% human serum (b). CCX025 inhibits the in vitro chemotaxis of primary mouse thymocytes to murine CCL25 (c). In an in vivo T-cell trafficking model in mice, CCX025 inhibited the recruitment of CD8+ T-cells to the small intestine of OT-1 mice given an oral challenge of ovalbumin (d)
Acknowledgment
This work was funded by ChemoCentryx, Inc.
Conflict of Interests
All authors are employees of ChemoCentryx, Inc.
Authors' Contribution
Karen Ebsworth, Matthew J. Walters, Robert D. Berahovich, Linda S. Ertl, Trevor T. Charvat, and Sreenivas Punna performed experiments; Matthew J. Walters, Robert D. Berahovich, James J. Campbell, Timothy J. Sullivan, and Pirow Bekker designed experiments and analyzed data; Pirow Bekker, Karen Ebsworth, and Matthew J. Walters wrote the paper.
References
- 1.Charo I. F., Ransohoff R. M. Mechanisms of disease: the many roles of chemokines and chemokine receptors in inflammation. The New England Journal of Medicine. 2006;354(6):610–621. doi: 10.1056/nejmra052723. [DOI] [PubMed] [Google Scholar]
- 2.Viola A., Luster A. D. Chemokines and their receptors: drug targets in immunity and inflammation. Annual Review of Pharmacology and Toxicology. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- 3.Mora J. R., Bono M. R., Manjunath N., et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424(6944):88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 4.Zabel B. A., Agace W. W., Campbell J. J., et al. Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine- mediated chemotaxis. Journal of Experimental Medicine. 1999;190(9):1241–1256. doi: 10.1084/jem.190.9.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papadakis K. A., Prehn J., Nelson V., et al. The role of thymus-expressed chemokine and its receptor CCR9 on lymphocytes in the regional specialization of the mucosal immune system. Journal of Immunology. 2000;165(9):5069–5076. doi: 10.4049/jimmunol.165.9.5069. [DOI] [PubMed] [Google Scholar]
- 6.Hadeiba H., Sato T., Habtezion A., Oderup C., Pan J., Butcher E. C. CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nature Immunology. 2008;9(11):1253–1260. doi: 10.1038/ni.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wendland M., Czeloth N., Mach N., et al. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(15):6347–6352. doi: 10.1073/pnas.0609180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vicari A. P., Figueroa D. J., Hedrick J. A., et al. TECK: A novel CC chemokine specifically expressed by thymic dendritic cells and potentially involved in T cell development. Immunity. 1997;7(2):291–301. doi: 10.1016/S1074-7613(00)80531-2. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel E. J., Campbell J. J., Haraldsen G., et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. Journal of Experimental Medicine. 2000;192(5):761–768. doi: 10.1084/jem.192.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wurbel M.-A., Philippe J.-M., Nguyen C., et al. The chemokine TECK is expressed by thymic and intestinal epithelial cells and attracts double- and single-positive thymocytes expressing the TECK receptor CCR9. European Journal of Immunology. 2000;30(1):262–271. doi: 10.1002/1521-4141(200001)30:1&lt;262::AID-IMMU262>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 11.Wurbel M.-A., Malissen M., Guy-Grand D., Malissen B., Campbell J. J. Impaired accumulation of antigen-specific CD8 lymphocytes in chemokine CCL25-deficient intestinal epithelium and lamina propria. The Journal of Immunology. 2007;178(12):7598–7606. doi: 10.4049/jimmunol.178.12.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walters M. J., Wang Y., Lai N., et al. Characterization of CCX282-B, an orally bioavailable antagonist of the CCR9 chemokine receptor, for treatment of inflammatory bowel disease. Journal of Pharmacology and Experimental Therapeutics. 2010;335(1):61–69. doi: 10.1124/jpet.110.169714. [DOI] [PubMed] [Google Scholar]
- 13.Rivera-Nieves J., Ho J., Bamias G., et al. Antibody blockade of CCL25/CCR9 ameliorates early but not late chronic murine ileitis. Gastroenterology. 2006;131(5):1518–1529. doi: 10.1053/j.gastro.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 14.Tubo N. J., Wurbel M. A., Charvat T. T., Schall T. J., Walters M. J., Campbell J. J. A systemically-administered small molecule antagonist of CCR9 acts as a tissue-selective inhibitor of lymphocyte trafficking. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.0050498.e50498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keshav S., Vaňásek T., Niv Y., et al. A randomized controlled trial of the efficacy and safety of CCX282-B, an orally-administered blocker of chemokine receptor CCR9, for patients with Crohn's diseas. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0060094.e60094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arijs I., De Hertogh G., MacHiels K., et al. Mucosal gene expression of cell adhesion molecules, chemokines, and chemokine receptors in patients with inflammatory bowel disease before and after infliximab treatment. American Journal of Gastroenterology. 2011;106(4):748–761. doi: 10.1038/ajg.2011.27. [DOI] [PubMed] [Google Scholar]
- 17.Linton L., Karlsson M., Grundström J., et al. HLA-DRhi and CCR9 define a pro-inflammatory monocyte subset in IBD. Clinical and Translational Gastroenterology. 2012;3, article e29 doi: 10.1038/ctg.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papadakis K. A., Prehn J., Moreno S. T., et al. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn's disease. Gastroenterology. 2001;121(2):246–254. doi: 10.1053/gast.2001.27154. [DOI] [PubMed] [Google Scholar]
- 19.Wurbel M.-A., McIntire M. G., Dwyer P., Fiebiger E. CCL25/CCR9 interactions regulate large intestinal inflammation in a murine model of acute colitis. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016442.e16442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mann E. R., Mccarthy N. E., Peake S. T. C., et al. Skin- and gut-homing molecules on human circulating gammadelta T cells and their dysregulation in inflammatory bowel disease. Clinical and Experimental Immunology. 2012;170(2):122–130. doi: 10.1111/j.1365-2249.2012.04649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eksteen B., Grant A. J., Miles A., et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. Journal of Experimental Medicine. 2004;200(11):1511–1517. doi: 10.1084/jem.20041035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boonstra K., van Erpecum K. J., van Nieuwkerk K. M. J., et al. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflammatory Bowel Diseases. 2012;18(12):2270–2276. doi: 10.1002/ibd.22938. [DOI] [PubMed] [Google Scholar]
- 23.Olsson R., Danielsson Å., Järnerot G., et al. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991;100(5):1319–1323. [PubMed] [Google Scholar]
- 24.Charvat T. T., Hu C., Melikian A., Novack A., Pennell A. N-(2-(hetaryl)aryl)arylsulfonamides and N-(2-(hetaryl)hetaryl)arylsulfonamides. PCT Patent Application WO2009038847, 2009.
- 25.Eksteen B., Adams D. H. GSK-1605786, a selective small-molecule antagonist of the CCR9 chemokine receptor for the treatment of Crohn's disease. IDrugs. 2010;13(7):472–481. [PubMed] [Google Scholar]
- 26.Jaen J. C., Walters M. J., Berahovich R. D., et al. CCR9 inhibition in the treatment of colonic inflammation. Gut. 2010;59:p. A100. [Google Scholar]
- 27.Panwala C. M., Jones J. C., Viney J. L. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. Journal of Immunology. 1998;161(10):5733–5744. [PubMed] [Google Scholar]
- 28.Wilk J. N., Bilsborough J., Viney J. L. The mdr1a−/− mouse model of spontaneous colitis: a relevant and appropriate animal model to study inflammatory bowel disease. Immunologic Research. 2005;31(2):151–159. doi: 10.1385/ir:31:2:151. [DOI] [PubMed] [Google Scholar]
- 29.Maxwell J., Brown W., Zhang Y., Rottman J. B., Budelsky A., Towne J. Tu1615 Modeling the distinct roles for IL-23 and IL-17 in inflammatory bowel disease using Mdr1a-/- mice. Gastroenterology. 2013;144(5, supplement 1):p. S806. doi: 10.1016/S0016-5085(13)62984-4. [DOI] [Google Scholar]
- 30.Stenstad H., Svensson M., Cucak H., Kotarsky K., Agace W. W. Differential homing mechanisms regulate regionalized effector CD8αβ + T cell accumulation within the small intestine. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(24):10122–10127. doi: 10.1073/pnas.0700269104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lichtenstein G. R., Rutgeerts P. Importance of mucosal healing in ulcerative colitis. Inflammatory Bowel Diseases. 2010;16(2):338–346. doi: 10.1002/ibd.20997. [DOI] [PubMed] [Google Scholar]
- 32.Reinecker H.-C., Steffen M., Witthoeft T., et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clinical and Experimental Immunology. 1993;94(1):174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saruta M., Yu Q. T., Avanesyan A., Fleshner P. R., Targan S. R., Papadakis K. A. Phenotype and effector function of CC chemokine receptor 9-expressing lymphocytes in small intestinal Crohn's disease. Journal of Immunology. 2007;178(5):3293–3300. doi: 10.4049/jimmunol.178.5.3293. [DOI] [PubMed] [Google Scholar]
- 34.Kühn R., Löhler J., Rennick D., Rajewsky K., Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–274. doi: 10.1016/0092-8674(93)80068-P. [DOI] [PubMed] [Google Scholar]
- 35.Apostolaki M., Manoloukos M., Roulis M., et al. Role of beta7 integrin and the chemokine/chemokine receptor pair CCL25/CCR9 in modeled TNF-dependent Crohn's disease. Gastroenterology. 2008;134(7):2025–2035. doi: 10.1053/j.gastro.2008.02.085. [DOI] [PubMed] [Google Scholar]
- 36.Wermers J. D., McNamee E. N., Wurbel M.-A., Jedlicka P., Riveranieves J. The Chemokine receptor CCR9 is required for the T-cell-mediated regulation of chronic ileitis in mice. Gastroenterology. 2011;140(5):1526–1535. doi: 10.1053/j.gastro.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cassani B., Villablanca E. J., Quintana F. J., et al. Gut-tropic T cells that express integrin α4β7 and CCR9 Are required for induction of oral immune tolerance in mice. Gastroenterology. 2011;141(6):2109–2118. doi: 10.1053/j.gastro.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walters M. J., Ebsworth K., Sullivan T. J., et al. CCR9 inhibition does not interfere with the development of immune tolerance to oral antigens. Immunology Letters. 2013;151(1-2):44–47. doi: 10.1016/j.imlet.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Wurbel M.-A., Malissen M., Guy-Grand D., et al. Mice lacking the CCR9 CC-chemokine receptor show a mild impairment of early T- and B-cell development and a reduction in T-cell receptor γσ+ gut intraepithelial lymphocytes. Blood. 2001;98(9):2626–2632. doi: 10.1182/blood.v98.9.2626. [DOI] [PubMed] [Google Scholar]
- 40.Pabst O., Ohl L., Wendland M., et al. Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. The Journal of Experimental Medicine. 2004;199(3):411–416. doi: 10.1084/jem.20030996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CCX025 is a potent inhibitor of T-cell migration. Increasing concentrations of CCX025 inhibit the chemotaxis of Molt-4 T-cells which constitutively express human CCR9, to human CCL25 in an in vitro migration assay. CCX025 was potent when the assay was run in buffer (a) or in 100% human serum (b). CCX025 inhibits the in vitro chemotaxis of primary mouse thymocytes to murine CCL25 (c). In an in vivo T-cell trafficking model in mice, CCX025 inhibited the recruitment of CD8+ T-cells to the small intestine of OT-1 mice given an oral challenge of ovalbumin (d)