

Abstract
Collagen-producing myofibroblast transdifferentiation is considered a crucial determinant in the formation of scar tissue in the lungs of patients with idiopathic pulmonary fibrosis (IPF). Multiple resident pulmonary cell types and bone marrow-derived fibrocytes have been implicated as contributors to fibrotic lesions due to the transdifferentiation potential of these cells into myofibroblasts. In this study, we assessed the expression of Wilms’ tumor 1 (WT1), a known marker of mesothelial cells, in various cell types in normal and fibrotic lungs. We demonstrate that WT1 is expressed by both mesothelial and mesenchymal cells in IPF lungs, but has limited or no expression in normal human lungs. We also demonstrate that WT1-positive cells accumulate in fibrotic lung lesions, using two different mouse models of pulmonary fibrosis and WT1 promoter-driven fluorescent reporter mice. Reconstitution of bone-marrow cells into a transforming growth factor-α transgenic-mouse model demonstrated that fibrocytes do not transform into WT1-positive mesenchymal cells, but do augment accumulation of WT1-positive cells in severe fibrotic lung disease. Importantly, the number of WT1-positive cells in fibrotic lesions were correlated with severity of lung disease as assessed by changes in lung function, histology, and hydroxyproline levels in mice. Finally, inhibition of WT1 expression was sufficient to attenuate collagen and other extracellular-matrix gene production by mesenchymal cells from both murine and human fibrotic lungs. Thus, the results of this study demonstrate a novel association between fibrocyte-driven WT1-positive cell accumulation and severe fibrotic lung disease.
INTRODUCTION
Pulmonary fibrosis represents a heterogeneous group of diseases in which fibrotic lesions are characterized by the accumulation of multiple mesenchymal cells involved in the excessive deposition of extracellular matrix (ECM) in the parenchyma and subpleural regions of the lung (1–4). Idiopathic pulmonary fibrosis (IPF) is a fatal fibrotic lung disease with an incidence of 4.6–7.4 people per 100,000 of the population (5, 6). Despite the clinical and public-health significance of IPF, the pathophysiology of this disease remains under-defined. The development of novel therapeutic approaches for IPF likely depends on a mechanistic understanding of the role of the multiple lung mesenchymal cells that accumulate and participate in IPF pathogenesis. Recent findings suggest the co-existence of multiple lung mesenchymal cells in fibrotic lung lesions, including fibrocytes, pericytes, mesothelial cells, fibroblasts, and myofibroblasts (7–9). However, the cellular mechanisms involved in the progressive accumulation of lung mesenchymal cells in multiple fibrotic lesions of the lung, including in the parenchyma, adventitia, and subpleural areas, are not well understood.
Histopathological analysis of IPF lungs demonstrates a predominant honeycombing pattern in subpleural/peripheral areas, the appearance of fibrotic foci, and reticular abnormalities (10, 11). The thickened peripheral surfaces of the lung contain clusters of inflammatory and lung-resident mesenchymal cells that exist in continuity with the established fibrosis, which is a characteristic histologic feature of IPF thought to be the root of the underlying disease process (3, 12). The thickening of the lung subpleura also is a predominant feature in pneumoconiosis caused by the inhalation of asbestos fibers and diffuse scleroderma (13, 14). Pulmonary function studies typically reveal a restrictive pattern due to reduced elasticity of the lungs with the thickened subpleura (3, 15). Currently, there have been no mechanisms identified to explain the pathogenesis of subpleural fibrosis (16). In mouse models, overexpression of either transforming growth factor-α (TGFα) or transforming growth factor-β (TGFβ) is sufficient to induce progressive pleura/subpleural fibrosis with severe impairment of pulmonary function (17, 18). TGFα transgenic mice develop progressive fibrosis in adventitia, parenchyma, and subpleural areas of the lung with histological features similar to IPF when the EGFR ligand, TGFα, was conditionally overexpressed in the lung epithelium using the Club cell (Clara cell) specific protein rtTA promoter (19). Results of adenoviral gene transfer of TGF-β1 into the pleural mesothelium in rats suggest that mesothelial cells may be important in the development of subpleural fibrosis (18). Recent studies of ours and others demonstrate the critical relationship between the EGFR and TGFβ pathways and the most prominent effects noted in the subpleural regions of the lung (20, 21).
Fibrocytes are unique bone marrow (BM)-derived mesenchymal progenitor cells that express a common leukocyte antigen, CD45, and a mesenchymal marker, type I collagen (Col1) (22, 23). Fibrocytes can be identified in tissues with active fibrosis and inflammation in multiple fibroproliferative diseases, including IPF (24–30). Several studies have demonstrated that the number of circulating fibrocytes in patients with IPF reflects fibrogenic activity and disease progression (28, 29). The relevance of fibrocytes to lung fibrosis also has been demonstrated using mouse models in which inhibition of fibrocyte recruitment and maturation was sufficient to reduce the fibrotic burden (23, 31, 32). However, fibrocyte-driven functions that are responsible for the initiation and maintenance of pulmonary fibrosis remain ill defined. Using a green fluorescent protein (GFP)-labeled BM-transplant model, we recently demonstrated that GFP-positive fibrocytes accumulate progressively in fibrotic lesions, including in the subpleura of TGFα-transgenic mice (8). However, the majority of myofibroblasts that accumulate in these lesions are derived from lung-resident fibroblasts, but not fibrocytes (7–9). In vitro co-culture studies combined with in vivo adoptive cell-transfer studies suggest that fibrocytes augment lung-resident mesenchymal-cell proliferation and accumulation in fibrotic lung lesions, including in the subpleural regions (8, 33). Although the new findings suggest that lung-resident mesenchymal cells contribute to the excessive ECM deposition in fibrotic regions, the molecular and cellular origins of this remain unknown (8, 9, 34).
The findings of the current study identify Wilms’ Tumor 1 (WT1)-positive cells as a sizable subset of lung-resident mesenchymal cells that progressively accumulate in the subpleural fibrotic lesions of both human IPF and mouse models of pulmonary fibrosis. Using a combination of fibrocyte adoptive cell transfers and imaging methods, we unequivocally demonstrate that fibrocytes augment the accumulation of WT1-positive cells in fibrotic lesions that is associated with severe lung disease in mouse models of pulmonary fibrosis. Moreover, inhibition of WT1 was sufficient to attenuate ECM production in lung mesenchymal cells of both human IPF and mouse models of pulmonary fibrosis.
METHODS
Mouse strains
The WT1CreERT2/+ transgenic mouse strain has been described previously (35). Heterozygous WT1CreERT2 mice (knock-in allele) are normal and can be used to track the genetic lineage of WT1-expressing cells (35). We generated WT1CreERT2/+ROSAmTmG/+ mice, a double-fluorescent Cre recombinase reporter mouse strain, by crossing WT1CreERT2/+ heterozygous and ROSAmTmG/+ heterozygous mice. Heterozygous WT1CreERT2/+ROSAmTmG/+ mice (knock-in allele) express CreERT2 under the WT1 gene locus. The generation of TGFα-overexpressing mice in an FVB/NJ inbred-strain background has been described previously (17). Homozygous Club cell (Clara cell) specific protein-rtTA (CCSP) mice were mated with heterozygous (TetO) 7-cmv TGFα mice to generate bitransgenic TGFα (CCSP/TGFα) mice. Female mice, 10–12 weeks old, were used in all experiments. Mice were housed under specific pathogen-free conditions, and all animal experimental protocols were approved by the Institutional Animal Use and Care Committee of the Cincinnati Children’s Hospital Research Foundation.
Human tissue samples
Human IPF and control non-IPF biopsies were obtained from adult patients who had undergone lung transplantation and paraffin-embedded. Lung samples from donor lungs with no lung disease were used as control non-IPF lung biopsies. IPF was diagnosed according to the American Thoracic Society consensus criteria (36). All tissues were acquired using research protocols and/or informed consent and approved by the University of Michigan Health System Institutional Review Board. All materials were de-identified to the research team.
Human and mouse primary lung mesenchymal cell cultures
Primary lung mesenchymal cell cultures were prepared as described previously (8). The lung tissue was collected into Dulbecco’s Modified Essential Medium (DMEM) supplemented to contain 10% FBS and 1% each of pencillin, streptamycin, and amphotericin. Each lung tissue sample was cut into 2 cm × 2 cm pieces, and each piece was finely minced and digested in 5 ml of DMEM containing collagenase (2 mg/ml), incubated at 37°C for 1 hr. Digested tissues were passed through a 100 μm filter, washed twice by centrifugation at 100 × g for 5 min and plated onto 100 mm tissue-culture plates in 10 ml DMEM, and incubated at 37°C, 5% CO2 to allow the cells to adhere and migrate away from the larger remaining tissue pieces. Unbound cells were removed on Day 2 of culture by washing cells with fresh DMEM. Adherent lung mesenchymal cells were continued in culture until confluence (1–2 wks).
Mouse models of TGFα- and bleomycin-induced fibrosis
TGFα expression was induced by administering food containing doxycycline (Dox; 62.5 mg/kg) to CCSP/TGFα mice. CCSP/− mice fed with Dox-treated food were used as control mice for Dox treatments as they develop no fibrosis due to the lack of a TGFα transgene. In a mouse model of blemycin-induced fibrosis, 10–12 week-old female FVB/NJ mice were administered bleomycin (0.05 mg) daily in 0.05 mL saline solution intradermally (i.d.) in the center of the shaved back (within a 50 mm radius) for 5 days per week for a total of 4 wks. At Day 28, mice were euthanized and lung samples were collected for further analysis. All animals received either bleomycin or saline for 4 wks. Pulmonary fibrosis in both models was evaluated by analysis of lung hydroxyproline concentration, histology and measurement of lung function, and visualization of gene transcripts and proteins by Western blot (8).
Adoptive cell-transfer studies
Adoptive cell-transfer studies were performed as described previously (8). In brief, fibrocytes (CD45+Col1+) of the lung were isolated by positive selection from lung mesenchymal-cell cultures of CCSP/TGFα mice on Dox for 4 wks using anti-CD45 magnetic beads (8). FACS analysis indicates that the purity of fibrocytes isolated by this technique is approximately 95% (8). Fibrocytes were resuspended in phosphate-buffered saline at two million cells per ml. FVB/NJ recipient mice were infused intravenously with 250 μl of sterile saline or fibrocytes at Day 14 of saline or bleomycin (i.d.) treatment. Bleomycin or saline injections were continued for an additional 2 wks (5 days/week), and mice were sacrificed for pulmonary fibrosis measurements on Day 28.
Tamoxifen treatments
WT1CreERT2/+ROSAmTmG/+ mice were i.d. treated with bleomycin for 4 wks to induce lung fibrosis or with saline as a control. Tamoxifen (H7904; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in ethanol and emulsified in sunflower oil (W530285; Sigma-Aldrich) at 25 mg/ml concentration. To determine WT1-expressing cells (green in color), mice were treated with Tamoxifen (2.5 mg/day) to activate Cre recombination via intraperitoneal injections at the end (Day 26 to 27) of bleomycin treatment and euthanized on Day 28.
Histology, hydroxyproline, and lung-function tests
Lungs were inflation-fixed using 4% paraformaldehyde and stained with Masson trichrome as previously described (21). Lung fibrosis was assessed by measuring the total weight of the right lung and determining hydroxyproline levels using a colorimetric assay as previously described (21). The lung-function measurements were performed using a computerized Flexi Vent system (SCIREQ, Montreal, Canada) as previously described (21).
Immunohistochemistry and subpleural thickness measurements
Formalin- or OCT-fixed lung tissue sections were prepared and stained with antibodies against WT1 [mouse monoclonal anti-human WT1, clone 6F-H2; Ventana, Tucson, AZ, USA and rabbit anti-mouse WT1, clone CAN-R9(IHC)-56-2; Abcam, Cambridge, MA, USA], vimentin (anti-human vimentin, ab137321; Abcam), αSMA (Clone 1A4, Dako, CA, USA), and pancytokeratin (rabbit anti-human pancytokeratin, AE1/AE3/PCR26; Ventana, Tucson, AZ, USA) as described previously (8). Pleural thickness was measured by histomorphometric measurement on stained lung sections. Ten randomly selected uniform fields (26.2 mm2) per lung section in the pleural regions of the lung were obtained for each animal using a Leica DM2700 M bright-field microscope (Leica Microsystems, Buffalo Grove, IL, USA). High-magnification images (40X) were captured with a 3CCD color video camera, and the number of WT1-positive and total cells were counted using MetaMorph imaging software (v6.2; Molecular Devices, Sunnyvale, CA, USA). Pleural thickness was measured using the measured-distance function of MetaMorph.
Confocal imaging
Lungs were embedded in OCT medium, and sections were prepared as described previously (8). Lung cells positive for WT1 [rabbit anti-WT1 antibody: clone CAN-R9 (IHC)-56-2; Abcam] were co-localized with vimentin, αSMA, and GFP by immunofluorescence staining using a rabbit anti-vimentin antibody (clone 3B4; Millipore, Billerica, MA, USA) and mouse monoclonal anti-αSMA antibody (clone 1A4 A5228; Sigma-Aldrich). Appropriate secondary antibodies conjugated with Alexa-Fluor® 488, Alexa-Fluor® 594, or Alexa-Fluor® 647 (Invitrogen®, Life Technologies, Grand Island, NY, USA), respectively, were used for co-immunostaining. Confocal images were collected using a Nikon AIR-A1 laser-scanning confocal microscope (Nikon, Melville, NY, USA). A z-stack of optical sections, 10 μm in total thickness, was captured from each lung tissue section and five 3D images were obtained per mouse. 3D-volume rendering was performed to quantify the number of GFP-, vimentin-, and WT1-positive cells in the lung lesions using the surface tool in Imaris (version 4.2.0; Bitplane, South Windsor, CT, USA). The morphological criteria used to count cells positive for any two different fluorescent signals was with a maximum gap size of 1 μm in diameter between two signals. Data are reported as ± SEM of cell number of experimental groups. The total five images per mouse were used to quantify stromal cells in CCSP/− and CCSP/TGFα chimera mice on Dox for 4 wks with 4–6 mice per group.
WT1 siRNA transfection studies
Primary human or mouse fibroblast cells were transfected with stealth WT1 small interfering RNA (siRNA) [human WT1 siRNA (Hss111388, Invitrogen) or mouse WT1 siRNA (MSS212628, Invitrogen) or stealth control siRNA (human - sc37007, Santa Cruz Biotechnology Inc., Dallas, TX, USA or mouse - 12935-200, Invitrogen)] using the Lipofectamine 3000 Transfection kit (Invitrogen) according to the manufacturer’s instructions. Primary lung-resident mesenchymal cells were separated from fibrocytes using anti-CD45 magnetic beads as described previously (8) and grown on 12-well plates to 90% confluence. Cells were transfected with siRNA using OptiMEM media containing no antibiotics. Transfected cells were harvested 72 h post transfection and used for RNA isolation and gene-expression analysis.
RNA preparation and RT-PCR
Total RNA was extracted from lung homogenates or cells using the RNAeasy Mini Kit (Qiagen Sciences, Valencia, CA, USA) as described previously (37). Extracted RNA was used in real-time polymerase chain reaction (RT-PCR) assays performed with the CFX384 Touch Real-Time PCR detection system (Bio-Rad, Hercules, California, USA). The relative quantities of mRNA for several genes were determined using iTaq™ universal SYBR green supermix (Bio-Rad) or Taqman® gene expression master mix (Applied Biosystems®, Life Technologies, Grand Island, New York, USA). Target-gene transcripts in each sample were normalized to hypoxanthine guanine phosphoribosyltransferase, 18s rRNA, or β-actin and expressed as a relative increase or decrease compared with control. All real-time primer sequences used are shown in Table 1.
Table I.
Primers and probes used for RT-PCR
| Gene | Forward | Reverse |
|---|---|---|
| Human Primers | ||
| β-ACTIN | CCAACCGCGAGAAGATGA | CCAGAGGCGTACAGGGATAG |
| WT1 | AGCTGTCCCACTTACAGATGC | CCTTGAAGTCACACTGGTATGG |
| Mouse Primers | ||
| HPRT | GCCCTTGACTATAATGAGTACTTCAGG | TTCAACTTGCGCTCATCTTAGG |
| WT1 | CAGATGAACCTAGGAGCTACCTTAAA | TGCCCTTCTGTCCATTTCA |
| MYCN | AGCACCTCCGGAGAGGATAC | CCACATCGATTTCCTCCTCT |
| IL-10 | CAGAGCCACATGCTCCTAGA | GTCCAGCTGGTCCTTTGTTT |
| TERT | AGAGCTTTGGGCAGAAGGA | GAGCATGCTGAAGAGAGTCTTG |
| THBS4 | AGACGCCTGTGATGACGAC | TGGGACAGTTGTCCAAAATG |
| COL1α | AGACATGTTCAGCTTTGTGGAC | GCAGCTGACTTCAGGGATG |
| COL3α | TCCCCTGGAATCTGTGAATC | TGAGTCGAATTGGGGAGAAT |
| COL5α | CTACATCCGTGCCCTGGT | CCAGCACCGTCTTCTGGTAG |
| FN1 | CGGAGAGAGTGCCCCTACTA | CGATATTGGTGAATCGCAGA |
| Human Probes | Catlog # | |
| 18s rRNA | Applied Biosystems® Life Technologies | |
| WT1 | Life Technologies™ | Hs01103751 |
| COL1A1 | Life Technologies™ | Hs00164004 |
| COL3A1 | Life Technologies™ | Hs00943809 |
| FN1 | Life Technologies™ | Hs00365052 |
Whole-transcriptome shotgun sequencing (RNA-Seq) and heat maps
The lungs of CCSP/− and CCSP/TGFα mice on Dox for 3 wks were collected in RNA-later solution (Invitrogen), and total RNA was prepared as described. From each experimental group, three lung-tissue samples were sequenced using an Illumina HiSeq-1000 Sequencer (Illumina Inc., San Diego, CA, USA) as described previously (37). To identify differentially expressed genes between the lungs of CCSP/− and CCSP/TGFα mice on Dox for 3 wks, statistical analysis was performed using the DESeq Bioconductor package (38) that utilizes a statistical model based on negative-binomial distribution of the read counts. Statistically significant genes were selected based on a P-value cut-off of 0.05 as well as a two-fold change (up or down), resulting in 1783 genes in total. Using this set of significant genes, hierarchical clustering on the log-transformed read counts normalized for different lengths of gene-coding regions (RPKM values) (39) was performed for the CCSP/− or CCSP/TGFα groups, and a heat map was generated. Genes of the WT1 network were identified using an analysis tool available in the Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA, USA). Complete RNA-Seq data are available at a gene expression omnibus or GEO database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66634; accession number: GSE66634).
Western-blot analysis
Immunoblotting and quantification was performed using the volume integration function on the Phosphor Imager software, Image quant 5.2 (Molecular Dynamics, Piscataway, NJ, USA) as described previously (37). Primary antibodies used included rabbit anti-WT1 antibody (1:1000; SC-192, Santa Cruz Biotechnology Inc.) and anti-glyceraldehyde 3-phosphate dehydrogenase antibody (1:1000, A300–641A, Bethyl Labs, Montgomery, TX, USA). Appropriate secondary antibody was conjugated with peroxidase (1:1000).
Statistics
All data were analyzed using Prism (version 5; GraphPad, La Jolla, CA, USA). One-way ANOVA with Sidak’s Multiple Comparison post-test was used to compare different experimental groups. Student’s t-test was used to compare between two experimental groups. Data were considered statistically significant for P values less than 0.05.
RESULTS
WT1-positive cells accumulate in the subpleural fibrotic lesions of TGFα mice
We previously have shown that the majority of mesenchymal cells in fibrotic lesions, including those in the subpleural regions, originates from lung-resident mesenchymal cells yet to be identified (8). To determine the lung-resident mesenchymal cells that accumulate in fibrotic lung lesions, total lung transcriptome analysis was performed on lung homogenates from CCSP/− control and CCSP/TGFα mice on Dox for 3 wks using next-generation sequencing (n=3). As shown using heat-map analysis, there were two clusters of differentially-expressed genes in the fibrotic lungs of the TGFα mice compared to control mice (Supplemental Figure 1). The lists of genes in Cluster 1 and Cluster 2 represent genes that are up regulated (926 genes) or down regulated (857 genes) by approximately two fold or more following TGFα overexpression for 3 wks. We analyzed the genes that were differentially expressed in fibrotic lungs to identify transcription factor-driven gene networks of lung mesenchymal cells that were selectively activated in TGFα mice compared to control mice using Ingenuity Pathway Analysis (37). The WT1-regulated gene network was markedly activated in the fibrotic lungs of TGFα mice (Supplemental Figure 1). WT1-network genes with the largest increases in Cluster 1 included IL10, MYCN, TERT, and THBS4 (Supplemental Figure 1), which are known to be involved in the maintenance of mesenchymal-cell proliferation and deposition of ECM proteins (40–44). To confirm increases in WT1-regulated genes, we quantified the transcripts for IL10, MYCN, TERT, and THBS4 using RT-PCR and observed significant in vivo increases in mice with TGFα overexpression for 4 wks (Supplementary Figure 1). We determined the kinetics of in vivo WT1 expression in mice with TGFα overexpression versus normal lungs and observed a progressive increase in WT1 transcripts between Day 4 and Week 4 during TGFα-induced fibrosis (Figure 1A). To identify whether WT1-expressing cells accumulate in the fibrotic lung lesions of TGFα mice, lung sections from CCSP/− control and CCSP/TGFα mice on Dox for 2 or 4 wks were immunostained for WT1 antigen. We observed increased staining for WT1 in the nucleus of cells localized in the subpleural fibrotic lesions of TGFα mice on Dox at Weeks 2 and 4, but limited or no immustaining in the lungs of control mice (Figure 1B). To further confirm increases in WT1, the lung subpleura of mice on Dox for 4 wks were minced and cultured for 5 days to isolate total lung mesenchymal-cell RNA, and WT1 transcripts were quantified. We found a significant increase in WT1 transcripts in the lung mesenchymal cells isolated from fibrotic lesions in TGFα mice compared to control mice (Figure 1C). We previously have shown that cultured lung mesenchymal cells contain two major cell subsets defined as fibrocytes (CD45+Col1+) and lung-resident mesenchymal cells (CD45−Col1+) based on surface expression of the hematopoietic marker CD45 (8). To assess whether WT1 was selectively expressed in lung-resident mesenchymal cells, we separated fibrocytes from lung-resident mesenchymal cells from the cultured lungs of CCSP/TGFα mice on Dox for 4 wks. We observed a significant increase in WT1 transcripts in the lung-resident mesenchymal cells compared to fibrocytes isolated from TGFα mice (Figure 1D). To substantiate our hypothesis that WT1 is selectively expressed in lung-resident mesenchymal cells, but not fibrocytes, we performed immunoblots for WT1 in fibrocytes and lung-resident mesenchymal cells isolated from the lung mesenchymal-cell cultures of CCSP/TGFα and CCSP/− mice. We found that WT1 was selectively induced in the lung-resident mesenchymal cells isolated from fibrotic lesions compared to control lungs, but there was no staining for WT1 in the fibrocytes (Figure 1E).
Figure 1. Wilms’ tumor 1 (WT1)-positive cells accumulate in the subpleural fibrotic lung lesions of TGFα mice.

(A) WT1 transcripts were quantified by RT-PCR in the lungs of CCSP/− or CCSP/TGFα transgenic mice fed doxycyclin (Dox) food for 4 days or 2, 3, and 4 wks (n=6). (B) Immunostaining of lung sections with anti-WT1 antibodies shows progressive accumulation of WT1-expressing cells in the subpleura of CCSP/TGF-α mice on Dox for 2 and 4 wks compared to CCSP/− mice on Dox for 4 wks. Images are representative of n=4 per group. Scale bar, 50 μm. (C) WT1 transcripts were quantified by RT-PCR in the total RNA of lung mesenchymal cell cultures from CCSP/− and CCSP/TGFα mice fed on Dox food for 4 wks. (D) The total lung mesenchymal cells were separated into fibrocytes (CD45+Col1+) and resident mesenchymal cells (CD45−Col1+) using ant-CD45 magnetic beads from lung mesenchymal-cell cultures of CCSP/− or CCSP/TGF-α mice on Dox for 4 wks, and WT1 transcripts were quantified by RT-PCR. (E) The lysates of fibrocytes (CD45+Col1+) and resident mesenchymal cells (CD45−Col1+) from CCSP/− or CCSP/TGF-α mice on Dox for 4 wks were blotted with anti-WT1 antibodies and quantified using the Phosphor Imager software, and values were normalized with GAPDH control. One-way ANOVA with Sidak’s multiple comparisons test was used to measure significant differences between groups. **P<0.005, ***P<0.0005, ****P<0.0001 Results are representative of two or more independent experiments.
WT1-positive cells accumulate in the subpleural fibrotic lesions of human IPF
We assessed whether increased WT1 expression detected in the mouse model is a feature of fibrosis in human IPF by analyzing lung sections from patients with IPF (n=6) and non-IPF patients (n=4). We performed double immunostainings on serial lung sections to visualize the localization patterns of WT1, vimentin, and cytokeratin. This allowed us to visualize WT1-positive cells that co-express mesothelial (cytokeratin) and/or mesenchymal (vimentin) cell markers. In contrast to normal lung, IPF lung tissue exhibited prominent staining for WT1 in cells populated in the thickened subpleural areas of the lung (Figure 2B). The high-magnification images demonstrated that WT1 was localized in the nuclear regions of lung cells of IPF lungs, but there was limited or no WT1 staining in the normal non-IPF lungs (Figure 2A). The antibody-control slides using mouse and rabbit IgG showed no detectable staining (data not shown). In IPF lungs, WT1-expressing cells were co-localized with staining for vimentin and/or cytokeratin (Figure 2B). In IPF lung tissues, the majority of mesothelial cells (cytokeratin-positive) were co-localized with WT1 in the nuclei, but their location was restricted to pleural surfaces with no staining for cytokeratin in the subpleural regions of IPF lungs (Figure 2B). However, the thickened subpleural region of the IPF lungs did display WT1-positive cells that co-localized with vimentin, but not cytokeratin (Figure 2B). Thus, the thickened pleura of IPF lungs, but not normal lung, had an accumulation of two major subsets of WT1-positive cells, where WT1 was expressed by both mesothelial and mesenchymal cells. Moreover, increased immunostaining for WT1 in both mesothelial and mesenchymal cells demonstrated overexpression of WT1 in IPF lungs.
Figure 2. Wilms’ tumor 1 (WT1)-positive cells accumulate in the subpleural fibrotic lesions of lungs in patients with idiopathic pulmonary fibrosis (IPF).

Mesothelial and mesenchymal cells are the major lung cell types that express WT1 in the subpleural fibrotic lesions of the human IPF lung. Serial lung sections from (A) non-IPF (n=4) and (B) IPF (n=6) patients were co-immunostained with antibodies against either cytokeratin (red, indicates mesothelial cells) and WT1 (brown) or vimentin (red, indicates mesenchymal cells) and WT1 (brown). All images were collected at 20X (scale bars, 100 μm) and 40X (scale bars, 20 μm) magnification. The dashed boxes in the 20X images indicate the areas highlighted in the 40X images.
To observe whether WT1 expression was increased during interstitial fibrotic lung diseases in humans, we extracted data from the mRNA expression catalogue available in the Lung Genomic Research Consortium database and confirmed WT1 overexpression in individuals with interstitial lung disease compared to controls (Supplementary Figure 2). To further substantiate that WT1 is overexpressed in lung-resident mesenchymal cells of human IPF lungs, we cultured the subpleural biopsies of human IPF and control non-IPF lungs (n=2). The subpleural pieces of the lungs were minced and cultured for 2 wks to obtain primary lung mesenchymal cells. We separated fibrocytes from lung-resident mesenchymal cells using anti-CD45 magnetic beads. WT1 transcripts were quantified in the lung-resident mesenchymal cells from primary cultures of IPF and control non-IPF lungs. Notably, WT1 transcripts were significantly increased in lung-resident mesenchymal cells of IPF compared to control non-IPF lungs (Supplementary Figure 2).
Fibrocytes do not transform into WT1-positive cells in pulmonary fibrosis
To further substantiate our findings that WT1-positive cells and fibrocytes are two distinct mesenchymal cell subsets that accumulate in vivo in subpleural fibrotic lesions, GFP-expressing BM cells were transferred into lethally irradiated recipient CCSP/− control and CCSP/TGFα mice (8). At 9 wks after BM transplantation, the majority of CD45+ cells in the lungs of both strains of mice were GFP postive (8). CCSP/− and CCSP/TGFα GFP chimera mice were then placed on Dox for 4 wks. Our previous findings have demonstrated an approximately 3- to 4-fold increase in GFP-positive fibrocytes, including in the subpleural fibrotic lesions, in CCSP/TGFα mice compared with controls (8). For this study, lung sections from chimeric mice were immunostained for WT1 and vimentin to construct 3D images and count cells based on GFP, WT1, and vimentin expression. Consistent with our previous findings, we observed a significant increase in vimentin-positive cells in CCSP/TGFα chimera mice compared to CCSP/− controls on Dox for 4 wks (Figure 3A and 3B). More than 20% of cells were co-localized with WT1, suggesting that WT1-positive cells are a sizable subset of total lung-resident mesenchymal cells in the subpleural fibrotic lesions of TGFα mice (Figure 3C). Notably, WT1 staining was restricted to the nuclear regions of GFP-negative cells, and there also was no staining for WT1 in the GFP-positive fibrocytes. Together, our immunostaining results for WT1 demonstrated that WT1-positive cells and fibrocytes are two distinct subsets of the lung mesenchymal cells that accumulate in the subpleural fibrotic lesions of the TGFα mice compared to control mice.
Figure 3. Fibrocytes do not transform into Wilms’ tumor 1 (WT1)-positive cells in the subpleural fibrotic lung lesions of TGFα Mice.
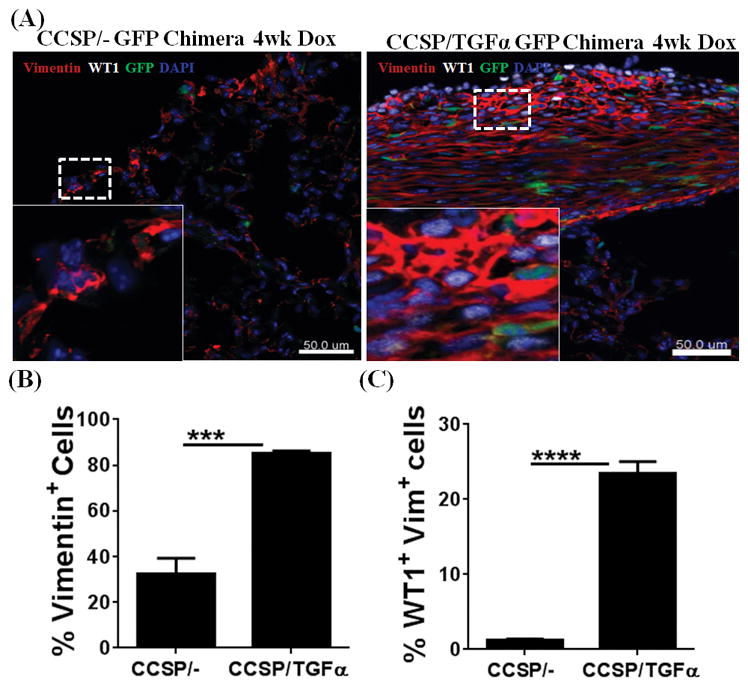
GFP-chimera mice were generated by transplanting 3×106 bone marrow (BM) cells from EGFP-transgenic mice into lethally-irradiated (11.75Gy) CCSP/− and CCSP/TGFα recipients. BM cells were harvested from the tibia and femur of the EGFP donor mice by flushing the bones with Dulbecco’s PBS under aseptic conditions and washing by centrifugation (5 min at 1,000 x g, 4°C). (A) Lung sections from CCSP/− GFP and CCSP/TGFα GFP chimera mice fed doxycyclin (Dox)-treated food for 4 wks were stained with antibodies for WT1 (white) and vimentin (red). The dashed box indicates the enlarged area. Images are representative of n=5 per group. Scale bar, 50 μm. (B) Quantification of the total vimentin-positive cells in the subpleural regions of CCSP/− GFP chimera and CCSP/TGFα chimeric mice using Imaris version 7.2.3 (n=5). (C) Quantification of the total WT1-positive cells from the subpleural regions of CCSP/− GFP chimera and CCSP/TGFα chimeric mice (n=5). Data shown are means ± SEM. Unpaired Student’s t-test was performed to measure the significance. ***P<0.0005, ****P<0.0001
WT1-positive cells accumulate in the lung during bleomycin-induced fibrosis
To substantiate our hypothesis that WT1-positive cells accumulate in the fibrotic lungs, we developed an alternative mouse model of bleomycin-induced pulmonary fibrosis. For these experiments, mice were injected i.d. with bleomycin for 4 wks. Lung sections stained with hemotoxylin and eosin demonstrated a significant increase in lung inflammation in the bleomycin-treated mice compared to saline-treated control mice (Supplementary Figure 3). Similarly, bleomycin invoked fibrotic responses in the airways, interstitium, and pleural regions of the lungs in bleomycin-treated mice (Supplementary Figure 3). Masson trichrome-stained lung sections revealed pleural thickening that was modest but consistent across the lung pleura during bleomycin-induced pulmonary fibrosis, which was associated with increased hydroxyproline levels (Supplementary Figure 3). In contrast, when bleomycin is administered via the intratracheal route, mice develop severe lung injury marked by excessive tissue inflammation and fibrosis, but with limited or no significant changes in pleural thickening and lung mechanics, including resistance and tissue elastance (45–47). Notably, mice injected i.d. with bleomycin for 4 wks displayed a decline in lung function with a greater than two-fold increase in resistance and tissue elastance compared to saline-treated control mice (Supplementary Figure 3). As observed in the TGFα model, thickened lung pleura was associated with a significant increase in the number of WT1-positive cells. However, WT1 expression was restricted to the cells localized in the lung pleura during bleomycin-induced pulmonary fibrosis (Figure 4A and B). We also generated Tamoxifen-inducible double transgenic WT1CreERT2/ROSA26-loxP-stop-loxP-tdTomato mice (WT1CreERT2/+ROSAmTmG/+) and used this genetic knock-in mouse model to identify genetic lineage of WT1-expressing cells. These mice express membrane red-fluorescent (“mT”) protein prior to Cre excision and membrane green-fluorescent (“mG”) protein following tomaxin-inducible CreERT2 activation (48). In support of our hypothesis, we observed increased GFP-expressing WT1-positive cells in the pleura of bleomycin-treated mice compared to saline-treated control mice (Figure 4C).
Figure 4. Wilms’ tumor 1 (WT1)-positive cells accumulate in the lungs during bleomycin-induced pulmonary fibrosis.

Bleomycin was administered via intradermal injections (100 μg/day; 5 days/week) for 4 wks and lungs were assessed for WT1-positive cells. (A) The lung sections were immunostained for WT1 antigen (brown), and the dotted box indicates the enlarged area. All images are representative of n=4 per group. Scale bar, 50 μm. (B) The number of WT1-positive cells were quantified for each lung section in bleomycin- and saline-treated control mice (n=4/group). Data shown are means ± SEM. Statistical significance between groups was measured using an unpaired Student’s t-test. *P<0.05, ***P<0.0005, ****P<0.0001 (C) WT1CreERT2/+ROSAmTmG/+ mice were injected intradermally with saline or bleomycin (100 μg/day; 5 days/wk) for 4 wks, and Cre-mediated recombination was induced by two doses of intraperitoneal Tamoxifen at Week 4 (2.5 mg/mice; Days 26–27). Images are representative of n=3 per group. The dashed box in the 20X images indicates the area highlighted in the 60X images. Scale bars, 200 μm.
Fibrocytes augment accumulation of WT1-positive cells in severe fibrotic lung disease
Previous studies from our laboratory and others have demonstrated that fibrocytes traffic to fibrotic lung lesions and increase collagen deposition and thickening of fibrotic lesions in the subpleura and adventitial lung regions of TGFα mice and other mouse models of pulmonary fibrosis (8, 32). To determine whether fibrocytes influence accumulation of WT1-positive cells in the progressive expansion of fibrotic lung lesions, adoptive cell-transfer experiments were performed. Fibrocytes (CD45+Col1+) were infused into the tail vein of CCSP/TGFα mice on Dox for 2 wks, and fibrotic lesions were assessed for the accumulation of WT1-positive cells at Day 7 after cell transfer. Transfer of fibrocytes resulted in significant increases in the number of WT1-positive cells in CCSP/TGFα mice compared with CCSP/− and CCSP/TGFα mice receiving saline (Figure 5A). Our previous studies demonstrated that transfer of fibrocytes augmented thickening of the subpleural fibrotic lesions and lung function decline in CCSP/TGFα mice infused with fibrocytes compared with CCSP/− and CCSP/TGFα mice that received saline (8). For this study, the number of WT1-positive cells in the subpleural regions were quantified using Metamorph image analysis software to determine whether this cell population was correlated with the thickening of the lung pleura. Fibrocyte infusion resulted in a significant increase in the number of WT1-positive cells in CCSP/TGFα mice compared with CCSP/− and CCSP/TGFα mice receiving saline (Figure 5B). Pearson correlation with linear regression analysis indicated a linear correlation between increased subpleural thickness and WT1-positive cell accumulations in CCSP/TGFα mice infused with either saline or fibrocytes (Figure 5C; r2=0.8198, P<0.005). Similarly, a linear correlation was observed between the number of WT1-positive cells in the lung subpleura and total lung hydroxyproline levels (Figure 5D; n=6/group, r2=0.6076, P<0.005). To determine whether fibrocyte-driven WT1-positive cell accumulation has any positive effect on the WT1-regulated gene network, total lung transcripts were analyzed using RT-PCR. WT1-regulated gene networks were markedly activated in the fibrotic lungs of TGFα mice infused with fibrocytes compared with CCSP/− and CCSP/TGFα mice that received saline (Figure 6A–E). In particular, transcripts of MYCN, IL10, Col1α, THBS4, and TERT were significantly elevated in CCSP/TGFα mice infused with either saline or fibrocytes (Figure 6A–E). Taken together, our data suggest that increased sub-pleural thickness is associated with an increased WT1-positive population, which in turn is associated with disease severity as assessed by collagen deposition, lung function, and the WT1-regulated gene network in TGFα mice.
Figure 5. Adoptive cell transfer of fibrocytes augments accumulation of Wilms’ tumor 1 (WT1)-positive cells in the subpleura of fibrotic lung lesions during TGFα-induced pulmonary fibrosis.

CCSP/− and CCSP/TGFα mice were fed with doxycyclin (Dox)-treated food for 14 days and fibrocytes (0.7×106 cells/mice) or saline was infused intravenously via the tail vein. After fibrocyte infusion, mice were continued on Dox for another 7 days and lungs were harvested for analysis. (A) Immunostaining of lung sections with anti-WT1 antibodies. Images are representative of n=4–6 per group. Scale bar, 50 μm. (B) The number of WT1-positive cells per total cells in the lung pleura/sub-pleura was calculated by counting WT1-positive cells and total cells in pleura/sub-pleural regions from five representative images per animal from CCSP/− and CCSP/TGF-α mice (n=4–6/group). Data shown are means + SEM. Statistical significance between groups was measured using One-way ANOVA with Sidak’s multiple comparison test. ****P<0.0001 (C) The correlation was calculated between the number of WT1-positive cells and subpleural thickness using Pearson correlation coefficient with linear regression analysis (r2=0.8198; P<0.0001; n=4–6 per group). (D) The correlation was calculated between the number of WT1-positive cells and hydroxyproline levels of the total lung using Pearson correlation coefficient with linear regression analysis (r2=0.6076; P<0.005; n=4–6 per group).
Figure 6. Adoptive cell transfer of fibrocytes augments Wilms’ tumor 1 (WT1)-regulated gene expression during TGFα induced fibrosis.
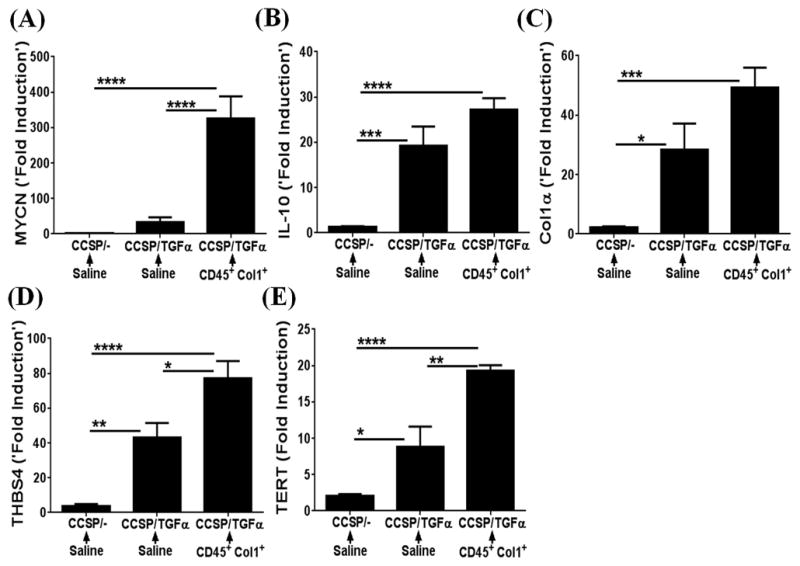
CCSP/− and CCSP/TGFα mice were placed on doxycycline (Dox)-treated food for 14 days and fibrocytes (0.7×106 cells/mice) or saline was infused intravenously via the tail vein. After fibrocyte infusion, mice were continued on Dox for another 7 days and lungs were harvested to quantify WT1-regulated network of genes by RT-PCR. The transcripts of WT1-regulated network genes (A) MYCN, (B) IL-10, (C) Col1α, (D) THBS4, and (E) TERT are shown as the fold induced obtained by normalizing the gene expression to a hypoxanthine guanine phosphoribosyl transferase (HPRT) control. Data shown are mean + SEM values (n=4–6/group). Statistical significance between groups was measured using One-way ANOVA with Sidak’s multiple comparison test. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001.
To further substantiate our hypothesis that fibrocytes contribute to WT1-positive cell accumulation and subpleural thickening, we intravenously transferred purified populations of TGFα-induced fibrocytes or saline on Day 14 into wild-type (FVB/NJ background) mice that were treated i.d. with bleomycin or saline for a total of 28 days. Bleomycin-treated mice that received TGFα-induced fibrocytes had an increased collagen deposition and accumulation of WT1-positive cells in the subpleural regions of the lung compared with mice treated with saline or bleomycin (Figure 7A and 7B). The total lung hydroxyproline levels were increased in bleomycin-treated mice compared with saline-treated mice, which was further augmented by the transfer of TGFα-induced fibrocytes (Figure 7C). Fibrocytes infused into saline-treated wild-type mice had no effect on collagen staining and hydroxyproline levels. We also observed a significant increase in resistance and elastance with the transfer of fibrocytes into mice during bleomycin-induced pulmonary fibrosis (Figure 7D and E). Notably, adoptive cell transfer of TGFα-induced fibrocytes into saline-treated mice was not sufficient to alter lung function or the number of WT1-positive cells in the lung pleura compared to i.d. saline-treated mice (Figure 7D–F). Moreover, similarly to the CCSP/TGFα model, we observed a linear correlation between the WT1-positive cell population in the lung pleura and total lung hydroxyproline levels (Figure 7G; r2=0.6125, P<0.05).
Figure 7. Adoptive cell transfer of fibrocytes augments accumulation of Wilms’ tumor 1 (WT1)-positive cells and fibrotic lung disease during bleomycin-induced pulmonary fibrosis.

FVB/NJ mice were injected intradermally with saline or bleomycin (100 μg/day; 5 days/week) for 2 wks. Mice were then infused with TGFα-induced fibrocytes isolated from mesenchymal-cell lung cultures of CCSP/TGFα mice fed doxycyclin (Dox)-treated food for 4 wks, and mice were continued on saline or bleomycin for another 2 wks. (A) Masson trichrome staining demonstrates increased collagen deposition in the subpleural areas and also interstitium of mice infused with fibrocytes and bleomycin-treated compared to saline- or bleomycin-treated mice. (B) The lung sections immunostained for WT1 antigen show increased accumulation of WT1-positive cells in the subpleural surfaces of mice infused with fibrocytes and bleomycin-treated compared to saline- or bleomycin-treated only mice. The dotted box indicates the enlarged area. (C) Total lung hydroxyproline levels were increased in mice infused with fibrocytes and bleomycin-treated compared to saline or bleomycin-treated only mice. (D) The resistance of airways was increased in mice infused with fibrocytes and bleomycin-treated compared to saline- or bleomycin-treated mice. (E) Tissue elastance was increased in mice infused with fibrocytes and bleomycin-treated compared to saline- or bleomycin-treated only mice. (F) The number of WT1-positive cells in the lung subpleura was significantly increased in mice infused with fibrocytes and bleomycin-treated compared to saline- or bleomycin-treated only mice. (G) The correlation was calculated between the number of WT1-positive cells and hydroxyproline levels of the lung using Pearson correlation coefficient with linear regression analysis (r2=0.6125; P<0.05). Data shown are means + SEM. Statistical significance was calculated using One-way ANOVA with Sidak’s multiple comparison. Data are cumulative of two independent experiments with similar results (n=6–8/group). *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001
WT1 is a critical regulator of ECM gene expression by lung mesenchymal cells
Myofibroblasts are the main effectors of fibrosis in all tissues, including the lung, and contribute to the excessive ECM deposition in the fibrotic lesions of human IPF (19, 49–52). To investigate WT1 expression in myofibroblasts that accumulate in the lung subpleura of human IPF lungs, dual stainings for WT1 and αSMA (myofibroblast marker) were performed on lung sections from IPF and control non-IPF lungs. Consistent with no staining for WT1 in normal lungs, the control non-IPF lungs stained negative for WT1 and had limited positive staining for αSMA in perivascular areas of the lung. In IPF lungs, WT1-positive cells were co-localized with cytoplasmic αSMA in the subpleural fibrotic lesions (Figure 8A). To investigate WT1 expression in myofibroblasts that accumulate in the lung subpleura of TGFα mice, dual stainings for WT1 and αSMA were performed on lung sections from CCSP/− and CCSP/TGFα transgenic mice on Dox for 6 wks, when fibrosis was established. Consistent with no staining for WT1 in normal lungs, the lung pleura of CCSP/− mice stained negative for WT1 and also had limited staining for αSMA. In CCSP/TGFα mice, WT1-positive cells were co-localized with cytoplasmic αSMA in the subpleural fibrotic lesions (Figure 8B). Importantly, CCSP/TGFα mice at 6 wks on Dox showed two distinct populations of WT1-positive cells. In particular, elongated and spindle-shaped cells that were positive for both αSMA and WT1 were predominatly localized in the mature surfaces of the subpleural regions, whereas WT1-positive cells with limited or no staining for αSMA were localized in the subpleural lesions that interface with lung parenchyma in CCSP/TGFα mice (Figure 8B). Together, the immunostaining results suggest that WT1 is expressed by both myofibroblasts and vimentin-positive mesenchymal cells of the lung subpleura of CCSP/TGFα transgenic mice and human IPF lungs.
Figure 8. Wilms’ tumor 1 (WT1) is expressed by myofibroblasts and is a critical regulator of extracellular matrix (ECM) gene expression.

(A) Lung sections of non-IPF and IPF patients were co-immunostained with antibodies against αSMA (red) and WT1 (brown). Images are representative of n=4 per group. Scale bar, 50 μm. (B) Immunofluorescence staining for WT1 and αSMA on lung sections of CCSP/− and CCSP/TGFα mice on Dox for 6 wks. The dashed box with the number one is used to indicate the subpleural area with cells positive for both WT1 and αSMA (WT1-positive myofibroblasts). The dashed box with the number two is used to indicate the subpleural area with the cells positive for WT1, but limited or no staining for αSMA. Images are representative of n=4 per group. Scale bar, 50 μm. (C) Primary lung-resident mesenchymal cells were isolated from the subpleural lung mesenchymal cell cultures of human IPF by negative selection using anti-CD45 magnetic beads and transfected with either control or WT1 siRNA for 72 h. The transcripts for WT1 and ECM genes, including Col1α, Col3α1, and FN1, are shown as the fold induced by normalizing to 18s rRNA control. Data shown are mean + SEM values (n=2). Statistical significance between groups was measured using an unpaired Student’s t-test. *P<0.05, **P<0.005 (D) Primary lung-resident mesenchymal cells were isolated from subpleural lung mesenchymal-cell cultures of CCSP/TGFα mice on Dox 4 wks by negative selection using anti-CD45 magnetic beads and transfected with either control or WT1 siRNA for 72 h. The transcripts for WT1 and ECM genes, including Col3α, Col5α, and FN1, are shown as the fold induced by normalizing to a hypoxanthine guanine phosphoribosyl transferase (HPRT) control. Data shown are mean + SEM values (n=4/group). Statistical significance between groups was measured using an unpaired Student’s t-test. *P<0.05, **P<0.005
To assess whether increased WT1 expression is merely associated with or actually plays an important role in regulating ECM gene expression and contributing to fibrosis, we performed an in vitro knockdown of WT1 and assessed changes in ECM gene transcripts. We cultured the subpleural fibrotic lesions of human IPF lung and separated lung-resident mesenchymal cells (CD45−Col1+) from fibrocytes by negative selection using anti-CD45 magnetic beads. Thus isolated primary lung-resident mesenchymal cells were transfected with WT1 or control siRNA for 72 h, and ECM gene expression was analyzed by RT-PCR. WT1 transcripts were effectively depleted in cells transfected with WT1-specific siRNA compared to control siRNA (Figure 8C). The major ECM genes, collagen 1α, collagen 3α, and FN1, were significantly decreased in human IPF cells transfected with WT1-specific siRNA compared to control siRNA (Figure 8C). To confirm that WT1 is a critical regulator of ECM genes in mesenchymal cells of TGFα mice, the lung pleura of TGFα mice on Dox for 4 wks were cultured to isolate lung-resident mesenchymal cells (CD45−Col1+) by negative selection using anti-CD45 magnetic beads. Thus isolated primary lung-resident mesenchymal cells were transfected with WT1 or control siRNA for 72 h, and ECM gene expression was analyzed by RT-PCR. WT1 transcripts were effectively depleted in cells transfected with WT1-specific siRNA compared to control siRNA (Figure 8D). The expression of major ECM genes, in particular collagen 3α, collagen 5α, and FN1, were significantly decreased in cells transfected with WT1-specific siRNA compared to control siRNA (Figure 8D).
DISCUSSION
Our study demonstrates that fibrocytes do accelerate subpleural thickening and pulmonary fibrosis when infused directly into the circulation of mouse models of TGFα- and bleomycin-induced fibrosis. Our mechanistic studies determined that fibrocytes contribute to the expansion of subpleural fibrotic lesions by activating WT1-positive cell accumulations in the fibrotic lung lesions of two different in vivo models of pulmonary fibrosis. Pulmonary mesothelial cells (PMC) are mesoderm-derived lung-resident mesenchymal cells that express both an epithelial cell-specific marker, cytokeratin, and a mesenchymal cell-specific marker, vimentin (53, 54). Previously, WT1 expression was thought to be limited to PMC of the lung (55); however, our immunostainings demonstrate that WT1 is overexpressed by both fibroblasts and myofibroblasts in human IPF lungs and in vivo models of pulmonary fibrosis. Notably, WT1 transcripts downregulation was sufficient to attenuate ECM gene expression in lung fibroblasts isolated from the fibrotic lungs of human IPF and TGFα mice. Thus, the findings from this study unequivocally demonstrate that WT1-positive fibroblasts and myofibroblasts are novel lung-resident mesenchymal cells in the fibrotic lungs of both human IPF and mouse models of pulmonary fibrosis. In the developing mouse lung, at embryonic stage E10.5, the majority of mesothelial cells has been shown to express WT1 (56). Later in life, mesothelial cells contribute to smooth-muscle cells in peri-vascular and peri-bronchial areas of the adult lung, but show no expression of WT1 (56). In agreement with this, we observed limited or no staining for WT1 in both normal adult human lungs and normal mouse lungs. A recent study by Karki and colleagues suggests that WT1 is necessary for the morphological integrity of the pleural membrane in normal lungs and the loss of WT1 contributes to IPF via mesothelial-to-mesenchymal transition (55). In contrast, the results of the present study suggest that WT1 transcripts are elevated in resident lung cells isolated from the subpleural lung mesenchymal cell cultures of human IPF compared to normal lungs. This is supported by the finding that WT1 expression occurs in PMC, fibroblasts, and myofibroblasts of human IPF lungs, but has limited or no expression in normal human lungs. Although these discrepancies in expression data for WT1 could be explained by variations in tissue sampling and also inflammation in subpleura and parenchymal areas of the lung in IPF. In support of, a recent study performed using 40 IPF subjects and 8 normal controls showed immune cell infiltrations and elevation of WT1 transcripts in IPF lungs compared to control samples (12).
Myofibroblasts are the major source for excessive ECM deposition in fibrotic lesions including in the lung pleura (19, 57, 58). For the first time, we demonstrate that myofibroblasts (αSMA positive) that accumulate in mature fibrotic lesions of the lung subpleura co-express WT1 in the nucleus in IPF lungs and TGFα mice on Dox for 6 wks. A recent study suggests that pleural mesothelial cells migrate into the lung parenchyma based on experiments for lineage tracking of WT1-positive cells with β-gal activation by Cre upon Tamoxifen injection (55). In particular, β-gal-stained WT1-positive cells were observed in large numbers on the pleural surface in control mice and migrated into the parenchyma with no significant change in number in the pleura within 4 hrs of TGFβ treatment (55). In contrast, our WT1 immunostainings suggest limited or no staining for WT1 on the pleural surfaces of adult normal mice. To further substantiate this finding, we used the heritable expression of the red fluorescent protein variant td-Tomato for lineage tags that are compatible with confocal analysis and require no staining with antibody. Further, the use of a fluorescent reporter has been shown to overcome limitations associated with X-gal staining using anti-βgal antibodies (7, 59). Notably, adult mice with reporter gene, when treated with both saline and Tamoxifen, had no GFP-positive cells in the lung pleura, whereas WT1-positive cells did accumulate in the lung pleural surfaces during bleomycin-induced pulmonary fibrosis. However, the lack of green cells in the parenchyma or adventitia of bleomycin-treated mice suggests that more careful evaluation of mesothelial origins in the progressive thickening of the lung subpleura and parenchyma is needed (55). Furthermore, a recent study on the adequacy of WT1-based epicardial fate mapping suggests that WT1creEGFP mediates sporadic and ectopic recombination resulting in highly variable patterns of green fluorescence when neither WT1 mRNA nor WT1 protein was detected (60). Our present study, using WT1creERT2 showed Cre activity by administration of Tamoxifen in the lung pleura, which was consistent with immunohistochemical detection of WT1-positive cells in bleomycin-treated mice compared to saline-treated control mice. Nonetheless, we do not know whether WT1-positive cells accumulate in the lung subpleura due to overexpression of WT1 by a specific growth factor or expansion of WT1-positive progenitors, or whether both of these contribute to the pool of WT1-positive cells. It is critical to identify the signals upregulated by TGFα or bleomycin that induce WT1 expression or the proliferation of WT1-positive progenitors in the subpleural fibrotic lung lesions. However, our findings do suggest that both mesothelial and mesenchymal cells upregulate WT1 in IPF and mouse models of pulmonary fibrosis. Recent fate-mapping studies using TGFα mice demonstrate limited transdifferentition of epithelial cells into myofibroblasts, which was also demonstrated with a mouse model of bleomycin-induced fibrosis (7, 19). Therefore, future studies are warranted that would examine mesothelial-to-myofibroblast transition in causing the progressive expansion of subpleural fibrotic lesions using complementary mouse models of pulmonary fibrosis.
Several clinical studies have demonstrated that the number of circulating fibrocytes in patients with IPF reflects fibrogenic activity and disease progression (24, 29, 61). Our recent findings demonstrated a progressive accumulation of BM-derived fibrocytes that expressed the monocyte-lineage marker CD14 and the tissue-invasive marker CD44 during TGFα-induced pulmonary fibrosis (8). To confirm that fibrocyte subsets of human IPF lung subpleura also express CD14 and CD44, we characterized fibrocyte subsets that selectively accumulate in the subpleural fibrotic lesions of human IPF lung using multicolor flow cytometry. Indeed, the major subsets of fibrocytes in the thickened lung subpleura of human IPF expressed both CD14 and CD44 (Supplemental Figure 4). In a recent study, we demonstrated that fibrocytes that accumulate in the lung subpleura have limited or no direct contribution to myofibroblasts, but do augment subpleural thickening and pulmonary fibrosis (8). Using a GFP-labeled BM transplant model, we also demonstrated that fibrocytes do not transform into WT1-positive lung-resident mesenchymal cells. This was further supported by immunoblots for WT1 in fibrocytes (CD45+Col1+) and lung-resident mesenchymal cells (CD45−Col1+) isolated from the lung mesenchymal cell cultures from TGFα transgenic mice on Dox for 4 wks. Overall, our results suggest that WT1-positive cells and fibrocytes are two distinct subsets that accumulate during the progressive thickening of the lung pleura. We therefore hypothesize that fibrocytes augment subpleural thickening and pulmonary fibrosis by altering WT1-positive cell accumulation during TGFα-induced fibrosis. Concurrently, using two different mouse models of pulmonary fibrosis, we have demonstrated that severe fibrotic lung disease is associated with fibrocyte-driven accumulation of WT1-positive cells. We provide data to suggest a previously unsuspected role for fibrocytes in causing WT1-positive cell accumulation as a downstream mediator of persistent lung fibrosis by production of collagen and other ECM proteins. In particular, knockdown of WT1 using siRNA resulted in a significant down regulation of ECM proteins, including Col 3α, Col 5α, and FN1. This phenomenon of WT1-regulated ECM gene expression is also observed in lung-resident fibroblasts of human IPF. Also, we have identified a network of WT1-regulated genes that show a progressive increase during TGFα-induced fibrosis in mice. In particular, WT1 has been shown to act as a transcription factor to regulate the transcription of multiple genes (MYCN, IL-10, TERT, and THBS4) involved in the diverse pro-fibrotic functions in fibroblasts, such as proliferation and enhanced survival under stress (40–44). In this study, we have revealed a unique role of fibrocytes as regulators that augment accumulation of WT1-positive cells, thereby increasing transcription of WT1-regulated genes in severe lung disease in mice. Our findings also support the likelihood that WT1 and its regulated network of genes play an important role in the pathogenesis of IPF. Interestingly, the blood of patients with interstitial lung disease is enriched with fibrocytes and IL-10 compared to healthy controls (62–64). Further, chronic overexpression of IL-10 has been shown to induce pulmonary fibrosis that involves fibrocyte recruitment and M2 activation via a CCL2/CCR2 axis (41, 65). Similarly, TERT mutations are the most common genetic defect found in familial pulmonary fibrosis (66, 67). TERT deficiency in mice has resulted in a significant protection against bleomycin-induced fibrosis, and fibroblasts displayed decreased proliferation and increased apoptosis rates with TERT deficiency (40). Additional studies involving fibrocytes, WT1, and WT1-regulated genes are now needed to identify the interplay among these cellular and molecular regulators of pulmonary fibrosis. In particular, studies are needed to establish whether WT1 expression in the subpleura of human IPF lungs indicates severity of fibrotic disease. Also, assessment of human IPF cells and their fiberoptic phenotypes with altered WT1 expression would facilitate establishment of the role of WT1 in the pathogenesis of IPF and identify the therapeutic relevance of targeting WT1 in IPF patients.
In summary, our study demonstrates accumulation of WT1-positive cells in the lung subpleura during progressive pulmonary fibrosis. Further, fibrocytes augment WT1-positive cell accumulations, and this supports an unique association of fibrocytes with severe lung disease in mice. Our findings demonstrate a positive association between accumulation of WT1-positive cells and subpleural thickening and collagen deposition. Furthermore, attenuation of WT1 expression resulted in a significant decrease in ECM gene transcripts by lung-resident fibroblasts of both murine and human IPF. The findings of this study elicit new questions and address limitations in the previous paradigm that WT1 expression is limited to mesothelial cells and functions as a negative regulator of mesothelial-to-myofibroblast differentiation in IPF (55). Moreover, our current findings provide impetus for future studies to identify the role of WT1 in mesenchymal cell phenotypes of human IPF and also test possible therapeutic options that will disrupt the functions of WT1 and fibrocytes in pulmonary fibrosis. Overall, our results provide novel insights into previously unrecognized roles for fibrocytes and WT1 in pulmonary fibrosis and the potential for these to be useful therapeutic targets.
Supplementary Material
Acknowledgments
Grant support: This research was supported by the National Institutes of Health grant, NIAMS 1R03AR062832 (SKM) and NHLBI U01HL111016 (ESW); by a Parker B. Francis Fellowship (SKM) and by American Heart Association grant 12SDG9130040 (SKM).
The authors thank the veterinary services at Cincinnati Children’s Hospital Medical Center for the care of mice used in this study and Cindy Davidson for help using MetaMorph imaging software. The authors also are grateful to Dr. A.P. Naren for his valuable suggestions and insights and J. Denise Wetzel, CCHMC Medical Writer, for editing of the manuscript.
Abbreviations used
- WT1
Wilms’ tumor 1
- IPF
idiopathic pulmonary fibrosis
- ECM
extracellular matrix
- CCSP
Club cell (Clara cell) specific protein-rtTA
- αSMA
alpha smooth muscle actin
- TGFα
transforming growth factor alpha
- TGFβ
transforming growth factor beta
- BM
bone marrow
- Dox
doxycycline
- DMEM
Dulbecco’s Modified Essential Medium
- i.d
intradermally
- siRNA
small interfering RNA
- GFP
green fluorescent protein
- PMC
pulmonary mesothelial cells
- Col1
type I collagen
References
- 1.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol. 2009;175:3–16. doi: 10.2353/ajpath.2009.081170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cool CD, Groshong SD, Rai PR, Henson PM, Stewart JS, Brown KK. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am J Respir Crit Care Med. 2006;174:654–658. doi: 10.1164/rccm.200602-205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutchison N, Fligny C, Duffield JS. Resident mesenchymal cells and fibrosis. Biochim Biophys Acta. 2013;1832:962–971. doi: 10.1016/j.bbadis.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navaratnam V, Fleming KM, West J, Smith CJ, Jenkins RG, Fogarty A, Hubbard RB. The rising incidence of idiopathic pulmonary fibrosis in the U. K Thorax. 2011;66:462–467. doi: 10.1136/thx.2010.148031. [DOI] [PubMed] [Google Scholar]
- 7.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madala SK, Edukulla R, Schmidt S, Davidson C, Ikegami M, Hardie WD. Bone marrow-derived stromal cells are invasive and hyperproliferative and alter transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2014;50:777–786. doi: 10.1165/rcmb.2013-0042OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleaveland KR, Velikoff M, Yang J, Agarwal M, Rippe RA, Moore BB, Kim KK. Fibrocytes are not an essential source of type I collagen during lung fibrosis. J Immunol. 2014;193:5229–5239. doi: 10.4049/jimmunol.1400753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DePianto DJ, Chandriani S, Abbas AR, Jia G, N’Diaye EN, Caplazi P, Kauder SE, Biswas S, Karnik SK, Ha C, Modrusan Z, Matthay MA, Kukreja J, Collard HR, Egen JG, Wolters PJ, Arron JR. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax. 2014 doi: 10.1136/thoraxjnl-2013-204596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DePianto DJ, Chandriani S, Abbas AR, Jia G, N’Diaye EN, Caplazi P, Kauder SE, Biswas S, Karnik SK, Ha C, Modrusan Z, Matthay MA, Kukreja J, Collard HR, Egen JG, Wolters PJ, Arron JR. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax. 2015;70:48–56. doi: 10.1136/thoraxjnl-2013-204596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz DA, Galvin JR, Yagla SJ, Speakman SB, Merchant JA, Hunninghake GW. Restrictive lung function and asbestos-induced pleural fibrosis. A quantitative approach. J Clin Invest. 1993;91:2685–2692. doi: 10.1172/JCI116507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider F, Gruden J, Tazelaar HD, Leslie KO. Pleuropulmonary pathology in patients with rheumatic disease. Archives of pathology & laboratory medicine. 2012;136:1242–1252. doi: 10.5858/arpa.2012-0248-SA. [DOI] [PubMed] [Google Scholar]
- 15.du Bois RM, Nathan SD, Richeldi L, Schwarz MI, Noble PW. Idiopathic pulmonary fibrosis: lung function is a clinically meaningful endpoint for phase III trials. Am J Respir Crit Care Med. 2012;186:712–715. doi: 10.1164/rccm.201206-1010PP. [DOI] [PubMed] [Google Scholar]
- 16.White ES, Mantovani AR. Inflammation, wound repair, and fibrosis: reassessing the spectrum of tissue injury and resolution. J Pathol. 2013;229:141–144. doi: 10.1002/path.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L741–749. doi: 10.1152/ajplung.00208.2003. [DOI] [PubMed] [Google Scholar]
- 18.Decologne N, Kolb M, Margetts PJ, Menetrier F, Artur Y, Garrido C, Gauldie J, Camus P, Bonniaud P. TGF-beta1 induces progressive pleural scarring and subpleural fibrosis. J Immunol. 2007;179:6043–6051. doi: 10.4049/jimmunol.179.9.6043. [DOI] [PubMed] [Google Scholar]
- 19.Hardie WD, Hagood JS, Dave V, Perl AK, Whitsett JA, Korfhagen TR, Glasser S. Signaling pathways in the epithelial origins of pulmonary fibrosis. Cell Cycle. 2010;9:2769–2776. doi: 10.4161/cc.9.14.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, Lee JY, Lee CM, Cho WK, Kang MJ, Koff JL, Yoon PO, Chae J, Park HO, Elias JA, Lee CG. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-beta-induced pulmonary fibrosis. J Biol Chem. 2012;287:41991–42000. doi: 10.1074/jbc.M112.356824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madala SK, Korfhagen TR, Schmidt S, Davidson C, Edukulla R, Ikegami M, Violette SM, Weinreb PH, Sheppard D, Hardie WD. Inhibition of the alphavbeta6 integrin leads to limited alteration of TGF-alpha-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2014;306:L726–735. doi: 10.1152/ajplung.00357.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 23.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson-Sjoland A, de Alba CG, Nihlberg K, Becerril C, Ramirez R, Pardo A, Westergren-Thorsson G, Selman M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–2140. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Wang CH, Huang CD, Lin HC, Huang TT, Lee KY, Lo YL, Lin SM, Chung KF, Kuo HP. Increased activation of fibrocytes in patients with chronic obstructive asthma through an epidermal growth factor receptor-dependent pathway. The Journal of allergy and clinical immunology. 2012;129:1367–1376. doi: 10.1016/j.jaci.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 26.Field JJ, Burdick MD, DeBaun MR, Strieter BA, Liu L, Mehrad B, Rose CE, Jr, Linden J, Strieter RM. The role of fibrocytes in sickle cell lung disease. PloS one. 2012;7:e33702. doi: 10.1371/journal.pone.0033702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaPar DJ, Burdick MD, Emaminia A, Harris DA, Strieter BA, Liu L, Robbins M, Kron IL, Strieter RM, Lau CL. Circulating fibrocytes correlate with bronchiolitis obliterans syndrome development after lung transplantation: a novel clinical biomarker. Ann Thorac Surg. 2011;92:470–477. doi: 10.1016/j.athoracsur.2011.04.065. discussion 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, Murray LA, Siner JM, Antin-Ozerkis DE, Montgomery RR, Reilkoff RA, Bucala RJ, Herzog EL. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. 2010;90:812–823. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, O’Byrne PM, Strieter RM, Kolb M. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588–594. doi: 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- 30.Moore BB, Kolb M. Fibrocytes and progression of fibrotic lung disease. Ready for showtime? Am J Respir Crit Care Med. 2014;190:1338–1339. doi: 10.1164/rccm.201411-2013ED. [DOI] [PubMed] [Google Scholar]
- 31.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleaveland KR, Moore BB, Kim KK. Paracrine functions of fibrocytes to promote lung fibrosis. Expert review of respiratory medicine. 2014;8:163–172. doi: 10.1586/17476348.2014.862154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, Gharib SA, Schnapp LM, Duffield JS. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188:820–830. doi: 10.1164/rccm.201212-2297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, Pu WT. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109–113. doi: 10.1038/nature07060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE, Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madala SK, Edukulla R, Phatak M, Schmidt S, Davidson C, Acciani TH, Korfhagen TR, Medvedovic M, Lecras TD, Wagner K, Hardie WD. Dual targeting of MEK and PI3K pathways attenuates established and progressive pulmonary fibrosis. PloS one. 2014;9:e86536. doi: 10.1371/journal.pone.0086536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 40.Liu T, Chung MJ, Ullenbruch M, Yu H, Jin H, Hu B, Choi YY, Ishikawa F, Phan SH. Telomerase activity is required for bleomycin-induced pulmonary fibrosis in mice. J Clin Invest. 2007;117:3800–3809. doi: 10.1172/JCI32369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barbarin V, Xing Z, Delos M, Lison D, Huaux F. Pulmonary overexpression of IL-10 augments lung fibrosis and Th2 responses induced by silica particles. Am J Physiol Lung Cell Mol Physiol. 2005;288:L841–848. doi: 10.1152/ajplung.00329.2004. [DOI] [PubMed] [Google Scholar]
- 42.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nature reviews Cancer. 2010;10:301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 43.Spinale FG. Cell-matrix signaling and thrombospondin: another link to myocardial matrix remodeling. Circulation research. 2004;95:446–448. doi: 10.1161/01.RES.0000142315.88477.42. [DOI] [PubMed] [Google Scholar]
- 44.Krady MM, Zeng J, Yu J, MacLauchlan S, Skokos EA, Tian W, Bornstein P, Sessa WC, Kyriakides TR. Thrombospondin-2 modulates extracellular matrix remodeling during physiological angiogenesis. The American journal of pathology. 2008;173:879–891. doi: 10.2353/ajpath.2008.080128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polosukhin VV, Degryse AL, Newcomb DC, Jones BR, Ware LB, Lee JW, Loyd JE, Blackwell TS, Lawson WE. Intratracheal bleomycin causes airway remodeling and airflow obstruction in mice. Experimental lung research. 2012;38:135–146. doi: 10.3109/01902148.2012.658595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. American journal of physiology Lung cellular and molecular physiology. 2008;294:L152–160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 47.Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 2010;207:535–552. doi: 10.1084/jem.20092121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 49.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. The American journal of pathology. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Habiel DM, Hogaboam C. Heterogeneity in fibroblast proliferation and survival in idiopathic pulmonary fibrosis. Frontiers in pharmacology. 2014;5:2. doi: 10.3389/fphar.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barkauskas CE, Noble PW. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. American journal of physiology Cell physiology. 2014;306:C987–996. doi: 10.1152/ajpcell.00321.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daste G, Serre G, Mauduyt MA, Vincent C, Caveriviere P, Soleilhavoup JP. Immunophenotyping of mesothelial cells and carcinoma cells with monoclonal antibodies to cytokeratins, vimentin, CEA and EMA improves the cytodiagnosis of serous effusions. Cytopathology: official journal of the British Society for Clinical Cytology. 1991;2:19–28. doi: 10.1111/j.1365-2303.1991.tb00380.x. [DOI] [PubMed] [Google Scholar]
- 54.LaRocca PJ, Rheinwald JG. Coexpression of simple epithelial keratins and vimentin by human mesothelium and mesothelioma in vivo and in culture. Cancer Res. 1984;44:2991–2999. [PubMed] [Google Scholar]
- 55.Karki S, Surolia R, Hock TD, Guroji P, Zolak JS, Duggal R, Ye T, Thannickal VJ, Antony VB. Wilms’ tumor 1 (Wt1) regulates pleural mesothelial cell plasticity and transition into myofibroblasts in idiopathic pulmonary fibrosis. FASEB J. 2014;28:1122–1131. doi: 10.1096/fj.13-236828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Que J, Wilm B, Hasegawa H, Wang F, Bader D, Hogan BL. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc Natl Acad Sci U S A. 2008;105:16626–16630. doi: 10.1073/pnas.0808649105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–1321. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- 58.Wynn TA. Integrating mechanisms of pulmonary fibrosis. The Journal of experimental medicine. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Current opinion in pulmonary medicine. 2012;18:517–523. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rudat C, Kispert A. Wt1 and epicardial fate mapping. Circ Res. 2012;111:165–169. doi: 10.1161/CIRCRESAHA.112.273946. [DOI] [PubMed] [Google Scholar]
- 61.Fujiwara A, Kobayashi H, Masuya M, Maruyama M, Nakamura S, Ibata H, Fujimoto H, Ohnishi M, Urawa M, Naito M, Takagi T, Kobayashi T, Gabazza EC, Takei Y, Taguchi O. Correlation between circulating fibrocytes, and activity and progression of interstitial lung diseases. Respirology. 2012;17:693–698. doi: 10.1111/j.1440-1843.2012.02167.x. [DOI] [PubMed] [Google Scholar]
- 62.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353:104–108. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 63.Alhamad EH, Cal JG, Shakoor Z, Almogren A, AlBoukai AA. Cytokine gene polymorphisms and serum cytokine levels in patients with idiopathic pulmonary fibrosis. BMC medical genetics. 2013;14:66. doi: 10.1186/1471-2350-14-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsoutsou PG, Gourgoulianis KI, Petinaki E, Germenis A, Tsoutsou AG, Mpaka M, Efremidou S, Molyvdas PA. Cytokine levels in the sera of patients with idiopathic pulmonary fibrosis. Respiratory medicine. 2006;100:938–945. doi: 10.1016/j.rmed.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 65.Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, Shanley TP. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L341–353. doi: 10.1152/ajplung.00122.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 67.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.