

Abstract
Asthma is a common inflammatory disease of airways, often associated with type2 responses triggered by allergens such as house dust mites (HDM). IL-25 is a key mucosal cytokine that may be produced by stressed epithelial cells; it rapidly activates innate lymphoid cells type2 (ILC2) to produce IL-13 and IL-5. When administered directly into lungs, IL-25 induces acute inflammation. However the mechanisms underlying IL-25-initiated inflammation and the roles of this cytokine in the context of HDM-induced allergic inflammation are not fully understood. We show here that lung resident conventional dendritic cells (cDCs) were direct targets of IL-25. IL-25-stimulated DCs rapidly induced mediators such as the chemokine CCL17, which in turn attracted IL-9-producing T cells. Importantly, these mechanisms also operated during HDM-induced allergic lung inflammation.
INTRODUCTION
Asthma is a chronic inflammatory disease of lungs, which can be acutely incited by allergens, among other insults. These triggers induce inflammatory mediators such as IL-25, IL-33 and/or TSLP, cytokines that tend to skew responses towards type2 (1). IL-25 (IL-17E) is a member of the IL-17 family of cytokines. Elevated levels of IL-25 have been correlated with poorly controlled asthma in patients (2, 3) and IL-25 contributes to allergen-induced allergic asthma-like inflammation in mice (4). Recently IL-25 has been implicated in rhinovirus-induced exacerbation of allergic lung inflammation in patients and mouse models (5).
Mucosal epithelial cells produce IL-25 upon encounter with helminthes, certain viruses and allergens (1, 5). Repeated exogenous pulmonary instillation of IL-25 is sufficient to cause an acute allergic asthma-like inflammation, in part due to rapid production of the type2 cytokines IL-13 and IL-5 by innate lymphoid type2 (ILC2) cells (6, 7). IL-25 may also target a not well-defined multi-potential myeloid precursor cell population (6), as well as Th9 and some NKT cells (8, 9).
In addition to IL-13 and IL-5, ILC2s may also transiently produce IL-9 after encounter with IL-25 (10, 11). IL-9 is the signature cytokine of Th9 cells, and has also been implicated in human asthma and allergen-induced lung inflammation in mice. Exposure to HDM during one week leads to the appearance of IL-9-producing T cells, prior to the arrival of Th2 cells (12, 13). IL-9 may help initiate and/or amplify type2 responses at mucosal sites, such as after infection with N.brasiliensis; it may promote accumulation of basophils and may act as a growth and survival factor of mast cells and ILC2s (10, 11, 13, 14).
IL-25 is the only member of the IL-17 cytokine family associated with type2 responses. It signals via a heteromeric receptor (common IL-17RA and proprietary IL-17RB) and the adaptor protein CIKS (a.k.a. Act1, Traf3ip2), which is also required for signaling by other IL-17 cytokines (15). Mice lacking this adaptor no longer respond to instillation of IL-25 into lungs (16, 17)
Despite much recent progress it remains not well understood how IL-25 initiates allergic lung inflammation and how it contributes in the context of allergen-induced inflammation. Herein we show that IL-25 targeted lung dendritic cells (DC) to induce mediators such as the chemokine CCL17, which was critical for rapid accumulation of IL-9-producing T cells. We further demonstrate that in an acute model of HDM-induced pulmonary inflammation, IL-9 production by T cells was also dependent on IL-25 signaling into DCs.
Materials and Methods
Mice
Ciksflox/− (18), Il17rbflox/− (Supplemental Figure 1A), Ccl17−/+ (kindly provided by I.Förster), Cd11c-Cre, Lck-Cre and LysM-Cre mice (Jackson Laboratories) (all on C57BL/6J backgrounds) were intercrossed to generate global (Ciks−/−, Il17rb−/−, and Ccl17−/−) and Cre-dependent conditional knockouts (see Supplemental Figures 1B–D) and littermate controls (flox/flox, or +/flox/Cre or +/−; all behaved like WT and are referred to as WT). Mice were bred and housed in NIAID Institute facilities and all experiments were done with approval of the NIAID Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
Experimentally-induced airway inflammation
Age-matched mice were anesthetized with isofluorane and challenged via intranasal (i.n.) inhalation of typically 25μl of PBS containing 1μg of rIL-25/mouse in single dose challenges, or 25μl containing 500ng per dose when challenged on 3 consecutive days (R&D Systems, Minneapolis, MN); PBS solvent was used in control inhalations. Alternatively, mice were challenged i.n. on days 0, 3 and 6 with 30μg per dose of house dust mites (HDM in saline, Greer Laboratories) or just saline for control inhalations.
Isolation of lung parenchymal cells
Mice were anesthetized with Avertin, exsanguinated, tracheae exposed and lungs lavaged with 0.5ml of PBS. Lungs were perfused with PBS and 1ml of Liberase DH: DNaseI (0.25:0.1 mg/ml; Roche) was installed through the trachea. Lung tissue was homogenized with gentleMACS (Miltenyi Biotech), following manufacturer’s protocol. Cells were ACK-lysed to remove erythrocytes, passed through 100μm strainer and centrifuged at 300xg for 5 min. Single cell suspensions were stained for flow cytometric analysis (see below). Alternatively, subpopulations were isolated with AutoMACSpro based on expression of CD11c, CD11b, MHC II, CD207, CD4 and/or CD45 (Miltenyi Biotec) following manufacturer’s instructions. Sorted cells were used for RNA analysis or stained for flow cytometric analysis.
Flow cytometric analysis
Antibodies used to stain total or sorted lung parenchymal cells are listed in Supplemental Figure 2C. For intracellular cytokine expression, bead-sorted CD45+ cells were cultured overnight in IMDM (Life Technologies)/10% FCS (Hyclone), stimulated (or not) for 5h with PMA/ionomycin and protein transport block (eBioscience), followed by surface and intracellular staining Stained cells were analyzed with a FACSCantoII 8-color (BD Biosciences) and data analyzed with FlowJo (Tree Star Inc.).
RT-PCR
RNA was extracted from whole lung tissue or sorted cells with addition of 1 mL Trizol (Invitrogen). RNA was purified from supernatant in 70% EtOH with RNeasy and cDNA synthesized with QuantiTect (Qiagen). Gene expression was quantified with Taqman kit (Applied Biosystems).
ELISA assays
Levels of IL-9 in BALFs (Biolegend) and CCL17 in lung tissue extracts (R&D System) were determined following the manufacturer’s instructions. Protein from the right lung lobule was extracted in 500 μl of PBS containing Protein inhibitor cocktail (Roche Life Science).
Statistical analysis
Comparisons between multiple groups were performed with two-ways or one-way Anova, with Bonferroni post-test analysis. Comparisons between two groups were performed with two-tailed T-tests. All statistical analyses were carried out with GraphPad Prism.
Results and Discussion
IL-25 rapidly induces inflammatory mediators in lung
A single intra-nasal administration of IL-25, but not PBS, induced expression of several inflammatory genes in whole lungs within 4h, as determined with an initial limited PCR array (not shown). In addition to genes known to be rapidly induced in ILC2s, such as that encoding IL-13 (4), we also noted genes not known to originate with ILC2s, such as that encoding the chemokine CCL17 (a.k.a. TARC) (6). Various cells, in particular dendritic cells (DCs) may produce CCL17 (19). We validated induced expression of Ccl17 with RT-PCR of whole lung tissue; mRNA was induced by 4h, and a trend already emerged by 2h, consistent with direct targeting by IL-25 (Figure 1A). Stimulated expression was dependent on CIKS, the intracellular adaptor protein required for signaling by the IL-25 receptor (Figure 1B) (16).
FIGURE 1.
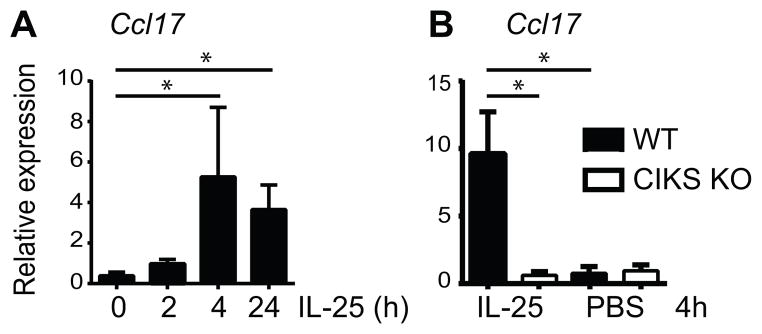
IL-25 rapidly induces Ccl17. Relative expression of Ccl17 mRNA in whole lung at times indicated after i.n. challenge with IL-25 and PBS in wild-type (WT) mice (A) and at 4h after challenge in WT and CIKS KO mice (B). Data shown as mean± sem, based on two independent experiments with a combined total n=5–6 mice/time point (A) or /genotype/condition (B). * p < 0.05
IL-25 directly targets lung-resident DCs
To identify the cell type(s) in which IL-25 induced CCL17, we administered IL-25 or PBS into lungs for 2h and 24h, bead-sorted lung cells into CD11c+, CD11c− CD11b+ and CD11c− CD11b− fractions and assessed mRNAs for CCL17, IL-13, IL-5 and IL-9 (Figure 2A). IL-25 failed to induce expression of these genes in CD11c− CD11b+ cells (includes monocytes, tissue macrophages; not shown). IL-5, IL-13 and IL-9 mRNAs were induced only in CD11c− CD11b− cells (includes ILC2s and T cells); expression of IL-5 and especially IL-13 was already increased by 2h and prominent by 24h, while that of IL-9 was delayed and only observed by 24h (not at 4h either; not shown). mRNA for CCL17 was induced exclusively in CD11c+ cells, starting by 2h and clearly up-regulated by 24h. We confirmed that ILC2 were the source of IL-5 and IL-13 in these experiments (not shown), as reported previously (20). Both ILC2’s and CD4+TCRβ+ T cells produced IL-9 by 24h after a single dose of IL-25 (Figure 2B). Induced expression of IL-9 in T cells required the IL-25 receptor in these cells (Supplemental Figure 2A), consistent the ability of IL-25 to induce IL-9 expression in Th9-polarized cells (9). The IL-9-producing T cells observed here were most likely innate or previously differentiated Th9 cells, given the rapid appearance of these cells after IL-25 challenge.
FIGURE 2.

IL-25 induces Ccl17 in lung DCs. (A) Relative expression of Ccl17, Il9, Il13 and Il5 in indicated cell populations, isolated at times shown after one challenge with IL-25 or PBS. (B) Relative expression of Il9 in WT lung cell populations shown, isolated 24h after one challenge as in (A). (C,D) Relative expression of Ccl17 in CD11c+ lung cells isolated from WT and CD11c Δ CIKS KO (C) or LysM Δ CIKS mice (D) after challenge as in (B). (E) Ccl17 expression in CD11c+, MHC II+ or MHC II− lung cells after challenge as in (B). (F) Flow cytometric analysis of lung cells of a CCL17-GFP mouse challenged as in (B) with markers, gates as indicated (representative of n=6 mice). (G) Relative expression of Ccl17 in WT cell populations shown, isolated after challenge as in (B). Data in (A–E, G) shown as mean ± sem, based on three independent experiments, and for each experiment cells were pooled from 3–5 mice/condition as well as /time point (A), /cell population (B,E,G) or /genotype (C,D).* p < 0.05.
We confirmed CCL17 as a direct target of IL-25 in CD11c+ cells, since IL-25-induced expression was largely abrogated in mice conditionally ablated of CIKS in these cells via CD11c-Cre (CD11c Δ CIKS mice) (Figure 2C). Under homeostatic conditions, CD11c+ lung cells are composed of alveolar macrophages, resident cDCs and a smaller number of monocyte-derived DCs/interstitial macrophages. We ruled out alveolar macrophages and monocyte-derived DCs/macrophages as primary sources, since IL-25 was still able to induce CCL17 in mice conditionally ablated of CIKS in these cells (but not in cDCs) via LysM-Cre (LysM Δ CIKS) (Figure 2D). To provide additional evidence that DCs were the primary source of CCL17, we separated CD11c+ cells into MHC IIhi and MHC II−/lo fractions. Basal and IL-25-induced expression of CCL17 was confined exclusively to MHC IIhi cells (Figure 2E). To further characterize the source of CCL17, we made use of reporter mice that carry eGFG in the CCL17 gene locus. As reported (21), eGFP+ cells were already present within the CD11c+ population after treatment with PBS; however, numbers of eGFP+ cells were notably increased upon treatment with IL-25 (Figure 2F, left panels). CCL17-eGFP+ cells were roughly equally divided between the two major resident lung cDCs, the CD103+ CD11b− and the CD103− CD11b+ populations (Figure 2F, right panels). We also bead-isolated CD207+ and CD207− CD11c+ cell fractions (CD207 is expressed on CD103+, not CD103− cDCs in lung) to confirm IL-25-induced expression of CCL17 in both cDC populations (Figure 2G). Finally we isolated CD11c+ cells from CCL17-eGFP reporter mice, stimulated them with IL-25 in vitro and observed induced expression of eGFP within 24h (not shown).
IL-25-induced expression of IL-9 by T cells requires IL-25 signaling into DCs
To assess biologic consequences of IL-25-signaling into DCs, we treated CD11c Δ CIKS mice and littermate controls with IL-25 (or PBS) on three consecutive days and analyzed inflammatory gene expression 24h later (Figure 3A). CCL17 induction was still largely dependent on IL-25/CIKS-mediated signaling in DCs. Unexpectedly IL-9 induction was also dependent on such signaling into DCs, even though DCs do not produce this cytokine (see above) (Figure 3A). IL-13 and IL-5 mRNA levels were partially reduced in IL-25-treated CD11c Δ CIKS mice, cytokines not expressed by DCs either (see above). Thus mediators induced by IL-25 in DCs were required for production of IL-9 and partially contributed to production of IL-13 and IL-5.
FIGURE 3. IL-25-induced inflammation is ameliorated in CD11c Δ CIKS mice.

(A) Relative expression of Ccl17, Il9, Il13 and Il5 in total lung cells of WT and CD11c Δ CIKS mice, 24h after 3 daily challenges with IL-25 or PBS. (B) Numbers of basophils (DX-5+ FcRεI+ c-Kit-), mast cells (DX-5- FcRεI+ c-Kit+) and ILC2s (Lin- St2+ Icos+) in total lung cells isolated from challenged mice as in (A). (C) Numbers of CD4+ TCRβ+ T cells in BAL fluid or total lung isolated from mice as in (A) after 4 daily challenges with IL-25 or PBS (D) Numbers of IL-9+ CD4+TCRβ+ T and ‘non-T’ cells (CD4- TCRβ-) in total lung and protein levels of IL-9 in BAL fluid and CCL17 in total lung in challenged mice as in (C). (E) CD4+TCRβ+ T cell numbers and IL-9 protein levels in BAL fluid and relative expression of Il9 and Il13 in total lung of WT and CCL17 KO mice challenged as in (A). Data shown as mean ± sem, based on three (A–D) or two (E) independent experiments with n=7–9 or n=6 mice/genotype/condition, respectively. * p < 0.05.
We investigated inflammatory cell accumulation after three treatments with IL-25. We observed increased numbers of ILC2s (Lin− ICOS+ CD25+), basophils (CD4− DX-5+ FcRεI+ c-Kit−) and mast cells (DX-5− FcεRI+ c-Kit+) in lungs of control mice (littermates), but these increases were notably smaller in CD11c Δ CIKS mice, especially for basophils and ILC2s (Figure 3B). The partial decrease in IL-13 and IL-5 in CD11c Δ CIKS mice noted above thus appears to have been due to lower numbers of ILC2s in these mice. We also observed increased numbers of eosinophils and CD4+ TCRβ+ T cells in wild-type mice, especially after one additional administration of IL-25; the increase in eosinophils was partially (not shown) and that in CD4 T cells largely prevented in CD11c Δ CIKS mice (Figure 3C).
We investigated for the cellular source of increased IL-9 production in these experiments. Induced production of IL-9 was seen only in CD4+ TCRβ+ T cells, not in CD4− TCRβ − cells (nonT cells) and only in wild-type, not CD11c Δ CIKS mice (Figure 3D). The notion that T cells and not ILC2s were the primary source of IL-9 after repeated IL-25 challenges was supported further by analysis of Rag1-deficient (KO). Despite the presence of ILC2s, IL-9 production was notably lower in Rag KO compared to wild-type mice (Supplemental Figure 2B). We additionally ruled out NKT cells as a major source of IL-9, since mice lacking NKTs (CD1d-deficient), were able to induce normal levels of IL-9 in response to IL-25 (not shown).
Along with increased numbers of IL-9-producing T cells, we also observed increased production of IL-9 and CCL17 proteins in the BAL fluid and in total lung tissue, respectively, in wild-type mice, but not CD11c Δ CIKS mice (Figure 3D). Thus increased IL-9 production by T cells required IL-25-signaling into DCs.
CCL17 is a chemokine that recruits Th2 and Th9 cells (19, 22). We therefore asked whether CCL17 might play a critical role in the appearance of IL-9-producing T cells in lungs after three challenges with IL-25. The IL-25-induced increases in T cells and IL-9 protein in BAL fluid were largely abrogated in mice deficient in CCL17 (CCL17 KO; Figure 3E). Furthermore, IL-25-induced expression of IL-9, but not IL-13 mRNA, was also decreased in CCL17 KO mice. The early appearance of IL-9-producing T cells thus largely depended on CCL17, which likely explains the requirement for IL-25 stimulation of DCs. It is possible that additional factors produced by DCs may also be required. It remains to be determined how IL-25-stimulated DCs contributed to increases in other cell types (see Figure 3B above); these effects may be mediated directly by DC-produced chemokines/mediators and/or indirectly by on-site production of survival/growth factors, such as IL-9.
IL-25 signaling in DCs promotes IL-9-producing T cells in the context of HDM-induced allergic lung inflammation
HDM is composed of multiple components that activate several potentially redundant inflammatory pathways. To determine whether IL-25, and specifically IL-25 signaling into DCs might uniquely contribute in the context of the HDM-induced model of asthma, we focused on IL-9 production by T cells as a readout. We challenged IL-25-receptor-deficient mice (IL-17RB KO) and their littermate controls (wild-type) i.n. with HDM or PBS on days 0, 3 and 6, and analyzed lung cells on day 7 for IL-9 expression in CD4+ TCRβ+ T cells (Figure 4A). Although intracellular IL-9 is difficult to detect (10, 23), we observed a significant increase in IL-9 in CD4+ TCRβ+ (Th9) cells in HDM- over PBS-challenged wild-type, but not IL-17RB KO mice. The IL-25 receptor was thus critical for accumulation of IL-9-producing T cells in this acute allergen-induced asthma model. To determine whether IL-9 production by T cells also required IL-25 stimulation of DCs, we challenged mice conditionally ablated of IL-17RB in DCs via CD11c-Cre (CD11c Δ RB) with HDM as above. Notably, production of IL-9 protein in T cells (Figure 4A) and IL-9 mRNA in lungs (Figure 4B) was largely abolished in mutant mice. To determine whether CCL17 was critical for the accumulation of IL-9+ T cells in the present model, we analyzed CCL17 KO mice. Loss of CCL17 prevented the accumulation of IL-9+ T cells and IL-9 mRNA after the HDM treatments (Figure 4A and 4B). IL-25-mediated signaling into DCs and CCL17 production were thus important for accumulation of IL-9+ T cells in the acute HDM-induced model of asthma. Therefore IL-25 stimulation of DCs contributes to lung inflammation not only when initiated by IL-25 itself, but also in the context of HDM-initiated inflammation.
FIGURE 4.

HDM-induced IL-9 production by T cells depends on IL-25 signaling to DCs. (A) Number of IL-9+ CD4+TCRβ+ cells in total lung of WT, and IL-17 RB KO, CD11c Δ RB or CCL17KO mice, isolated 24h after last of 3 challenges with HDM or PBS. Individual mouse data from three independent experiments shown. (B) Relative expression of Il9 in total lungs of WT and CD11c Δ RB or CCL17KO mice after challenge as in (A). Data shown as mean± sem, based on two independent experiments, each with 3–4 mice/genotype/condition. * p < 0.05.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health
We thank Dr. I. Förster for CCL17-GFP mice.
Footnotes
Disclosures
The authors have no financial conflict of interest
References
- 1.Divekar R, Kita H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr Opin Allergy Clin Immunol. 2014;15:98–103. doi: 10.1097/ACI.0000000000000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seys SF, Grabowski M, Adriaensen W, Decraene A, Dilissen E, Vanoirbeek JA, Dupont LJ, Ceuppens JL, Bullens DM. Sputum cytokine mapping reveals an ‘IL-5, IL-17A, IL-25-high’ pattern associated with poorly controlled asthma. Clin Exp Allergy. 2013;43:1009–1017. doi: 10.1111/cea.12125. [DOI] [PubMed] [Google Scholar]
- 3.Tang W, Smith SG, Beaudin S, Dua B, Howie K, Gauvreau G, O’Byrne PM. IL-25 and IL-25 Receptor Expression on Eosinophils from Subjects with Allergic Asthma. Int Arch Allergy Immunol. 2013;163:5–10. doi: 10.1159/000355331. [DOI] [PubMed] [Google Scholar]
- 4.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2014;16:45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 5.Beale J, Jayaraman A, Jackson DJ, Macintyre JD, Edwards MR, Walton RP, Zhu J, Ching YM, Shamji B, Edwards M, Westwick J, Cousins DJ, Hwang YY, McKenzie A, Johnston SL, Bartlett NW. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med. 2014;6:256ra134. doi: 10.1126/scitranslmed.3009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saenz SA, Siracusa MC, Monticelli LA, Ziegler CG, Kim BS, Brestoff JR, Peterson LW, Wherry EJ, Goldrath AW, Bhandoola A, Artis D. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med. 2013;210:1823–1837. doi: 10.1084/jem.20122332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen BC, Budelsky AL, Baptist AP, Schaller MA, Lukacs NW. Interleukin-25 induces type 2 cytokine production in a steroid-resistant interleukin-17RB+ myeloid population that exacerbates asthmatic pathology. Nat Med. 2012;18:751–758. doi: 10.1038/nm.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terashima A, Watarai H, Inoue S, Sekine E, Nakagawa R, Hase K, Iwamura C, Nakajima H, Nakayama T, Taniguchi M. A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med. 2008;205:2727–2733. doi: 10.1084/jem.20080698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angkasekwinai P, Chang SH, Thapa M, Watarai H, Dong C. Regulation of IL-9 expression by IL-25 signaling. Nat Immunol. 2010;11:250–256. doi: 10.1038/ni.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Licona-Limon P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limon I, Ishigame H, Hao L, Herbert DR, Flavell RA. Th9 Cells Drive Host Immunity against Gastrointestinal Worm Infection. Immunity. 2013;39:744–757. doi: 10.1016/j.immuni.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, Panzer U, Helmby H, Stockinger B. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. 2013 doi: 10.1084/jem.20130071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones CP, Gregory LG, Causton B, Campbell GA, Lloyd CM. Activin A and TGF-beta promote T(H)9 cell-mediated pulmonary allergic pathology. J Allergy Clin Immunol. 2012;129:1000–1010. e1003. doi: 10.1016/j.jaci.2011.12.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stassen M, Schmitt E, Bopp T. From interleukin-9 to T helper 9 cells. Ann N Y Acad Sci. 2012;1247:56–68. doi: 10.1111/j.1749-6632.2011.06351.x. [DOI] [PubMed] [Google Scholar]
- 14.Kearley J, Erjefalt JS, Andersson C, Benjamin E, Jones CP, Robichaud A, Pegorier S, Brewah Y, Burwell TJ, Bjermer L, Kiener PA, Kolbeck R, Lloyd CM, Coyle AJ, Humbles AA. IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am J Respir Crit Care Med. 2011;183:865–875. doi: 10.1164/rccm.200909-1462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, Chen A, Ren NY, Wang H, Siebenlist U. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swaidani S, Bulek K, Kang Z, Liu C, Lu Y, Yin W, Aronica M, Li X. The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. J Immunol. 2009;182:1631–1640. doi: 10.4049/jimmunol.182.3.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pisitkun P, Ha HL, Wang H, Claudio E, Tivy CC, Zhou H, Mayadas TN, Illei GG, Siebenlist U. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity. 2012;37:1104–1115. doi: 10.1016/j.immuni.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mikhak Z, Strassner JP, Luster AD. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J Exp Med. 2013;210:1855–1869. doi: 10.1084/jem.20130091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vercelli D, Gozdz J, von Mutius E. Innate lymphoid cells in asthma: when innate immunity comes in a Th2 flavor. Curr Opin Allergy Clin Immunol. 2013 doi: 10.1097/ACI.0000000000000023. [DOI] [PubMed] [Google Scholar]
- 21.Alferink J, Lieberam I, Reindl W, Behrens A, Weiss S, Huser N, Gerauer K, Ross R, Reske-Kunz AB, Ahmad-Nejad P, Wagner H, Forster I. Compartmentalized production of CCL17 in vivo: strong inducibility in peripheral dendritic cells contrasts selective absence from the spleen. J Exp Med. 2003;197:585–599. doi: 10.1084/jem.20021859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jabeen R, Goswami R, Awe O, Kulkarni A, Nguyen ET, Attenasio A, Walsh D, Olson MR, Kim MH, Tepper RS, Sun J, Kim CH, Taparowsky EJ, Zhou B, Kaplan MH. Th9 cell development requires a BATF-regulated transcriptional network. J Clin Invest. 2013;123:4641–4653. doi: 10.1172/JCI69489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, Sparwasser T, Helmby H, Stockinger B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol. 2011;12:1071–1077. doi: 10.1038/ni.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.