

Abstract
Oxygenated guaiane-type sesquiterpenes, xylaguaianols A–D (1–4), an iso-cadinane-type sesquiterpene isocadinanol A (5), and an α-pyrone 9-hydroxyxylarone (6), together with five known sesquiterpenes (7–11), and four known cytochalasins (12–15) were isolated from a culture broth of Xylaria sp. NC1214, a fungal endophyte of the moss Hypnum sp. The structures of all compounds were elucidated by the analysis of their spectroscopic data and relative configurations of 1–5 were determined with the help of NMR NOESY experiments. Cytochalasins C (12), D (13), and Q (14) were investigated for their cytotoxic activity against five tumor cell lines. Cytochalasin D showed significant cytotoxicity against all five cell lines, with IC50s ranging from 0.22 to 1.44 μM, whereas cytochalasins C and Q exhibited moderate, but selective cytotoxicity.
Keywords: Xylaria sp, fungal endophyte, Hypnum sp, xylaguaianols, sesquiterpenes, α-pyrone, cytochalasins
1. Introduction
Endophytic fungi constitute rich sources of secondary metabolites with novel structures and biomedical potential (Schulz et al., 2002; Gunatilaka, 2006: Zhang et al., 2006). Xylaria species, often isolated as endophytes from decaying plant tissue, occur worldwide from arctic to tropical regions (Schüffler et al, 2007). Many Xylaria species are known to produce diverse secondary metabolites (Song et al., 2014), which include succinic acid derivatives (Klaiklay et al., 2012), cytochalasins (Espada et al., 1997), α-pyrones (Schüffler et al., 2007), terpenoids (Li et al., 2010; Wu et al., 2014; Yan et al., 2011), xanthones (Davis and Pierens, 2006; Healy et al., 2004), cyclopeptides (Lin et al., 2001; Wu et al., 2011), and lactones (Jimenez-Romero et al., 2008). In an ongoing search for bioactive natural products from fungal endophytes, a cytotoxic EtOAc extract of Xylaria sp. NC1214, a fungal endophyte of the moss Hypnum sp., cultured in potato dextrose broth (PDB) containing 0.25 mM CuSO4 (Paranagama et al., 2007) was investigated. Fractionation of this extract resulted in the isolation of fifteen metabolites including four cytotoxic cytochalasins. Herein reported are the isolation and structure elucidation of four new oxygenated guaiane-type sesquiterpenes (1–4), a rare bicyclic sesquiterpene (5) and a new α-pyrone (6), as well as the known sesquiterpenes, epi-guaidiol A (7) (Xu et al., 2009), hydroheptelidic acid (8) (Calhoun et al., 1992), gliocladic acid (9) (Itoh et al., 1982), bullatantriol (10) (Sung et al., 1992), and 1β,4β,7α-trihydroxyeudesmane (11) (Sung et al., 1992), and four known cytochalasins, cytochalasins C (12), D (13), Q (14), and R (15) (Edwards et al., 1989). Also reported herein is the cytotoxic activity of cytochalasins 12–14 against five sentinel cancer cell lines.
2. Results and discussion
Xylaguaianols A (1) and B (2) (Figure 1) were determined to have the same molecular formula, C15H28O4, on the basis of their HRMS and 13C NMR spectroscopic data, indicating that they are sesquiterpenes. Comparison of their 1H and 13C NMR spectroscopic data (Tables 1 and 2, respectively) with those reported for bicyclic sesquiterpenes (Wu et al., 2014) suggested that 1 and 2 are tetrahydroxy derivatives of guaiane-type sesquiterpenes. The 13C and DEPT NMR spectra of xylaguaianol A (1) (Table 2) indicated fifteen carbons consisting of three primary, six secondary (of which one was oxygenated; δC 68.8), three tertiary, and three oxygenated quaternary carbons (δC 75.3, 77.6 and 81.9). The analysis of the 1H NMR and HSQC data (Table 1) established that 1 possessed three tertiary methyl groups (δH 1.08 s, δC 33.3; δH 1.18 s, δC 24.5; δH 1.25 s, δC 26.6), six methylene groups of which one is oxygenated (δH 3.28−3.33 m and 3.59−3.65 m, δC 68.8), and three methine groups (δH 1.95 m, δC 33.4; δH 2.17 m, δC 49.3; δH 2.86 m, δc 55.0). Detailed analysis of 1H−1H COSY and HMBC spectra (Figure 2) confirmed that 1 contained a guaiane-type sesquiterpene skeleton bearing four hydroxyl groups. Careful inspection of the HMBC spectrum suggested the presence of long-range correlations of H-15 to C-4 (δC 81.9), H-14 to C-10 (δC 75.3), H-13 to C-11 (δC 77.6), and H-13 to C-12 (δC 68.8) confirming that these four hydroxyls were attached to C-4, C-10, C-11 and C-12. These data indicated that 1 had the same overall skeletal structure as that of 4β-hydroxyxylaranol recently encountered in an endophytic fungus of a mangrove plant (Zeng et al., 2015). However, comparison of their [α]D and NMR data suggested that they are not identical but may be stereoisomers. The relative configuration of 1 was assigned by the NOESY experiment combined with MM2 energy-minimized three-dimensional molecular modeling. The NOE correlations of H-1 with H-5/H-14, and H-5 with H-14/H-15 established that they are on the same face of the molecule (Figure 3). Additional NOEs were observed between H-6α and 4-OH/H-12/H-13, indicating that the protons at C-6α, C-12 and C-13 as well as the hydroxyl at C-4 were on the same side of the ring system. Thus, the structure of xylaguaianol A was established as pseudo-1β,5β,7β(H)-guaiane-4α,10α,11,12-tetraol (1). The stereoconfiguration at C-11 of 1 was defined by comparison with the data for xylaguaianol B (2) as described below.
Fig. 1.
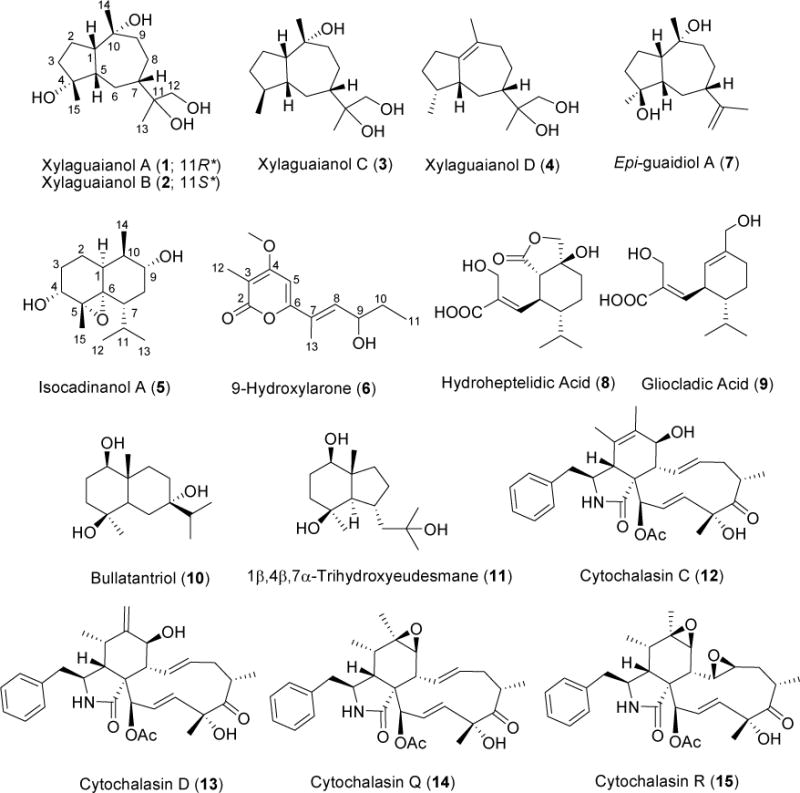
Structures of metabolites 1–15.
Table 1.
1H NMR (400 MHz) spectroscopic data for compounds 1–4 (acetone-d6, δ in ppm, J in Hz).
| position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 2.86 m | 2.90 m | 2.05 m | – |
| 2α | 1.50 m | 1.53 m | 1.50 m | 2.31 m |
| 2β | 1.71 m | 1.70 m | 1.64 m | 2.16 m |
| 3α | 1.63 m | 1.67 m | 1.66 m | 1.35 m |
| 3β | 1.91 m | 1.93 m | 1.21 m | 1.64 m |
| 4 | – | – | 1.97 m | 2.08 m |
| 5 | 2.17 m | 2.24 m | 1.88 m | 2.38 m |
| 6α | 2.17 m | 2.13 m | 1.77 m | 0.81 m |
| 6β | 1.12 m | 1.20 m | 0.90 m | 1.76 m |
| 7 | 1.95 m | 1.99 m | 1.89 m | 1.67 m |
| 8α | 2.12 m | 2.08 m | 1.82 m | 1.13 m |
| 8β | 1.45 m | 1.44 m | 1.14m | 2.05 m |
| 9α | 1.85 m | 1.70 m | 1.88 m | 2.08 m |
| 9β | 1.72 m | 1.53 m | 1.37m | 2.08 m |
| 12a | 3.28–3.33 m | 3.17–3.22 m | 3.29–3.36 m | 3.35–3.40 m |
| 12b | 3.59–3.65 m | 3.41–3.50 m | 3.40–3.50 m | 3.47–3.52 m |
| 13 | 1.25 s | 1.23 s | 0.98 s | 1.00 s |
| 14 | 1.08 s | 1.07 s | 1.10 s | 1.63 d (1.2) |
| 15 | 1.18 s | 1.20 s | 0.93 d (6.8) | 0.88 d (7.2) |
| 12-OH | 3.50–3.56 m | 3.54–3.60 m | ||
| 13-OH | 3.12 s | |||
| 14-OH | 3.03 s | |||
| 15-OH | 3.18 s | 3.19 s |
Table 2.
13C NMR (100 MHz) spectroscopic data for compounds 1–4 (acetone-d6, δ in ppm).
| position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 55.0, CH | 54.9, CH | 56.6, CH | 142.0, C |
| 2 | 26.2, CH2 | 26.3, CH2 | 26.8, CH2 | 30.9, CH2 |
| 3 | 38.9, CH2 | 39.0, CH2 | 31.7, CH2 | 33.7, CH2 |
| 4 | 81.9, C | 81.8, C | 40.0, CH | 39.9, CH |
| 5 | 49.3, CH | 49.3, CH | 47.9, CH | 47.3, CH |
| 6 | 36.0, CH2 | 36.3, CH2 | 22.3, CH2 | 29.6, CH2 |
| 7 | 33.4, CH | 32.2, CH | 44.9, CH | 50.3, CH |
| 8 | 25.9, CH2 | 25.8, CH2 | 25.0, CH2 | 27.6, CH2 |
| 9 | 29.7a, CH2 | 28.9a, CH2 | 34.3, CH2 | 35.8, CH2 |
| 10 | 75.3, C | 75.2, C | 74.0, C | 129.7, C |
| 11 | 77.6, C | 77.6, C | 75.9, C | 75.2, C |
| 12 | 68.8, CH2 | 70.2, CH2 | 69.2, CH2 | 69.0, CH2 |
| 13 | 26.6, CH3 | 23.8, CH3 | 19.3, CH3 | 21.0, CH3 |
| 14 | 33.3, CH3 | 33.2, CH3 | 31.9, CH3 | 22.5, CH3 |
| 15 | 24.5, CH3 | 24.6, CH3 | 16.8, CH3 | 15.6, CH3 |
Overlapping with signals due to the solvent.
Fig. 2.
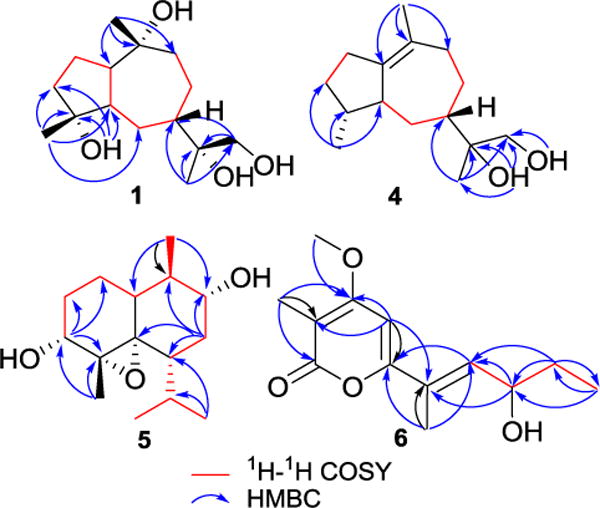
Key HMBC and 1H–1H COSY correlations for compounds 1, 4, 5, and 6.
Fig. 3.
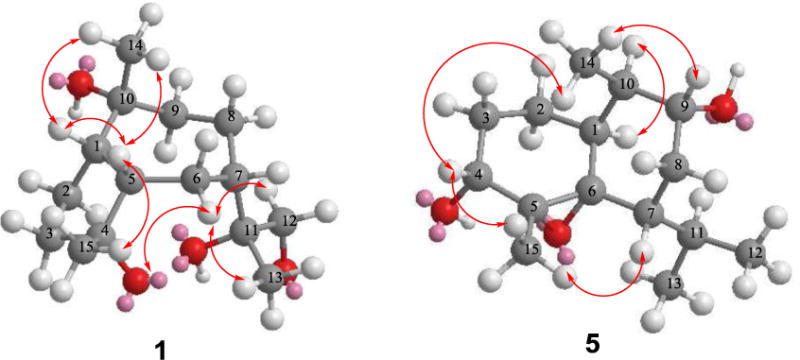
Key NOE correlations for compounds 1 and 5.
Analysis of the 1H (Table 1), 13C (Table 2) and 2D NMR data of xylaguaianol B (2) suggested that it shared similar structural features with those of 1. In addition, the NOESY data of 2 indicated that its bicyclic ring system had the same relative configuration as in 1. Careful comparison of the 13C NMR chemical shifts of 1 and 2 suggested that the major differences between these existed at C-7 (δC 33.4 for 1, δC 32.2 for 2), C-12 (δC 68.8 for 1, δC 70.2 for 2) and C-13 (δC 26.6 for 1, δC 23.8 for 2), indicating that 1 and 2 were a pair of epimers of R/S configurations at C-11. Careful examination of the 1H and 13C NMR data recently reported for a series of guaiane-type sesquiterpenes epimeric at C-11, whose absolute configurations have been determined by X-ray crystallographic analysis (Wu et al., 2014), suggested that the 13C NMR data may be used to define the absolute stereochemistry at C-11 for this type of sesquiterpenes. The reported data indicated that the γ-gauche interaction (Whitesell et al., 1987) between C-6/C-8 and 11-OH/C-12/C-13 resulted in the difference of chemical shifts for these carbons, especially for C-13. The 7R/11R epimer always exhibited δC for C-13 at a higher field compared to that of the corresponding 7R/11S epimer. It was found that the δC for C-13 of 2 (23.8) was at a higher field than that of 1 (26.6) and this allowed us to distinguish the 11R* and 11S* configurations for 1 and 2, respectively, by assuming 7S*-configuration of 1 and 2. Thus, xylaguaianol A and xylaguaianol B were identified as 1β,5β,7β(H)-guaiane-4α,10α,11R*,12-tetraol (1) and 1β,5β,7β(H)-guaiane-4α,10α,11S*,12-tetraol (2), respectively.
Xylaguaianol C (3), obtained as a colorless oil, was determined to have the molecular formula C15H28O3 by HRESIMS and NMR data, suggesting two degrees of unsaturation. The 13C NMR data (Table 2) established the presence of fifteen sp3 carbons of which three were oxygenated (δC 69.2, 74.0, 75.9). Further analysis of the 1H (Table 1), 13C (Table 2), and 1H−1H COSY NMR data together with its molecular formula suggested the presence of a trihydroxylated guaiane-type sesquiterpene skeleton in 3. The HMBC correlations of H-13 to C-11 and C-12, and H-14 to C-10 confirmed that three of these hydroxyls were located at C-10, C-11 and C-12. The NOESY correlations of H-1 with H-14/H-5 and H-5 with H-15 indicated that these protons were co-facial. Additional correlations of H-8α with H-12/H-13 were also observed, suggesting that these protons were on the same side of the guaiane-ring. However, with the data available it was not possible to define the configuration at C-11 of 3, and because attempted crystallization failed, X-ray crystallography could not be applied for this purpose. Thus, the structure xylaguaianol C was determined as 1β,4α,5β,7β(H)-guaiane-10α,11,12-triol (3).
Xylaguaianol D (4), obtained as a colorless oil, was assigned the molecular formula C15H26O2 by a combination of HRESIMS and NMR data, suggesting three degrees of unsaturation. The 13C NMR data of 4 (Table 2) demonstrated the presence of fifteen signals consisting of thirteen sp3 carbons including two oxygenated (δC 69.0 and 75.2) and two quaternary sp2 carbons (δC 129.7 and 142.0). Comparison of the 1H and 13C NMR data of 4 (Tables 1 and 2, respectively) with those of 1–3 combined with its 1H−1H COSY data established the presence of a double bond bearing guaiane-type sesquiterpene ring system in 4. This double bond was located between C-1 and C-10 based on strong HMBC correlations observed for methyl protons at δ 1.63 (H-14) and two olefinic carbons at δC 129.7 (C-1) and 142.0 (C-10) (Figure 2). The NOE correlations of H-4 with H-5 and H-6α with H-15/H-12 suggested that these protons are on the same side of the molecule. Thus, xylaguaianol D was identified as 4β,5β,7β(H)-1,10-dehydroxyguaiane-11,12-diol (4).
Isocadinanol A (5), obtained as a colorless oil, was determined to have the molecular formula C15H26O3 by its HRESIMS and NMR data, suggesting three degrees of unsaturation. The 13C NMR spectrum of 5 revealed the presence of fifteen carbons suggesting that it was a sesquiterpene. Analysis of 1H and HSQC NMR data indicated that 5 possessed a tertiary methyl (δH 1.31 s, δC 27.8), three secondary methyls [δH 0.86 d (J = 6.4 Hz), δC 21.0; δH 0.97 d (J = 6.8 Hz), δC 21.2; δH 1.05 d (J = 6.4 Hz), δC 14.9], three methylenes [δH 1.08 m and 1.54 m, δC 19.7; δH 1.13 m and 1.67 m, δC 37.2; δH 1.66 m and 2.12 ddd (J = 13.2, 4.4, 2.4 Hz), δC 34.8], and six methines of which two were oxygenated [δH 2.92 dd (J = 16.0, 1.6 Hz), δC 68.3; δH 3.43 m, δC 71.3]. These data also indicated that 5 contained two oxygenated quaternary carbons of an oxirane moiety (δC 65.9 and 66.4). The presence of the fragments –CH(CH3)CH(O)CH2CHCH(CH3)2 and –CH(O)CH2CH2CH– in 5 was deduced by the analysis of its 1H–1H COSY data and the connectivity between these two fragments and with the two epoxy quaternary carbons and a tertiary methyl was established by the analysis of its HMBC data (Figure 2). These data suggested that 5 contained an isocadinane ring derived from a cadinane-like ring system in which the methyl group at C-4 has undergone rearrangement to C-5. The relative configuration of 5 was determined based on its NOESY spectrum combined with MM2 energy-minimized three-dimensional molecular modeling. The NOE correlation of H-1 with H-10 confirmed that these had the same relative configuration. Additional NOEs observed between H-9/H-14, H-14/H-4, H-4/H-15, and H-15/H-7 (Figure 3) indicated that H-1, hydroxyls at C-4 and C-9, the C-5(6) epoxy group and the isopropyl group at C-7 were all on the same side of the ring system. Thus, the structure of isocadinanol A was established as 1α,7β,10α(H)-5α(6α)-epoxy-isocadinane-4α,9α-diol (5).
9-Hydroxyxylarone (6), obtained as a white amorphous solid, was assigned the molecular formula C13H18O4 by a combination of HRESIMS and NMR data, indicating five degrees of unsaturation. Inspection of its 1H, 13C and HMBC NMR data suggested the presence of thirteen carbons consisting of four methyl carbons of which one was oxygenated, one methylene carbon, three methine carbons of which two were aromatic and/or olefinic and five sp2 quaternary carbons. Analysis of 1H NMR and HSQC data demonstrated the presence of three tertiary methyls of which one was oxygenated (δH 1.93 s, δC 8.7; δH 1.94 s, δC 13.0; δH 3.90 s, δC 56.2), one secondary methyl [δH 0.92 t (J = 7.2 Hz), δC 9.6], one methylene (δH 1.55–1.75 m, δC 30.4) and three methines [δH 4.43 dt (J = 8.6, 6.6 Hz), δC 70.0; δH 6.18 s, δC 92.9; δH 6.49 dd (J = 8.8, 1.2 Hz), δC 135.8]. Further analysis of the HMBC spectrum (Figure 2) indicated the existence of an α-pyrone system in 6 and comparison of the 1H and 13C NMR data of 6 with the known α-pyrone, xylarone (Schüffler et al., 2007), established that they were structurally related, except for the presence of an oxymethine moiety (δH 4.43 m, δC 70.0) in 6 instead of the methylene moiety in xylarone. These findings together with HRMS data suggested that 6 was a hydroxylated derivative of xylarone. The HMBC correlations of H-10 and H-11 to C-9 and H-9 to C-7 and C-8 suggested that this hydroxyl group was at C-9. However, lack of sufficient material precluded us from performing additional studies to determine its configuration. Thus, the structure of this metabolite was elucidated as 9-hydroxyxylarone (6).
Metabolites 7–15 were identified as epi-guaidiol A (7) (Xu et al., 2009), hydroheptelidic acid (8) (Calhoun et al., 1992), gliocladic acid (9) (Itoh et al., 1982), bullatantriol (10) (Sung et al., 1992), 1β,4β,7α-trihydroxyeudesmane (11) (Sung et al., 1992), cytochalasin C (12) (Edwards et al., 1989), cytochalasin D (13) (Edwards et al., 1989), cytochalasin Q (14) (Edwards et al., 1989), and cytochalasin R (15) (Edwards et al., 1989), respectively, by comparison of their MS and NMR spectroscopic data with those reported. Since many cytochalasins have been reported to be cytotoxic (Van Goietsenoven et al., 2011; Xu et al., 2015), cytochalasins C (12), D (13) and Q (14) encountered in this study were tested against a panel of five human tumor cell lines including prostate adenocarcinoma (PC-3M), non-small cell lung cancer (NCI-H460), CNS glioma (SF-268), breast adenocarcinoma (MCF-7), and metastatic breast adenocarcinoma (MDA-MB-231). As depicted in Table 3, cytochalasin D (13) exhibited strong cytotoxicity against all five cell lines with moderate selectivity to NCI-H460. Cytochalasins C (12) and Q (14) showed moderate cytotoxicity against all tumor cell lines except MCF-7.
Table 3.
Cytotoxicity data for cytochalasins C (12), D (13) and Q (14).a
| Compound | Tumor Cell Lineb | ||||
|---|---|---|---|---|---|
|
| |||||
| PC-3M | NCI-H460 | SF-268 | MCF-7 | MDA-MB-231 | |
| 12 | 1.65 ± 0.33 | 1.06 ± 0.03 | 0.96 ± 0.08 | > 5 | 1.72 ± 0.27 |
| 13 | 1.03 ± 0.08 | 0.22 ± 0.02 | 0.43 ± 0.06 | 1.44 ± 0.26 | 1.01 ± 0.08 |
| 14 | 1.53 ± 0.12 | 1.51 ± 0.16 | 1.31 ± 0.08 | > 5 | 1.32 ± 0.13 |
| doxorubicin | 0.25 ± 0.02 | 0.05 ± 0.01 | 0.45 ± 0.07 | 0.32 ± 0.09 | 0.67 ± 0.11 |
Results are expressed as IC50 value ± standard deviation in μM. Doxorubicin and DMSO were used as positive and negative controls.
Cell lines used: PC-3M = metastatic human prostate adenocarcinoma; NCI-H460 = human non-small cell lung cancer; SF-268 = human CNS glioma; MCF-7 = human breast adenocarcinoma; MDA-MB-231 = metastatic human breast adenocarcinoma.
3. Conclusion
Four new oxygenated guaiane-type sesquiterpenes, xylaguaianols A–D (1–4), a rare bicyclic sesquiterpene, isocadinanol A (5), a new α-pyrone, 9-hydroxyxylarone (6), together with five known sesquiterpenes 7–11, and four known cytochalasins 12–15, were isolated from the cytotoxic EtOAc extract of the fungus, Xylaria sp. NC1214, endophytic in the moss Hypnum sp. Their structures were determined on the basis of extensive spectroscopic analyses. When tested for cytotoxic activity against five human tumor cell lines, cytochalasin D (13) was found to be the most potent amongst cytochalasins C (12), D (13), and Q (14) with selectivity for non-small cell lung carcinoma cell line NCI-H460 (IC50 = 0.22 μM). These cytotoxicity data suggested that cytochalasins warrant further investigation as potential anticancer agents.
4. Experimental
a. General experimental procedures
1D and 2D NMR spectra were recorded on a Bruker Avance III 400 NMR instrument at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Chemical shift values (δ) are given in parts per million (ppm), and the coupling constants are in Hz. Low-resolution and high-resolution MS were recorded on Shimadzu LCMS-DQ8000α and JEOL HX110A spectrometers, respectively. Optical rotations were measured at 25 °C with a JASCO Dip-370 digital polarimeter using MeOH or acetone as solvent. UV spectra were recorded in MeOH using a Shimadzu UV-1601 UV-Vis spectrometer. Column chromatography (CC) was performed using Baker silica gel 40 μm flash chromatography packing (J. T. Baker) or Sephadex LH-20 (25−100 μm; GE Healthcare). Analytical and preparative thin-layer chromatography (TLC) were performed on pre-coated 0.20 mm thick plates of silica gel 60 F254 (Merck). HPLC purifications were carried out on a 10 × 250 mm Phenomenex Luna 5 μm C18 column with a Waters Delta Prep system consisting of a PDA 996 detector. MM2 energy minimizations of possible conformations of compounds were performed using CambridgeSoft Chembio3D Ultra.
b. Fungal material
Endophyte NC1214 was isolated on 2% malt extract agar from surface-sterilized, photosynthetic tissue of a freshly collected sample of Hypnum sp. (Hypnaceae; Hypnum moss) obtained from a granite bald in mixed deciduous forest in the southern Appalachian Mountains near Highlands, North Carolina (for isolation details, see U’Ren et al., 2012a). The strain was accessioned as a living mycelial voucher at the Robert L. Gilbertson Mycological Herbarium (MYCO-ARIZ, NC1214). Total genomic DNA was isolated from fresh mycelium and the nuclear ribosomal internal transcribed spacers and 5.8s gene (ITS rDNA; ca. 600 base pairs [bp]) and the adjacent portion of the nuclear ribosomal large subunit (LSU rDNA) was amplified as single fragment by PCR (U’Ren et al., 2012b). The product (1147 bp) was visualized on a 1% agarose gel, cleaned, normalized, and sequenced as described previously (U’Ren et al., 2012b). Sequencing reads were assembled, bases were called, and quality scores were assigned by phred (Ewing and Green, 1998) and phrap (Ewing et al., 1998) through Mesquite (Maddison and Maddison, 2011), followed by manual editing in Sequencher (Gene Codes Corp.). The resulting bidirectional sequence has been deposited in GenBank (accession JQ761854.1). Phylogenetic analyses of >2400 endophytic fungi and related strains from lichens for a related study (Chen et al., 2015), coupled with rich taxon sampling from GenBank and focusing only on the conserved 5.8S and LSUrDNA sequences, placed the isolate within Xylariaceae (Xylariales, Sordariomycetes, Pezizomycotina, Ascomycota). Multilocus analyses with sampling across the Xylariaceae (U’Ren et al., 2012) indicated that NC1214 was part of a clade consisting of endophytes and endolichenic fungi that is sister to the clade containing Xylaria coccophora. Pending description of this novel clade, we refer to this strain conservatively as Xylaria sp. NC1214.
c. Fermentation, extraction and isolation
A seed culture of Xylaria sp. NC1214 grown on PDA for 2 weeks was used for inoculation. Mycelia were scraped out and vortexed with sterile PDB (90 mL) and filtered through a 100 μm sterile filter to separate spores and/or fragments of hyphae from the mycelia. Absorbance of the filtrate was measured (at 600 nm) and adjusted to 0.6. This filtrate was used to inoculate 10 × 2.0 L Erlenmeyer flasks, each holding 1.0 L of the medium (PDB) containing 0.25 mM aq. CuSO4 and incubated at 160 rpm and 28 °C. After 14 days, mycelia were separated by filtration, and the filtrate was extracted with EtOAc (3 × 5 L). Evaporation of the EtOAc extract under reduced pressure afforded the crude extract (3.24 g). A portion (3.04 g) of this extract was partitioned between MeOH:H2O (80:20) and hexanes and the resulting MeOH:H2O (80:20) fraction was diluted with H2O to MeOH:H2O (50:50) and extracted (x 3) with CHCl3. The CHCl3 extracts were combined and evaporated under reduced pressure to obtain the CHCl3 fraction (2.78 g). The MeOH:H2O (80:20) fraction after extraction with CHCl3 was evaporated to afford the aq. MeOH fraction (0.20 g). A portion of the above CHCl3 fraction (2.73 g) was subjected to Sephadex LH-20 (100 g) gel-permeation chromatography and eluted sequentially with 350 mL each of hexanes:CH2Cl2 (4:1), CH2Cl2:acetone (4:1), CH2Cl2:acetone (2:3), and MeOH, respectively. Evaporation of these yielded fractions A (0.60 g), B (1.97 g), C (0.11 g), and D (0.02 g), respectively. Fraction A (0.59 g) was subjected to chromatography over a column of RP C-18 and eluted with a gradient of MeOH:H2O (60:40, 70:30, 80:20, 90:10, 100:0; each 120 mL) to afford five fractions A1–A5, respectively. Fraction A1 (135.4 mg) was subjected to silica gel chromatography and eluted with CHCl3:MeOH (from 98:2 to 90:10) to give five fractions A1–1 to A1–5. The major fraction A1–3 (50.3 mg) was further fractionated by C-18 (20 g) CC, eluted with a gradient initially of MeOH:H2O (50:50) to MeOH, followed by HPLC purification [C-18; MeOH:H2O (55:45)] to afford 6 (0.5 mg; Rf 0.44, CHCl3–MeOH, 14:1) and 12 (2.4 mg; Rf 0.69, CHCl3–MeOH, 14:1). Fraction A2 (386.8 mg) was further fractionated by silica gel CC using CHCl3–MeOH (from 98:2 to 90:10) as the eluent to give 15 sub-fractions A2–1 to A2–15. Evaporation of fractions A2–8, A2–11, and A2–12 under reduced pressure yielded 7 (0.6 mg, Rf 0.26, CHCl3–MeOH, 14:1), 1 (0.8 mg; Rf 0.34, CHCl3–MeOH, 10:1) and 3 (1.8 mg; Rf 0.30, CHCl3–MeOH, 10:1), respectively. Further fractionation of A2–1 (78.2 mg) by silica gel (12 g) CC and elution with CHCl3–MeOH (from 99:1 to 95:5) followed by prep-HPLC purification [C-18; MeOH:H2O (55:45)] afforded 14 (25.3 mg, Rt 20.0 min) and 15 (0.7 mg, Rt 38.1 min). Purification of fraction A2–7 (21.4 mg) by prep-HPLC [C-18; MeOH:H2O (60:40)] afforded 5 (2.0 mg; Rf 0.26, CHCl3–MeOH, 14:1). Further fractionation of fraction B (1.97 g) by a RP C-18 CC and elution with a gradient of MeOH:H2O (50:50, 60:40, 70:30, 80:20, 90:10, 100:0; each 350 mL) afforded six fractions B1–B6, respectively. Fraction B1 (279.4 mg) was further separated using silica gel CC and eluted with CHCl3–MeOH (from 98:2 to 85:15) followed by purification RP C-18 CC [MeOH:H2O (50:50 to 70:30)] to give 11 (2.4 mg; Rf 0.32, CH2Cl2–acetone, 2:1). Fraction B2 (762.2 mg) was separated by silica gel CC and eluted with CHCl3–MeOH (from 98:2 to 85:15) to give 13 sub-fractions, B2–1 to B2–13. Fraction B2–5 (45.6 mg) was separated by C-18 (10 g) CC and eluted with MeOH:H2O (50:50 to 70:30) to afford 2 (2.0 mg; Rf 0.40, CH2Cl2–acetone, 5:2). B2–9 (32.0 mg) was further purified over a column (10 g) of RP C-18 and eluted with MeOH:H2O (50:50 to 70:30) to afford 10 (2.3 mg; Rf 0.30, CH2Cl2–acetone, 2:1). Fraction B3 (494.6 mg) was separated by silica gel (80 g) CC and eluted with CHCl3–MeOH (from 98:2 to 85:15) to give 18 sub-fractions, B3–1 to B3–18. Evaporation of B3–4 under reduced pressure afforded 13 (137.1 mg; Rf 0.47, CHCl3–MeOH, 5:1). Fraction B3–13 (23.5 mg) was further separated by RP C-18 CC and eluted with MeOH:H2O (70:30 to 80:20) to give 4 (1.6 mg; Rf 0.23, CH2Cl2–acetone, 1:1). In order to isolate the polar metabolites of this fungal strain, the MeOH:H2O (50:50) fraction and the fractions C−D obtained from the CHCl3 fraction resulting from solvent–solvent partitioning of the original EtOAc extract which had similar TLC profiles were combined (0.33 g) and separated by C18 RP prep-HPLC with solvent gradients of 10% to 58% aqueous MeCN (0−30 min), 58% aqueous MeCN to MeCN (30−35 min), and 100% MeCN (35−40 min), yielding 8 (148.0 mg; Rt 17.8 min) and 9 (43.2 mg; Rt 23.6 min).
3.4. Spectroscopic data of metabolites
3.4.1. Xylaguaianol A (1)
Colorless oil; [α]25D +30.2 (c 0.08, MeOH); for 13C NMR and 1H NMR spectroscopic data, see Tables 1 and 2; APCIMS m/z 277 [M−H2O+Na] + (25), 237 [M−2H2O+H]+ (20), 219 [M−3H2O+H]+ (100); positive HRESIMS m/z 255.1956 [M−H2O+H]+ (calcd for C15H27O3, 255.1960).
3.4.2. Xylaguaianol B (2)
Colorless oil; [α]25D +1.2 (c 0.12, MeOH); for 13C NMR and 1H NMR spectroscopic data, see Tables 1 and 2; positive APCIMS m/z 277 [M−H2O+Na] + (20), 237 [M−2H2O+H]+ (34), 219 [M−3H2O+H]+ (100), 201 [M−4H2O+H]+ (55); positive HRESIMS m/z 255.1954 [M−H2O+H]+ (calcd for C15H27O3, 255.1960).
3.4.3. Xylaguaianol C (3)
Colorless oil; [α]25D +22.2 (c 0.16, MeOH); for 13C NMR and 1H NMR spectroscopic data, see Tables 1 and 2; positive APCIMS m/z 257 [M+H]+ (10), 221 [M−2H2O+H]+ (85), 207 [M−H2O−CH2OH]+ (100), 203 [M−3H2O+H]+ (68); positive HRESIMS m/z 279.1924 [M+Na]+ (calcd for C15H28O3Na, 279.1931).
3.4.4. Xylaguaianol D (4)
Colorless oil; [α]25D +14.7 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 212 (3.7) nm; for 13C NMR and 1H NMR spectroscopic data, see Tables 1 and 2; positive APCIMS m/z 233 [M−C2H4+Na]+ (100); positive HRESIMS m/z 261.1876 [M+Na]+ (calcd for C15H26O2Na, 261.1831).
3.4.5. Isocadinanol A (5)
Colorless oil; [α]25D +10.1 (c 0.10, MeOH); 1H NMR (400 MHz, CDCl3) δ 3.43 (1H, m, H-9), 2.92 (1H, dd, J = 16.0, 1.6 Hz, H-4), 2.12 (1H, ddd, J = 13.2, 4.4, 2.4 Hz, H-8α), 1.74 (1H, m, H-11), 1.67 (1H, m, H-3α), 1.66 (1H, m, H-8β), 1.54 (1H, m, H-2α), 1.44 (1H, m, H-1), 1.34 (1H, m, H-10), 1.31 (3H, s, H-15), 1.13 (1H, m, H-3β), 1.08 (1H, m, H-2β), 1.05 (3H, d, J = 6.4 Hz, H-14), 0.97 (3H, d, J = 6.8 Hz, H-13), 0.91 (1H, m, H-7), 0.86 (3H, d, J = 6.4 Hz, H-12); 13C NMR (100 MHz, CDCl3) δ 71.3 (CH, C-9), 68.3 (CH, C-4), 66.4 (C, C-6), 65.9 (C, C-5), 49.5 (CH, C-7), 43.9 (CH, C-10), 37.7 (CH, C-1), 37.2 (CH2, C-3), 34.8 (CH2, C-8), 27.8 (CH3, C-15), 27.5 (CH, C-11), 21.2 (CH3,C-13), 21.0 (CH3, C-12), 19.7 (CH2, C-2), 14.9 (CH3, C-14); positive APCIMS m/z 201 [M−3H2O+H]+ (38), 219 [M−2H2O+H]+ (100), 237 [M−H2O+H]+ (70); HRESIMS m/z 277.1773 [M+Na]+ (calcd for C15H28O3Na, 277.1774).
3.4.6. 9-Hydroxyxylarone (6)
White amorphous solid; [α]25D +8.5 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 309.5 (1.8), 225.5 (3.9) nm; 1H NMR (400 MHz, CDCl3) δ 6.49 (1H, dd, J = 8.8, 1.2 Hz, H-8), 6.18 (1H, s, H-5), 4.43 (1H, dt, J = 8.6, 6.6 Hz, H-9), 3.90 (3H, s, OCH3-4), 1.94 (1H, s, H-13), 1.93 (3H, s, H-12), 1.55–1.75 (2H, m, H-10), 0.92 (3H, t, J = 7.2 Hz, H-11); 13C NMR (100 MHz, CDCl3) δ 164. 1 (C, C-4), 163.8 (C, C-2), 159.1 (C, C-6), 135.8 (CH, C-8), 127.7 (C, C-7), 102.8 (C, C-3), 92.9 (CH, C-5), 70.0 (CH, C-9), 56.2 (CH3, OCH3-4), 30.4 (CH2, C-10), 13.0 (CH3, C-13), 9.6 (CH3,C-11), 8.7 (CH3, C-12); positive APCIMS m/z 239 [M+H]+ (100); positive HRESIMS m/z 239.1271 [M+H]+ (calcd for C13H19O4, 239.1278).
3.5. Cytotoxicity Assay
The resazurin-based colorimetric (AlamarBlue) assay was employed for evaluation of cytotoxicity in vitro of samples against human prostate adenocarcinoma (PC-3M), human non-small cell lung (NCI-H460), human CNS glioma (SF-268), human breast (MCF-7), and human metastatic breast adenocarcinoma (MDA-MB-231) as described previously (Wijeratne et al., 2012; Wijeratne et al., 2014). Doxorubicin and DMSO were used as positive and negative controls, respectively.
Supplementary Material
Acknowledgments
The authors thank NCI (R01 CA90265), NIGMS (P41 GM09060), NSF (DEB-0640996 to AEA), and the China Scholarship Council (Fellowship to H. Wei) for financial support for this work, and Drs. F. Lutzoni and J. Miadlikowska (Duke University) for their help with the collection of the fungal strain.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.phytochem.2015. XX.XXX.
References
- Calhoun LA, Findlay JA, Miller JD, Whitney NJ. Metabolites toxic to spruce budworm from balsam fir needle endophytes. Mycol Res. 1992;96:282–286. [Google Scholar]
- Chen KH, Miadlikowska J, Molnár K, Arnold AE, U’Ren JM, Gaya E, Gueidan C, Lutzoni F. Phylogenetic analyses of eurotiomycetous endophytes reveal their affinities to Chaetothyriales, Eurotiales, and a new order–Phaeomoniellales. Mol Phylogen Evol. 2015;85:117–30. doi: 10.1016/j.ympev.2015.01.008. [DOI] [PubMed] [Google Scholar]
- Davis RA, Pierens GK. 1H and 13C NMR assignments for two new xanthones from the endophytic fungus Xylaria sp. FRR 5657. Magn Reson Chem. 2006;44:966–968. doi: 10.1002/mrc.1872. [DOI] [PubMed] [Google Scholar]
- Edwards RL, Maitland DJ, Whalley AJS. Metabolites of the higher fungi. Part 24 Cytochalasin N, O, P, Q, and R New cytochalasins from the fungus Hypoxylon terricola Mill. J Chem Soc, Perkin Trans. 1989;1:57–65. [Google Scholar]
- Espada A, Rivera-Sagredo A, De La Fuente JM, Hueso-Rodriguez JA, Elson SW. New cytochalasins from the fungus Xylaria hypoxylon. Tetrahedron. 1997;53:6485–6492. [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Gunatilaka AAL. Natural products from plant-associated microorganisms: distribution, structural diversity, bioactivity, and implications of their occurrence. J Nat Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy PC, Hocking A, Tran-Dinh N, Pitt JI, Shivas RG, Mitchell JK, Kotiw M, Davis RA. Xanthones from a microfungus of the genus Xylaria. Phytochemistry. 2004;65:2373–2378. doi: 10.1016/j.phytochem.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Takahashi S, Arai M. Structure of gliocladic acid. J Antibiot. 1982;35:541–542. doi: 10.7164/antibiotics.35.541. [DOI] [PubMed] [Google Scholar]
- Jimenez-Romero C, Ortega-Barria E, Arnold AE, Cubilla-Rios L. Activity against Plasmodium falciparum of lactones isolated from the endophytic fungus Xylaria sp. Pharm Biol. 2008;46:700–703. [Google Scholar]
- Klaiklay S, Rukachaisirikul V, Sukpondma Y, Phongpaichit S, Buatong J, Bussaban B. Metabolites from the mangrove-derived fungus Xylaria cubensis PSU-MA34. Arch Pharm Res. 2012;35:1127–1131. doi: 10.1007/s12272-012-0701-y. [DOI] [PubMed] [Google Scholar]
- Li Y, Hu Z, Lu C, Shen Y. Four new terpenoids from Xylaria sp. 101. Helv Chim Acta. 2010;93:796–802. [Google Scholar]
- Lin Y, Wu X, Feng S, Jiang G, Zhou S, Vrijmoed LLP, Jones EBG. A novel N-cinnamoylcyclopeptide containing an allenic ether from the fungus Xylaria sp. (strain# 2508) from the South China Sea. Tetrahedron Lett. 2001;42:449–451. [Google Scholar]
- Maddison WP, Maddison DR. Mesquite. 2011 www.mesquiteproject.org.
- Paranagama PA, Wijeratne EMK, Gunatilaka AAL. Uncovering biosynthetic potential of plant-associated fungi: Effect of culture conditions on metabolite production by Parasphaerosphaeria quadriseptata and Chaetomium chiversii. J Nat Prod. 2007;70:1939–1945. doi: 10.1021/np070504b. [DOI] [PubMed] [Google Scholar]
- Schulz B, Boyle C, Draeger S, Römmert AK, Krohn K. Endophytic fungi: a source of novel biologically active secondary metabolites. Mycol Res. 2002;106:996–1004. [Google Scholar]
- Schüffler A, Sterner O, Anke H. Cytotoxic α-pyrones from Xylaria hypoxylon. Z Naturforsch. 2007;62c:169–172. doi: 10.1515/znc-2007-3-403. [DOI] [PubMed] [Google Scholar]
- Song F, Wu S, Zhai Y, Xuan Q, Wang T. Secondary metabolites from the genus Xylaria and their bioactivities. Chem Biodivers. 2014;11:673–694. doi: 10.1002/cbdv.201200286. [DOI] [PubMed] [Google Scholar]
- Sung TV, Steffan B, Steglich B, Klebe G, Adam G. Sesquiterpenoids from the roots of Homalomena aromatica. Phytochemistry. 1992;31:3515–3520. doi: 10.1016/0031-9422(92)83719-f. [DOI] [PubMed] [Google Scholar]
- U’Ren JM, Lutzoni F, Miadlikowska J, Laetsch AD, Arnold AE. Host and geographic structure of endophytic and endolichenic fungi at a continental scale. Amer J Botany. 2012a;99:898–914. doi: 10.3732/ajb.1100459. [DOI] [PubMed] [Google Scholar]
- U’Ren JM, Arnold AE. Multigene phylogenetic analysis of the Xylariaceae: what are the roles of previously unknown endophytic and endolichenic fungi? Inoculum. 2012b;63:50. [Google Scholar]
- Van Goietsenoven G, Mathieu V, Andolfi A, Cimmino A, Lefranc F, Kiss R, Evidente A. In vitro growth inhibitory effects of cytochalasins and derivatives in cancer cells. Planta Med. 2011;77:711–717. doi: 10.1055/s-0030-1250523. [DOI] [PubMed] [Google Scholar]
- Whitesell JK, Minton MA. Stereochemical analysis of alicyclic compounds by C-13 NMR spectroscopy. Chapman and Hall Ltd; New York: 1987. [Google Scholar]
- Wijeratne EMK, Bashyal BP, Liu MX, Rocha DD, Gunaherath GMKB, U’Ren JM, Gunatilaka MK, Arnold AE, Whitesell L, Gunatilaka AAL. Geopyxins A–E, ent-kaurane diterpenoids from endolichenic fungal strains Geopyxis aff. majalis and Geopyxis sp. AZ0066: structure–activity relationships of geopyxins and their analogues. J Nat Prod. 2012;75:361–369. doi: 10.1021/np200769q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijeratne EMK, Xu Y, Schrez-Shouval R, Marron MT, Rocha DD, Liu MX, Costa-Lotufo LV, Santagata S, Lindquist S, Whitesell L, Gunatilaka AAL. Structure-activity relationships for withanolides as inducers of the cellular heat-shock response. J Med Chem. 2014;57:2851–2863. doi: 10.1021/jm401279n. [DOI] [PubMed] [Google Scholar]
- Wu S, He J, Li X, Huang R, Song F, Chen Y, Miao C. Guaiane sesquiterpenes and isopimarane diterpenes from an endophytic fungus Xylaria sp. Phytochemistry. 2014;105:197–204. doi: 10.1016/j.phytochem.2014.04.016. [DOI] [PubMed] [Google Scholar]
- Wu W, Dai H, Bao L, Ren B, Lu J, Luo Y, Guo L, Zhang L, Liu H. Isolation and structural elucidation of proline-containing cyclopentapeptides from an endolichenic Xylaria sp. J Nat Prod. 2011;74:1303–1308. doi: 10.1021/np100909y. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhang H, Wan X, Zou Z. Complete assignments of 1H and 13C NMR data for two new sesquiterpenes from Cyperus rotundus L. Magn Reson Chem. 2009;47:527–531. doi: 10.1002/mrc.2416. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bashyal BP, Liu MX, Espinosa-Artiles P, U’Ren JM, Arnold AE, Gunatilaka AAL. Cytotoxic cytochalasins and other metabolites from Xylariaceae sp FL0390, a fungal endophyte of Spanish moss. Nat Prod Commun. 2015 In press. [PubMed] [Google Scholar]
- Yan S, Li S, Wu W, Zhao F, Bao L, Ding R, Gao H, Wen H, Song F, Liu H. Terpenoid and phenolic metabolites from the fungus Xylaria sp. associated with termite nests. Chem Biodivers. 2011;8:1689–1700. doi: 10.1002/cbdv.201100026. [DOI] [PubMed] [Google Scholar]
- Zhang H, Song Y, Tan R. Biology and chemistry of endophytes. Nat Prod Rep. 2006;23:753–771. doi: 10.1039/b609472b. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Gu H, Zuo W, Zhang L, Bai H, Guo Z, Proksch P, Mei W, Dai H. Two new sesquiterpenoids from endophytic fungus J3 isolated from Mangrove Plant Ceriops tagal. Arch Pharm Res. 2015;38:673–676. doi: 10.1007/s12272-014-0448-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.