

Abstract
Rhodesain, the major cathepsin L-like cysteine protease in the protozoan Trypanosoma brucei rhodesiense, the causative agent of African sleeping sickness, is a well-validated drug target. In this work, we used a fragment-based approach to identify inhibitors of this cysteine protease, and identified inhibitors of T. brucei. To discover inhibitors active against rhodesain and T. brucei, we screened a library of covalent fragments against rhodesain and conducted preliminary SAR studies. We envision that in vitro enzymatic assays will further expand the use of the covalent tethering method, a simple fragment-based drug discovery technique to discover covalent drug leads.
Keywords: Rhodesain, Trypanosomes, Covalent Fragments, Cysteine protease
Graphical Abstract
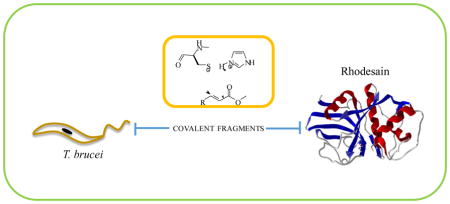
Fragment-based drug discovery (FBDD) is a powerful method to discover drug leads and has been widely adopted in both academia and industry 1. FBDD can be used to explore chemical diversity space with libraries which are smaller in size, producing drug leads with high ligand-binding efficiency 2. It also provides a rational path to high-affinity lead compounds that possess drug like properties3. The fragment-based approach is particularly well-suited for target-based drug discovery because the structural requirements for inhibition or inactivation can be used to guide the choice of pharmacophore and other structural motifs during drug design 4.
The human African trypanosomiasis (sleeping sickness), a disease caused by the kinetoplastid protozoans Trypanosoma brucei rhodeseinse and Trypanosoma brucei gambiense, is one of the neglected tropical diseases (NTDs). The disease is fatal if untreated, and current treatment options available for the disease are ineffective and have well-documented adverse effects. It is important to note that the incidence of sleeping sickness has decreased in the last decade, although the need to develop new and effective drugs remains a key objective in controlling and eradicating the disease 5. A promising drug candidate in clinical trials for human African trypanosomiasis is the nitroimidazole fexinidazole. Fexinidazole is also being developed as a potential treatment for Chagas Disease 6, 7. The major cathepsin L-like cysteine protease in T. brucei rhodesiense, rhodesain, is a validated drug target. The cysteine protease is essential for the survival and infectivity of the parasite. Its important role in the ability of the parasite to proliferate has been investigated by several groups 8–10.
Steverding and co-workers have also shown that pharmacological inhibition of rhodesain is lethal to T. brucei 11. A number of potent inhibitors of rhodesain that also have antitrypanosomal activity have been reported 8. Among them are peptide-based covalent inhibitors with Michael acceptors such as 1 and 2 and thiosemicarbazone based inhibitors for example 3 and 4. Furthermore, a peptide derived covalent inhibitor of its homologue in Trypanosoma cruzi, K777, was recently a promising pre-clinical drug candidate for the treatment of Chagas disease (American trypanosomiasis), which highlights the importance of rhodesain as a drug target 12, 13.
This provides the rationale that covalent or non-covalent inhibitors of rhodesain may be advanced into the drug development pipeline against African sleeping sickness. To discover rhodesain inhibitors, we employed the irreversible tethering method to discover covalent inhibitors of cysteine proteases14. In this method, a mixture of cysteine reactive electrophilic fragments were incubated with the cysteine protease, allowing the best binding fragments to covalently and irreversibly modify the catalytic cysteine of the protease, and the covalent cysteine protease-inhibitor complexes were subsequently detected using mass spectrometry methods. The fragments can subsequently be elaborated into drug leads while retaining the original Michael acceptor electrophile. The originally developed method requires mass spectrometry to identify fragment hits, and this requirement limits the widespread use of this technology. We thought to expand the method and asked if the electrophilic fragments can be screened individually in enzymatic assays to identify weak and irreversible fragment inhibitors. Notably, such an approach would contradict current practices in academia and industry, in which reactive compounds are removed from compound screening collections. We envisioned that if successful, this strategy would substantially expand the use of irreversible tethering in laboratory settings in which mass spectrometry services are not available. Thus, a previously made library of 200 electrophilic fragments was screened for inhibitor activity against rhodesain in in vitro enzymatic assays 14, 15. The active hits were then investigated for their antitrypanosomal activity and cytotoxicity to human Hep G2 cells. The initial screen of the fragment library (10 μM) against rhodesain in 384-well assay plates led to the identification of seven positive hits (SI Figure 1). These seven compounds caused ≥ 85% inhibition of rhodesain. A follow-up assay using 200 μL reaction mixtures in 96-well plates to confirm the activity of the active compounds led to the identification of compounds 5 and 7 as hits, the other five compounds caused < 40% inhibition of rhodesain at 10 μM (Table 1). Fragments 5 and 7 were recently reported as inhibitors of papain, a cysteine protease structurally and biochemically related to rhodesain14. Fragments 5 and 7 seem to be better inhibitors of rhodesain than papain judging by the kinact/KI values, however, it is important to note that the optimal assay conditions for the two cysteine proteases are different, and the assay conditions may influence the observed inactivation constants. Since K777 has a vinyl sulfone electrophile as a Michael acceptor, we investigated if vinyl sulfone analogues of 5 and 7 were capable of inhibiting rhodesain16. We have found that compound 6, which is the vinyl sulfone analogue of 5, was also active in our assays, but it was less reactive towards the cysteine protease than the acrylate (5) (Table 1). However, compound 8, the vinyl sulfone analog of 7, was inactive in the enzymatic assay, suggesting that unique structural features of 7 drive its activity against rhodesain.
Table 1.
Antitrypanosomal activity, cytotoxicity and protease inhibitory data of active acrylates and their vinyl sulfone analogues.
| Compound | Rhodesain Kinact/KI (M−1s−1) | T. brucei IC50 (μM) | Hep G2 IC50 (μM) |
|---|---|---|---|
| 5 | 18.32 | 30.31 ± 1.92 | >150 |
| 6 | 3.47 | 30.16 ± 0.86 | 69± 13.68 |
| 7 | 13.22 | 43.36± 2.50 | >150 |
| 8 | inactive | 29.52± 2.83 | 76± 13.17 |
| 9 | inactive | >50 | >150 |
| 10 | inactive | >50 | >150 |
In addition, we investigated if the previously reported14 papain inhibitor 9 can inhibit rhodesain, and found that it was not active at inhibiting rhodesain. The vinyl sulfone analogue of 9 (10) was then synthesized and tested, and it was also inactive towards rhodesain. We then determined the time dependent inhibition constant kinact/KI of compounds 5, 7 and 6, which were 18.32, 13.22 and 3.47 M−1s−1, respectively (Table 1). Taken together, our preliminary SAR analysis indicates that a combination of specific structural features of the fragment and the electrophile make inhibitors of rhodesain. Crystal structure analysis of the fragment-rhodesain complexes is underway, and it should provide a structural ratonale for extensive SAR studies. Although the identified covalent inhibitors of rhodesain are fragments, and therefore need to be optimized further to increase their potency and selectivity, we asked if any of those compounds have antitrypanosomal activity. We rationalized that those fragments that display antitrypanosomal activity, yet are not toxic to human hepatocellular carcinoma (Hep G2), could be further optimized into inhibitors that display selective toxicity to T. brucei but not to human cells.
Therefore, compounds 5–10 were tested for growth inhibitory activity on T. brucei as well as their cytotoxicity on Hep G2 cells. compounds 5 and 7 have emerged as promising lead structures, since both compounds were potent at inhibiting rhodesain in vitro (kinact/KI values 18.32 and 13.22 respectively), and displayed selective toxicity to T. brucei without being toxic to HepG2 cells (Table 1). Importantly, compound 8 was inactive in our in vitro assays, despite the fact that it was active in the antitrypanosomal assay. This indicates that 8 may be reactive towards one or more other catalytic cysteines in T. brucei, although the weak selectivity index of 8 makes it a less desired lead compound.
In conclusion, an electrophilic fragment library was evaluated for inhibitory activity against the cathepsin-L like cysteine protease rhodesain. The unique feature of this approach is that reactive compounds were screened in an enzymatic assay in a 384 well plate format to identify specific hits, which stands in sharp contrast to the currently accepted dogma in the pharmaceutical industry that reactive compounds must be excluded from all HTS screens, because reactive compounds can display promiscuous reactivity toward their protein targets. Our results show that in fact it is possible to screen a library of cysteine reactive fragments in enzymatic assays in a 384 well plate format if the library of the cysteine reactive fragments is properly designed 14. Furthermore, the non-peptidic nature of the identified inhibitors of rhodesain could result in better pharmacokinetic properties of the covalent rhodesain inhibitor drug leads. Furthermore, current known covalent inhibitors of rhodesain have two electron withdrawing groups present at the Michael acceptor site, which can increase the number of off-target effects for such inhibitors. In contrast, our fragment libraries have only one electron-withdrawing group at the Michael acceptor site, which should reduce the electrophilicity and non-specific reactivity of these fragments. We envision that fragments that contain other electrophiles can be assembled and tested against other cysteine proteases either using mass spectrometry or enzymatic assays in the 96 or 384 well plate format, which will significantly expand the use of the irreversible tethering technology. Further optimization of the identified rhodesain inhibitor fragments into potent and selective lead compounds will be reported in the near future. Although compounds 5 and 7 were also previously identified as papain hits, we believe that we can achieve reasonable selectivity for rhodesain amongst other papain-family cysteine proteases upon growth of the fragment into a drug lead, similar to how selectivity amongst ATP competitive kinase inhibitors is acheived.
Supplementary Material
Figure 1.
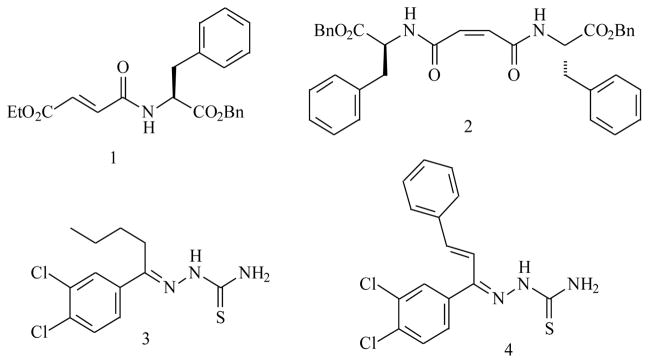
Inhibitors of rhodesain that have antitrypanosomal activity.
Figure 2.

Inhibitors of rhodesain from this study.
Figure 3.

Pseudo-first order and second-order inhibition plots for compounds 5, 6 and 7.
Scheme 1.
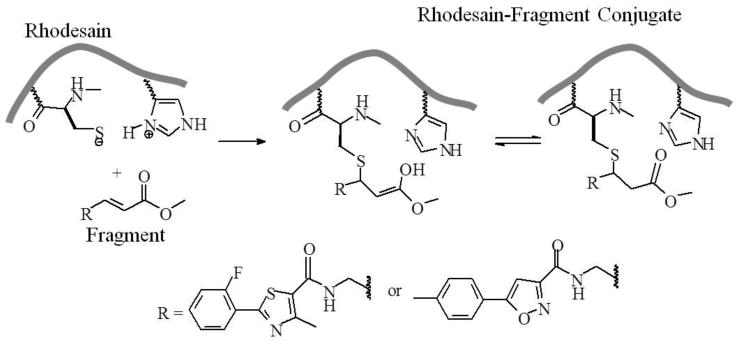
Overview of rhodesain-fragment conjugation.
Acknowledgments
This work was supported in part by the US National Institutes of Health (SC2GM109782 to I.V.O.), Chemistry of Life Processes Institute Lambert Fellowship (Z.X.), the ACS Medicinal Chemistry Fellowship (S.G.K.) and Northwestern University. A.S. is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts. We thank Rama Mishra and the Center for Molecular Innovation and Drug Discovery for assisting with the initial design of the library of electrophilic fragments.
Footnotes
Supplementary material: chemical synthesis, bioassay, compound characterization data, activity data and structures of fragments are provided as supporting material.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Rees D, Congreve M, Murray C, Carr R. Nat Rev Drug Discovery. 2004;3:660. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 2.Congreve M, Chessari G, Tisi D, Woodhead A. J Med Chem. 2008;51:3661–3663. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 3.Tsai J, Lee J, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass N, Sproesser K, Li L, Smalley K, Fong D, Zhu Y, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim S, Schlessinger J, Zhang K, West B, Powell B, Habets G, Zhang C, Ibrahim P, Hirth P, Artis D, Herlyn M, Bollag G. PNAS. 2008;105:3046. [Google Scholar]
- 4.Scott D, Coyne A, Hudson S, Abell CF. Biochem. 2012;51:4991. doi: 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- 5. [Accessed 02/26/2015];World Health Organization Report. 2015 http://apps.who.int/iris/bitstream/10665/152781/1/9789241564861_eng.pdf?ua=1.
- 6.Nagle A, Khare S, Kumar A, Supek F, Buchynskyy A, Mathison C, Chennamaneni N, Pendem N, Buckner F, Gelb M, Molteni V. Chem Reviews. 2014;114(22):11332–11333. doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaiser M, Bray MA, Cal M, Bourdin TB, Torreele E, Brun R. Antimicrob Agents Chemother. 2011;55:5602. doi: 10.1128/AAC.00246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ettari R, Tamborini L, Angelo IC, Micale N, Pinto A, De Micheli C, Conti P. J Med Chem. 2013;56:5637. doi: 10.1021/jm301424d. [DOI] [PubMed] [Google Scholar]
- 9.Mott B, Ferreira R, Simeonov A, Jadhav A, Ang K, Leister W, Shen M, Silveira J, Doyle P, Arkin M, McKerrow J, Inglese J, Austin C, Thomas C, Shoichet B, Maloney D. J Med Chem. 2010;53:52. doi: 10.1021/jm901069a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ettari R, Tamborini L, Angelo IC, Grasso S, Schirmeister T, Lo Presti L, De Micheli C, Pinto A, Conti P. ChemMedChem. 2013;8:2070. doi: 10.1002/cmdc.201300390. [DOI] [PubMed] [Google Scholar]
- 11.Steverding D, Sexton DW, Wang X, Gehrke SS, Wagner GK, Caffrey CR. Int J Parasitol. 2012;42:481. doi: 10.1016/j.ijpara.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 12.McKerrow JH, Doyle PS, Engel JC, Podust LM, Robertson SA, Ferreira R, Saxton T, Arkin M, Kerr ID, Brinen LS, Craik CS. Mem Inst Oswaldo Cruz. 2009;104:263. doi: 10.1590/s0074-02762009000900034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choy JW, Bryant C, Calvet CM, Doyle PS, Gunatilleke SS, Leung SS, Ang KK, Chen S, Gut J, Oses-Prieto JA, Johnston JB, Arkin MR, Burlingame AL, Taunton J, Jacobson MP, McKerrow JM, Podust LM, Renslo AR. Beilstein J Org Chem. 2013;9:15. doi: 10.3762/bjoc.9.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kathman S, Xu Z, Statsyuk AA. J Med Chem. 2014;57:4970. doi: 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The fragment library includes compounds that satisfy the following criteria: molecular weight (MW) ≤ 350 Da, AlogP ≤ 3, hydrogen-bond acceptors ≤ 3, hydrogen-bond donors ≤ 3, rotatable bonds ≤ 3, and polar surface area ≤ 80. The library has a diversity index of 0.75.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.