

Abstract
Noroviruses (NoVs) are a leading cause of epidemic and sporadic cases of acute gastroenteritis worldwide. Oysters are well recognized as the main vectors of environmentally transmitted NoVs, and disease outbreaks linked to oyster consumption have been commonly observed. Here, to quantify the genetic diversity, temporal distribution, and circulation of oyster-related NoVs on a global scale, 1,077 oyster-related NoV sequences deposited from 1983 to 2014 were downloaded from both NCBI GenBank and the NoroNet outbreak database and were then screened for quality control. A total of 665 sequences with reliable information were obtained and were subsequently subjected to genotyping and phylogenetic analyses. The results indicated that the majority of oyster-related NoV sequences were obtained from coastal countries and regions and that the numbers of sequences in these regions were unevenly distributed. Moreover, >80% of human NoV genotypes were detected in oyster samples or oyster-related outbreaks. A higher proportion of genogroup I (GI) (34%) was observed for oyster-related sequences than for non-oyster-related outbreaks, where GII strains dominated with an overwhelming majority of >90%, indicating that the prevalences of GI and GII are different in humans and oysters. In addition, a related convergence of the circulation trend was found between oyster-related NoV sequences and human pandemic outbreaks. This suggests that oysters not only act as a vector of NoV through environmental transmission but also serve as an important reservoir of human NoVs. These results highlight the importance of oysters in the persistence and transmission of human NoVs in the environment and have important implications for the surveillance of human NoVs in oyster samples.
INTRODUCTION
Norovirus (NoV) is known as the leading cause of nonbacterial acute gastroenteritis in humans and can infect people of all ages across the world (1). As a member of the family Caliciviridae, NoV is a nonenveloped, positive-sense, single-stranded RNA virus with a linear genome that contains three open reading frames (ORFs) (2). The genus Norovirus currently contains at least 6 distinct genogroups (genogroup I [GI], GII, GIII, GIV, GV, and GVI), each of which has been subdivided into multiple genotypes (3, 4). GI, GII, and GIV strains have been detected in humans and are further subdivided into 9, 22, and 2 genotypes, respectively (3, 4). One important genotype, genogroup II genotype 4 (GII.4), has been recognized recently as the predominant cause of major viral gastroenteritis epidemics worldwide (5–7) and contains a number of genetic variants (4, 8).
Over the past 20 years, new epidemic variants of GII.4 have emerged every 2 to 3 years (9) and usually have become the dominant strains in every season (7, 10, 11). For example, the Yerseke 2006a variant emerged in 2006, disappeared in 2008, and was replaced by the Den Haag 2006b variant (see Fig. S1 in the supplemental material). The New Orleans 2009 variant was the major player in the worldwide NoV outbreaks from 2010 to 2013 (see Fig. S1) and was found in most oyster-related outbreaks (12–14). A relatively high worldwide incidence of NoV outbreaks was observed in late 2012. Molecular data on these outbreaks in Australia, France, New Zealand, and Japan are accessible via NoroNet and suggest that they were related to the emergence of a new variant of NoV GII.4, termed Sydney 2012 (11, 15). The Sydney 2012 variant was also associated with oyster-related outbreaks during 2012, and this epidemic trend has continued up to the present.
Oysters are known as the main vectors for human NoV transmission in the environment (16, 17). Furthermore, because oysters typically grow in coastal water potentially contaminated by human activities (18) and are often consumed half-cooked or raw (12, 19), this food presents a high risk for viral infections and is frequently involved in NoV outbreaks (20, 21). NoVs appear to be able to persist in oyster tissues for weeks and cannot be effectively removed during commercial depuration (22–24).
In oyster-related NoV outbreaks, multiple virus strains have frequently been observed both in infected patients and in the corresponding oysters (20, 21). For example, from 1998 to 2009, contamination by multiple NoV strains was observed in 65% of reported outbreaks (24). In contrast to the pattern in non-oyster-related outbreaks, GI NoVs are proportionately more common than GII NoVs in oyster-related outbreaks (24). It has been suggested that GI NoVs are also more concentrated in oysters and have greater persistence in oyster tissues than GII strains (25, 26).
Outbreaks of NoV gastroenteritis occur throughout the year but are most prevalent in the cold seasons (spring or winter) (27, 28). The ability of oysters to bioaccumulate NoVs may also be affected by certain environmental conditions, such as water temperature, salinity, chlorophyll a concentration, and even the presence of phytoplankton (29, 30).
In order to gain a better understanding of how NoVs are transmitted via oysters in the environment, we examined the genetic variants associated with oyster-related NoV outbreaks. In the present study, all oyster-related NoV sequences deposited in NCBI GenBank and the NoroNet outbreak database from 1983 to 2014 were downloaded and were subjected to genotyping and phylogenetic analyses, and the genetic diversity and temporal-geographical distribution of oyster-related NoVs are reported. The results of this study will help investigators to develop strategies to prevent NoV contamination of oysters and NoV transmission through oysters and will ultimately improve our understanding of the pathogenicity of this widespread disease.
MATERIALS AND METHODS
Sequence collection and selection criteria.
As shown in Fig. 1, oyster-related NoV sequences were collected from three independent sources: (i) from the GenBank nucleotide database, by searching with a combination of the key words “oysters AND norovirus” and “shellfish AND norovirus,” in July 2014; (ii) from the “Norovirus Molecular Platform” within the RIVM NoroNet outbreak database (http://www.rivm.nl/en/Topics/N/NoroNet), by searching with a combination of the key words “food item,” “shellfish,” and “oyster”; and (iii) from the PubMed and Google Scholar literature databases (studies published between 1995 and 2014), by searching with a combination of the terms “norovirus,” “oyster,” “shellfish,” and “caliciviridae” included in the titles, key words, and abstracts (studies reported in languages other than English were excluded).
FIG 1.

Flow chart of sequence collection strategy.
All sequences were included for analysis if they met one of the following criteria: (i) they were obtained from oyster samples, or (ii) they were detected in human samples linked to oyster-related gastroenteritis outbreaks.
All oyster-related NoV sequences were downloaded in FASTA format. The bioinformatics software Geneious was used to construct sequence files and to edit the corresponding information for each sequence in a uniform format, e.g., sequence name or accession number, sequence length, genotype, host, sample source, submission time, isolation time, and geographic area (for details, see Table S1 in the supplemental material). Duplicate sequences were screened and were removed by comparing the accession numbers of the query sequences.
Genotyping and phylogenetic analysis.
The genogroups, genotypes, and GII.4 variants of the NoV sequences were determined using the RIVM Norovirus Genotyping Tool (http://www.rivm.nl/mpf/norovirus/typingtool) (31). This tool is designed to identify the norovirus genotypes or GII.4 variants by using phylogenetic analysis methods (31).
RESULTS
Sequence retrieval and quality control.
A total of 445 and 158 sequences were obtained from the GenBank nucleotide database by searching with “oyster AND norovirus” and “shellfish AND norovirus” as key words, respectively. Upon further examination of the sequence source and description, 9 and 109 sequences from these two sequence data sets were excluded from subsequent analysis (Fig. 1). From the RIVM NoroNet database, there were 247 sequences that met the study criteria, and 89 were removed after quality control (Fig. 1). The literature search yielded 159 citations that described 385 NoV sequences (Fig. 1), while only 14 research articles (8.8%) were about oyster-related NoV sequences (n = 302). All sequences selected and screened from these three independent sources were combined, and duplicate sequences were removed. In total, 670 sequences were used for subsequent study (Fig. 1). Surprisingly, after a BLASTN search was performed through the NCBI nr database, five sequences from the RIVM NoroNet database were identified as partial bacterial 16S rRNA gene sequences (see Table S2 in the supplemental material) and were excluded from genotyping. It is not clear why these sequences were included in the NoroNet database; they should be removed, because they are inaccurate. As a precaution, we recommend a quality control assay to confirm the novoviral nature of the unassigned sequences deposited in this database prior to further analysis.
Length distribution and genomic location.
The lengths of these 665 sequences ranged from 60 to 3,814 nucleotides (nt). Most were shorter than 400 nt (>95%); of these, 36 sequences (5.41%) were shorter than 80 nt. Six sequences were longer than 1,000 nt. All NoV sequences were located on the region of ORF1 or ORF2, or in the ORF1–ORF2 overlap region (see Fig. S2 in the supplemental material). Of all 665 sequences, 150 (22.56%) belonged to ORF1, and 508 (76.39%) belonged to ORF2. The remaining 7 sequences were located in the ORF1–ORF2 overlap region.
Genetic diversity.
The genotypes of 665 NoV nucleotide sequences were analyzed. More than 65.71% of the sequences were identified as genogroup II (GII), and 33.53% were identified as GI. Five sequences, which were detected in Hong Kong markets, belonged to GIV.
On the basis of ORF1 sequences (polymerase), 25 genotypes of NoV were identified. Ten genotypes belonged to GI (GI.P1, GI.P2, GI.P3, GI.P4, GI.P6, GI.P7, GI.P8, GI.P11, GI.Pa, and GI.Pb), and 15 genotypes belonged to GII (GII.P2, GII.P3, GII.P4, GII.P5, GII.P6, GII.P7, GII.P8, GII.P12, GII.P16, GII.P18, GII.P21, GII.P22, GII.Pb, GII.Pe, and GII.Pg). In GI, the predominant genotype was GI.P2, which accounted for as much as 41.51% of sequences, followed by GI.Pb (13.21%), GI.P1 (11.32%), and GI.P4 (11.32%) (Fig. 2A). In GII, GII.P4, which accounted for 32.63% of sequences, was the predominant genotype, followed by GII.Pg (13.68%) and GII.Pe (8.42%) (Fig. 2C). GII.P4 (31 of 157 sequences) was the most prevalent genotype among all ORF1 NoV sequences.
FIG 2.

Genetic diversity and prevalences of oyster-related NoVs based on ORF1 (A and C) or ORF2 (B and D). (A and B) Genogroup I; (C and D) genogroup II.
Twenty-three genotypes of NoV were identified on the basis of ORF2 sequences (capsid). Seven genotypes belonged to GI (GI.1, GI.2, GI.3, GI.4, GI.5, GI.6, and GI.7), and 16 genotypes belonged to GII (GII.1, GII.2, GII.3, GII.4, GII.5, GII.6, GII.7, GII.8, GII.9, GII.12, GII.13, GII.14, GII.16, GII.18, GII.20, and GII.21). The predominant genotype in GI was GI.4, which accounted for as much as 45.03% of sequences, followed by GI.3 (13.45%), GI.1 (11.11%), and GI.6 (8.77%) (Fig. 2B). In GII, GII.4 was the predominant genotype (29.28%), followed by GII.3 (16.23%) and GII.6 (9.57%) (Fig. 2D). GII.4 (101 of 515 sequences) was the most prevalent genotype among all ORF2 NoV sequences.
Prevalence of GII.4 variants.
A high diversity of GII.4 variants was found among all the NoV GII.4 sequences (n = 132) deposited from 1983 to 2014. Based on ORF1 sequences, 31 sequences were grouped into 5 variant groups, comprising the 2002 (Farmington Hills 2002), 2002CN (Lanzhou 2002), 2006a (Yerseke 2006a), 2006b (Den Haag 2006b), and 2010 (New Orleans 2009) variants (Fig. 3A). The 2010 variant was predominant, accounting for nearly 25.80% of the GII.4 sequences (n = 31). More than one-third of the sequences (14 of 31) could not be assigned to any known variant. Based on ORF2 sequences, 101 sequences were clustered into 6 variant groups, comprising the 2002 (Farmington Hills 2002), 2003 (Japan 2001), 2004 (Hunter 2004), 2006b (Den Haag 2006b), 2010 (New Orleans 2009), and 2012 (Sydney 2012) variants (Fig. 3B). The 2010 variants were predominant for ORF2 as well as for ORF1 sequences, accounting for nearly 20.79% of the GII.4 sequences (n = 101), followed by the 2012 (17.82%) and 2006b (13.86%) variants. Thirty-nine sequences could not be assigned to any known variant.
FIG 3.
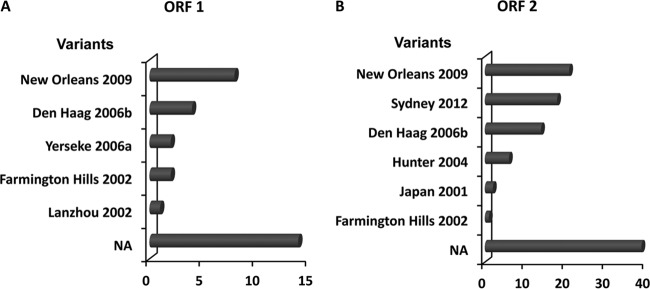
Diversity of NoV GII.4 variants based on ORF1 and ORF2. NA, GII.4 sequences not assigned to any known variants. The horizontal axis represents the numbers of sequences of GII.4 variants.
Global distribution.
These 665 oyster-related NoV sequences were obtained from 20 countries, which can be further grouped into four regions: Asia (China, Hong Kong, Japan, South Korea, Taiwan, and Thailand), Europe (Denmark, Finland, France, Germany, Ireland, Italy, the Netherlands, Spain, Sweden, and the United Kingdom), North America (Canada and the United States), and Oceania (Australia and New Zealand). Overall, the numbers of sequences in these regions were unevenly distributed, and certain countries were overrepresented (Fig. 4). Most sequences were reported from Asian (48.12%) and European (42.86%) countries and regions (Fig. 4). The number of sequences from Japan was the highest (35.04%), followed by those from Ireland (18.20%) and France (15.34%). Obviously, the majority of the NoV sequences were reported from coastal countries and regions (Fig. 4), such as Japan, New Zealand, France, Australia, and Ireland. Interestingly, no NoV sequences were obtained from inland countries, such as most African, South American, and Central Asian countries (Fig. 4).
FIG 4.

Geographical distributions of oyster-related NoVs from 1983 to 2014. The numbers of sequences distributed in different regions are shown in different colors. (Map template from Digital Vector Maps.)
With regard to genotypic prevalence, multiple NoV genotypes were found in an individual country or region (Table 1). Nearly one-half of oyster-related NoV genotypes (26 of 53) were detected in France, and this phenomenon was also very prominent in Japan (21 of 53), Hong Kong (16 of 53), Ireland (15 of 53), and Denmark (14 of 53) (Table 1). However, a large number of genotypes were detected only in a single geographical region—for example, GI.P8, GII.Pb, and GIV in Hong Kong, GI.Pa, GII.P16, and GII.P22 in New Zealand, and GI.P6 and GI.P11 in South Korea. Four genotypes—GII.9, GII.16, GII.18, and GII.20—were detected only in Australia, Japan, Taiwan, and Canada, respectively (Table 1).
TABLE 1.
Distribution of genotypes of oyster-related NoVs in different geographical regionsa
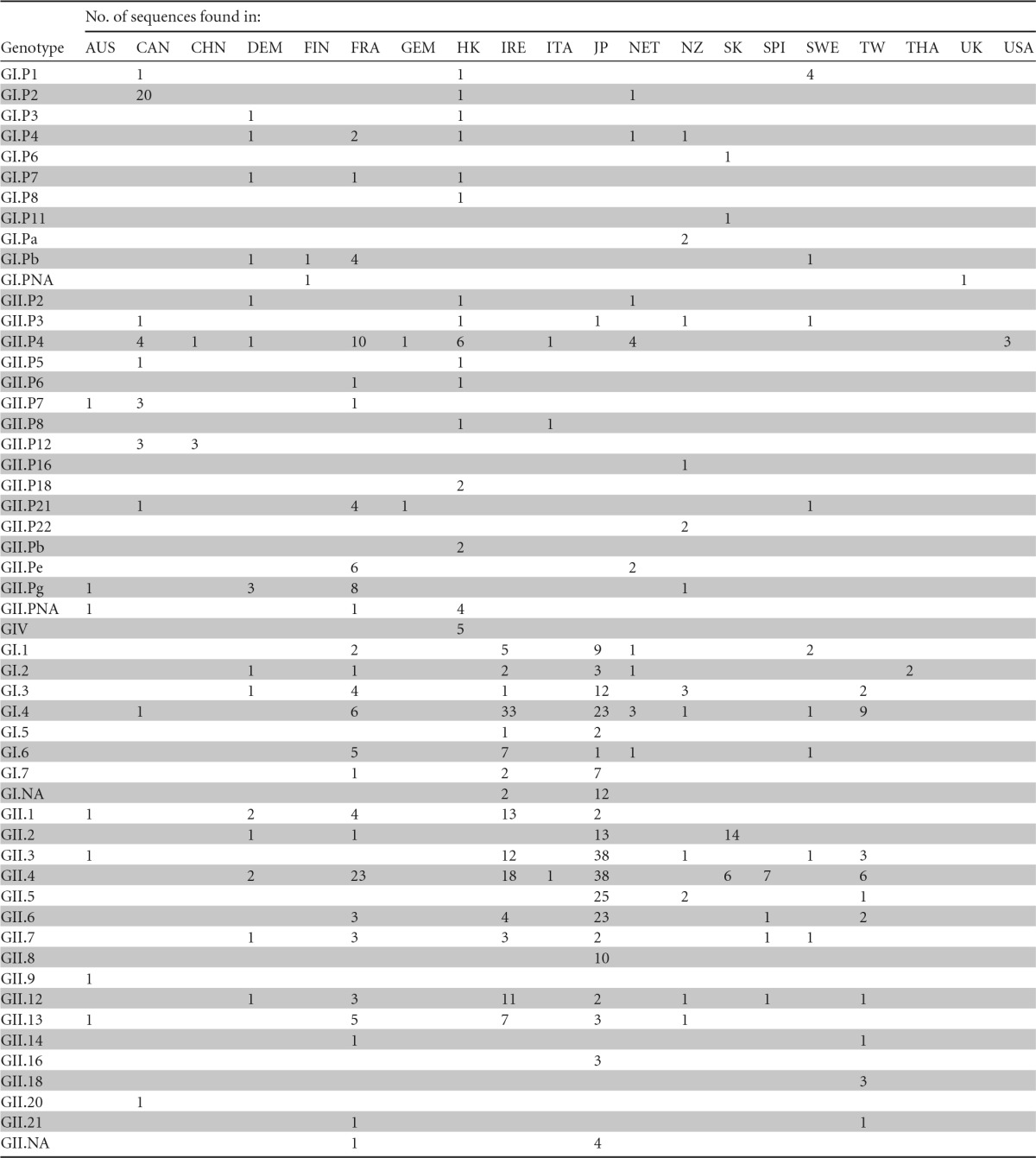
AUS, Australia; CAN, Canada; CHN, China; DEM, Denmark; FIN, Finland; FRA, France; GEM, Germany; HK, Hong Kong; IRE, Ireland; ITA, Italy; JP, Japan; NET, the Netherlands; NZ, New Zealand; SK, South Korea; SPI, Spain; SWE, Sweden; TW, Taiwan; THA, Thailand; UK, United Kingdom; USA, United States.
Yearly distribution.
Figure 5A shows the yearly distribution of oyster-related NoV sequences detected worldwide from 1983 to 2014. The number of sequences rose every 2 to 3 years from 2000 to 2014, increased significantly from 1994 to 2003, and reached a peak in 2003. Although this trend declined slightly during 2004 to 2009 (Fig. 5A), the number of sequences then increased dramatically and reached the highest peak in 2010. Almost 20% of oyster-related NoV sequences were documented in that year (n = 132). Another peak was found in 2012 (Fig. 5A).
FIG 5.

Yearly distribution of oyster-related NoV sequences from 1983 to 2014. (A) Distribution of the genotypes. The six dominant genotypes—GII.4, GI.4, GII.3, GII.6, GII.2, and GII.5—are shown in different colors. (B) Distribution of GII.4 variants. Different GII.4 variants are shown in different colors. The total number of sequences detected in a peak year is given above the peak.
As the predominant genotype, GII.4 also showed a similar trend during this time. For example, the total number of GII.4 sequences peaked in 2003, 2005, 2010, and 2013 as well (Fig. 5B). However, the circulation of GII.4 strains changed every 2 to 3 years due to their replacement by a newly emergent variant (see Fig. S3 in the supplemental material). The Farmington Hills 2002 variant emerged in 2002 and persisted to 2004, until the Hunter 2004 variant emerged in 2005. This pattern of regular replacement was also found with the Yerseke variant in 2006, the Den Haag variant in 2006, the New Orleans variant in 2009, and the Sydney variant in 2012 (Fig. 5B; see also Fig. S3).
DISCUSSION
GIV detected in Hong Kong oysters.
Little is known about the origin of the GIV NoVs, partly due to a lack of sequence data. Only 108 GIV NoV sequences, including 4 full-length GIV.1 genomes and 1 full-length GIV.2 genome, are available in GenBank (32). Of these, only one GIV NoV sequence (GenBank accession no. GU726163) was isolated from oyster samples, in Hong Kong in 2008.
In this study, another four NoV sequences (GenBank accession no. AY219906, AY219909, AY219910, and AY219911), which also originated from Hong Kong in 2002, were finally identified as GIV NoV sequences. In the original study, however, these four NoV sequences were defined as unclassified genetic clusters because they exhibited >25% divergence from previously published NoV sequences and from each other (19). To further confirm their associations, phylogenetic trees based on 73 nt (partial ORF1 sequence) were constructed and showed that these 4 sequences (GenBank accession no. AY219911, AY219906, AY219910, and AY219909) were clustered with GIV.2 sequences and with GenBank accession no. GU726163 (GIV.1) (see Fig. S4 in the supplemental material). In addition, these five sequences represented the first record of GIV NoVs in oyster samples (19). In contrast to GI and GII NoVs, GIV strains are rarely discovered in oyster samples but are usually identified in humans (GIV.1) (32, 33), dogs (GIV.1) (34–36), cats (GIV.2) (37), and lions (GIV.2) (38). The observation of relatively few GIV strains in oysters may be due to the fact that most routine screening and diagnostic studies on oysters so far have focused primarily on GI and GII NoVs (18, 23, 39, 40). The mechanisms by which oysters accumulate this rare genogroup of NoVs remain to be elucidated.
Unassigned sequences.
Genotyping results showed that more than 4% of NoV sequences (27 of 655), including 2 GI.PNA, 6 GII.PNA, 14 GI.NA, and 5 GII.NA sequences, could not be assigned to any known genotype (Table 1). One reason for this is that some relatively short sequences (>95% of sequences were shorter than 400 nt) could not be genotyped confidently using phylogenetic methods (see Fig. S2 in the supplemental material). In addition, some of these unassigned sequences may represent novel genotypes or variants that have not been documented thus far. Also, the possibility that some sequences may result from PCR bias cannot be ruled out completely.
Oysters accumulate diverse NoV genotypes.
It has been shown that histo-blood group antigens (HBGAs) on cells of the human gastrointestinal tract function as receptors or coreceptors for NoV infection (41, 42). Human susceptibility to NoV infection is determined by the types of ABH or Lewis HBGAs (43). Interestingly, no single HBGA can bind to all NoV genotypes, and there is also variability in binding profiles among strains of the same genotype, e.g., GII.4 strains (41).
Oysters selectively accumulate NoV strains using their HBGA analogues (24, 30, 44–46). Recently, various studies have shown that oysters, including Virginia oysters (Crassostrea virginica), Pacific oysters (Crassostrea gigas), and Kumamoto oysters (Crassostrea sikamea) (45), express only type A- and/or type O-like HBGAs in their gastrointestinal tissues (24, 25, 30, 44–47). However, the results from this study show that >80% of human NoV genotypes were detected in oyster samples or oyster-related outbreaks (Fig. 2). In addition, a recent report (48) showed that GIV.2 strains that are rarely discovered in oyster samples bind to HBGAs with a specificity very similar to that of GI.1, which is known to accumulate efficiently in oysters through HBGA attachment. Given this information, how is it possible that oysters with limited HBGAs (type A and/or O) can accumulate such diverse NoV genotypes? Unlike humans, oysters can aggregate a broad range of NoV genotypes. However, given that they act as the transmission vector rather than the natural host of NoVs (49), the requirement for an efficient binding capacity seems unnecessary.
In addition to HBGA-specific recognition, the binding of NoVs to oyster tissues may also involve nonspecific interactions (24, 50). However, this nonspecific attachment is assumed to be less efficient than HBGA-mediated binding (24), and the persistence of NoVs in oysters is possibly shorter in response to nonspecific interactions (30). Accordingly, it seems unlikely that a high number of NoVs is maintained in oysters via nonspecific binding mechanisms. Given that the diagnostic limitation of NoVs is normally 10 to 1,000 copies per g of oyster tissue (23, 40, 51), it is possible to detect NoVs only if there is a high abundance in the oyster samples tested. Taking these considerations together, the possibility of nonspecific attachment seems an unlikely cause of the diverse NoV genotypes observed in oysters.
Alternatively, there may be other binding ligands or vectors in oyster tissues that contribute to NoV enrichment in oysters. Recently, a group of commensal enteric bacteria expressing HBGA-like carbohydrates were shown to be associated with norovirus binding and infection (52). Furthermore, the sialic acid-containing ligand present in all oyster tissues may also contribute to the binding or retention of NoV particles (25, 30). Interestingly, sialic acids are distributed largely on the surfaces of certain enteric bacteria as well (53, 54). Thus, it is reasonable to speculate that HBGA-expressing bacteria may play key roles as binding partners in the enrichment of NoVs in oysters (55). While this hypothesis remains speculative and requires further evaluation, it potentially helps to explain the diversity of NoV genotypes found in oysters with limited HBGAs (types A and O).
Difference in prevalence between oyster-related and human-related NoV genotypes.
Almost all human NoV genogroups, genotypes (except for certain rare genotypes), or variants are found in oyster samples (Fig. 2). However, the prevalence of each genogroup detected in oyster-related outbreaks is clearly distinct from that in non-oyster-related human NoV outbreaks. It has been shown that approximately 8% of all NoV sequences collected worldwide from 4,228 human outbreaks in 23 countries and regions during 2010 and 2013 represent GI and GII and that GII accounts for nearly 92% of these sequences (15). In contrast, in this study, more than 65.71% of oyster-related NoV sequences were classified as GII, and 33.53% of the sequences belonged to GI. In addition, a difference in the prevalence of genotypes was observed. For example, GII.4, the predominant genotype, accounts for 19.85% (132 of 665) of oyster-related NoV sequences (Fig. 2). However, nearly 68.17% of NoV sequences were identified as belonging to genotype GII.4 in human outbreaks (15).
Similar observations were also documented in the work of Le Guyader and colleagues (24, 25, 30). This suggests that some GI genotypes, such as GI.1 strains, accumulate efficiently and are retained through an HBGA A-like ligand present in the oyster gut (25, 30), while GII strains accumulate less due to a sialic acid-containing ligand expressed in all tissues that contributes to their retention in the gills and leads to their destruction (or elimination) by an unknown mechanism (24, 25, 30).
Does the difference in prevalence between GI and GII oyster-related NoV sequences result from the disparity in the abundances of NoV genotypes in the water where oysters live? Fortunately, various studies have compared the genetic profiles of NoV genogroups or genotypes in different environments (56, 57). These studies detected much higher concentrations of GII than of GI NoVs in the waters tested; however, GI NoVs were enriched to a greater degree than GII strains (56, 57). Accordingly, oysters do not act just as passive filters of human NoVs in the environment but rather direct the specific selection and persistence of different NoVs (26). These observations may help us to understand the discrepancy in GI/GII distribution between oyster-related and non-oyster-related human outbreaks.
Geographical distribution of NoVs.
Almost all the oyster-related NoV sequences were obtained from the regions of North America, Europe, Asia, and Oceania; there were no sequences derived from the tropical regions, such as Africa, South America, and most of Southeast Asia (Fig. 4). Coincidentally, epidemiological studies also show that NoV outbreaks often occurred during cold seasons (below 10°C) (27, 28, 58), and rare outbreaks took place in tropical regions (17, 59). It is well known that in tropical regions, seasonal temperature remains relatively constant at approximately 18°C throughout the year (http://en.wikipedia.org/wiki/Tropics). Recently, it has been shown that the capacity of oysters to bind to NoVs can be affected by environmental conditions, especially water temperature (29, 30). High water temperature may regulate or inhibit the expression of oyster HBGAs or suppress the spread of NoVs. Accordingly, few oyster-related NoV outbreaks occur in tropical regions. However, it should be noted that the surveillance systems in some tropical regions, i.e., most African countries, may not be as efficient as those in more-developed countries, which may lead to the underreporting of NoV outbreaks.
Oyster-related NoVs are found mainly in coastal regions worldwide. Interestingly, the major regions of oyster aquaculture are also located on the coastlines of Northeast Asia (China and Japan), North America (Canada and the United States), and most of Europe and Oceania (Australia and New Zealand) (http://www.fao.org/figis/geoserver/factsheets/species.html; http://www.fao.org/fishery/species/3514/en). In addition, NoV outbreaks associated with oyster consumption have been well documented (12–14, 21, 60). For example, in our previous study, approximately 90% of human NoV sequences were discovered in coastal regions and likely resulted from the consumption of NoV-contaminated oysters (61). Therefore, the circulation of NoVs between the environment and humans depends largely on the cultivation and consumption of oysters.
Yearly distribution trends.
Human pandemic NoV outbreaks occurred in 2002, 2004, 2006, 2009, and 2012 (10, 62). In oyster-related samples, NoV sequences peaked in 2003, 2005 to 2006, 2010, and 2012 (Fig. 5A). This indicates that oysters not only act as the transmission vector of human NoVs in the environment but also are an important reservoir of NoVs. That is to say, once a new NoV genotype or variant emerges from the human host, oysters can accumulate this strain, thus preserving it for a period. The oyster then essentially waits for the opportunity to bring the virus back to the host. However, it is still not known whether this adaptation of oysters for the accumulation of NoVs provides both organisms some type of benefit in a symbiotic relationship. Future studies should focus on the molecular interactions between oysters and various types of NoVs.
Limitations of this study.
The analysis of data collected from an international surveillance database (NoroNet) and published literature is potentially associated with various kinds of biases (59). NoroNet is the only source of surveillance data used in this study. This database is generated by an informal network that shares virologic, epidemiologic, and molecular data on NoVs (59). Although NoroNet now includes laboratories in countries outside Europe, the majority of the data is submitted by European countries (http://www.rivm.nl/en/Topics/N/NoroNet). This may partially explain why there are few oyster-related NoV sequences from some countries and regions, especially the United States and Australia (Fig. 4). Recently, several state-based NoV outbreak surveillance networks have also been developed, e.g., CaliciNet (United States), EpiSurv (New Zealand), and OzFoodNet (Australia), but most of these databases are not openly accessible. Thus, in order to avoid data bias, most of the sequences analyzed in this study were downloaded from both the NCBI and the NoroNet database; fewer than one-quarter of the sequences used in this study (153 of 665) were retrieved from the NoroNet database alone.
Due to the lack of a reliable authorized method for the detection of NoVs in oyster and clinical samples (4), both the lengths and the genome-targeting regions of these viral sequences differ greatly in databases (see Fig. S2 and Table S1 in the supplemental material). In addition, the target regions for genotyping are either polymerase (ORF1) or VP1 (ORF2), or both ORF1 and ORF2. Therefore, the genotyping of NoV sequences may produce biases (59). In 2013, an international group of NoV experts proposed a dual typing system based on complete capsid (VP1) and partial polymerase (1,300 nt) sequences (4). However, this system could not be used in this study, because the sequences analyzed were obtained from 1983 to 2014. For stringency, every sequence in this study was genotyped using the RIVM online autotyping tool, which is specialized for the genotyping of NoVs.
In conclusion, the global geographical distribution and genetic diversity of oyster-related NoVs deposited in databases from 1983 to 2014 were analyzed. A high degree of genetic diversity was observed for oyster-related NoVs, and almost all the human NoV genotypes were found in oyster-related NoV sequences. These sequences were widely but unevenly distributed geographically, and most of them were detected in coastal regions. A higher frequency of GI strains was found in oyster-related than in human-related NoV sequences, while the yearly distributions of oyster-related sequences and human outbreak sequences were similar, indicating that oysters may act as a reservoir of NoVs in the environment. The results presented in this study contribute to the understanding of the environmental transmission of NoVs via oysters and may also be helpful for the evaluation and implementation of appropriate measures for monitoring NoV infections throughout the world.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NoroNet network and the anonymous contributors for collecting and sharing the NoV sequences.
This work was partially supported by the National Natural Science Foundation of China (41376135), the National High Technology Research and Development Program of China (863 Program; 2014AA093506), the Doctoral Fund of the Ministry of Education of China (20133104110006), the Innovation Program of the Shanghai Municipal Education Commission (14ZZ144), Shanghai, China, and the Special Fund for the Development of Science and Technology of Shanghai Ocean University (2015).
We thank the three anonymous reviewers for insightful comments on the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01729-15.
REFERENCES
- 1.Glass RI, Parashar UD, Estes MK. 2009. Norovirus gastroenteritis. N Engl J Med 361:1776–1785. doi: 10.1056/NEJMra0804575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xi J, Graham DY, Wang K, Estes MK. 1990. Norwalk virus genome cloning and characterization. Science 250:1580–1583. doi: 10.1126/science.2177224. [DOI] [PubMed] [Google Scholar]
- 3.Zheng D-P, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS. 2006. Norovirus classification and proposed strain nomenclature. Virology 346:312–323. doi: 10.1016/j.virol.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Kroneman A, Vega E, Vennema H, Vinjé J, White PA, Hansman G, Green K, Martella V, Katayama K, Koopmans M. 2013. Proposal for a unified norovirus nomenclature and genotyping. Arch Virol 158:2059–2068. doi: 10.1007/s00705-013-1708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siebenga JJ, Vennema H, Renckens B, de Bruin E, van der Veer B, Siezen RJ, Koopmans M. 2007. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J Virol 81:9932–9941. doi: 10.1128/JVI.00674-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindesmith LC, Donaldson EF, LoBue AD, Cannon JL, Zheng D-P, Vinjé J, Baric RS. 2008. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med 5:e31. doi: 10.1371/journal.pmed.0050031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siebenga JJ, Vennema H, Zheng D-P, Vinjé J, Lee BE, Pang X-L, Ho EC, Lim W, Choudekar A, Broor S. 2009. Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001–2007. J Infect Dis 200:802–812. doi: 10.1086/605127. [DOI] [PubMed] [Google Scholar]
- 8.Bull RA, White PA. 2011. Mechanisms of GII.4 norovirus evolution. Trends Microbiol 19:233–240. doi: 10.1016/j.tim.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Xia M, Tan M, Huang P, Zhong W, Pang XL, Lee BE, Meller J, Wang T, Jiang X. 2010. Genetic and phenotypic characterization of GII-4 noroviruses that circulated during 1987 to 2008. J Virol 84:9595–9607. doi: 10.1128/JVI.02614-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eden J-S, Tanaka MM, Boni MF, Rawlinson WD, White PA. 2013. Recombination within the pandemic norovirus GII.4 lineage. J Virol 87:6270–6282. doi: 10.1128/JVI.03464-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Beek J, Ambert-Balay K, Botteldoorn N, Eden J, Fonager J, Hewitt J, Iritani N, Kroneman A, Vennema H, Vinjé J. 2013. Indications for worldwide increased norovirus activity associated with emergence of a new variant of genotype II.4, late 2012. Euro Surveill 18:8–9. http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20345. [PubMed] [Google Scholar]
- 12.Alfano-Sobsey E, Sweat D, Hall A, Breedlove F, Rodriguez R, Greene S, Pierce A, Sobsey M, Davies M, Ledford S. 2012. Norovirus outbreak associated with undercooked oysters and secondary household transmission. Epidemiol Infect 140:276–282. doi: 10.1017/S0950268811000665. [DOI] [PubMed] [Google Scholar]
- 13.McIntyre L, Galanis E, Mattison K, Mykytczuk O, Buenaventura E, Wong J, Prystajecky N, Ritson M, Stone J, Moreau D. 2012. Multiple clusters of norovirus among shellfish consumers linked to symptomatic oyster harvesters. J Food Prot 75:1715–1720. doi: 10.4315/0362-028X.JFP-12-113. [DOI] [PubMed] [Google Scholar]
- 14.Iritani N, Kaida A, Abe N, Kubo H, Sekiguchi JI, Yamamoto SP, Goto K, Tanaka T, Noda M. 2014. Detection and genetic characterization of human enteric viruses in oyster-associated gastroenteritis outbreaks between 2001 and 2012 in Osaka City, Japan. J Med Virol 86:2019–2025. doi: 10.1002/jmv.23883. [DOI] [PubMed] [Google Scholar]
- 15.van Beek J, Kroneman A, Vennema H, Koopmans M. 2013. Noronet report, April 2013. National Institute for Public Health and the Environment, Bilthoven, The Netherlands: http://www.rivm.nl/en/Documents_and_publications/Common_and_Present/Publications/Centre_for_Infectious_Disease_Control/Noronet_updates/NoroNet_update_april_2013. [Google Scholar]
- 16.Mathijs E, Stals A, Baert L, Botteldoorn N, Denayer S, Mauroy A, Scipioni A, Daube G, Dierick K, Herman L. 2012. A review of known and hypothetical transmission routes for noroviruses. Food Environ Virol 4:131–152. doi: 10.1007/s12560-012-9091-z. [DOI] [PubMed] [Google Scholar]
- 17.Bitler E, Matthews J, Dickey B, Eisenberg J, Leon J. 2013. Norovirus outbreaks: a systematic review of commonly implicated transmission routes and vehicles. Epidemiol Infect 141:1563–1571. doi: 10.1017/S095026881300006X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowther JA, Gustar NE, Powell AL, Hartnell RE, Lees DN. 2012. Two-year systematic study to assess norovirus contamination in oysters from commercial harvesting areas in the United Kingdom. Appl Environ Microbiol 78:5812–5817. doi: 10.1128/AEM.01046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng PK, Wong DK, Chung TW, Lim WW. 2005. Norovirus contamination found in oysters worldwide. J Med Virol 76:593–597. doi: 10.1002/jmv.20402. [DOI] [PubMed] [Google Scholar]
- 20.Nishida T, Kimura H, Saitoh M, Shinohara M, Kato M, Fukuda S, Munemura T, Mikami T, Kawamoto A, Akiyama M. 2003. Detection, quantitation, and phylogenetic analysis of noroviruses in Japanese oysters. Appl Environ Microbiol 69:5782–5786. doi: 10.1128/AEM.69.10.5782-5786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webby R, Carville K, Kirk M, Greening G, Ratcliff R, Crerar S, Dempsey K, Sarna M, Stafford R, Patel M. 2007. Internationally distributed frozen oyster meat causing multiple outbreaks of norovirus infection in Australia. Clin Infect Dis 44:1026–1031. doi: 10.1086/512807. [DOI] [PubMed] [Google Scholar]
- 22.Nappier SP, Graczyk TK, Schwab KJ. 2008. Bioaccumulation, retention, and depuration of enteric viruses by Crassostrea virginica and Crassostrea ariakensis oysters. Appl Environ Microbiol 74:6825–6831. doi: 10.1128/AEM.01000-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terio V, Martella V, Moschidou P, Di Pinto P, Tantillo G, Buonavoglia C. 2010. Norovirus in retail shellfish. Food Microbiol 27:29–32. doi: 10.1016/j.fm.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 24.Le Guyader FS, Atmar RL, Le Pendu J. 2012. Transmission of viruses through shellfish: when specific ligands come into play. Curr Opin Virol 2:103–110. doi: 10.1016/j.coviro.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maalouf H, Schaeffer J, Parnaudeau S, Le Pendu J, Atmar RL, Crawford SE, Le Guyader FS. 2011. Strain-dependent norovirus bioaccumulation in oysters. Appl Environ Microbiol 77:3189–3196. doi: 10.1128/AEM.03010-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Guyader S, Atmar R, Maalouf H, Le Pendu J. 2013. Shellfish contamination by norovirus: strain selection based on ligand expression? Clin Virol 41:3–18. [Google Scholar]
- 27.Dey SK, Phathammavong O, Okitsu S, Mizuguchi M, Ohta Y, Ushijima H. 2010. Seasonal pattern and genotype distribution of norovirus infection in Japan. Pediatr Infect Dis J 29:e32–e34. doi: 10.1097/INF.0b013e3181d742bf. [DOI] [PubMed] [Google Scholar]
- 28.Iritani N, Kaida A, Kubo H, Abe N, Goto K, Ogura H, Seto Y. 2010. Molecular epidemiology of noroviruses detected in seasonal outbreaks of acute nonbacterial gastroenteritis in Osaka City, Japan, from 1996–1997 to 2008–2009. J Med Virol 82:2097–2105. doi: 10.1002/jmv.21915. [DOI] [PubMed] [Google Scholar]
- 29.Maalouf H, Pommepuy M, Le Guyader FS. 2010. Environmental conditions leading to shellfish contamination and related outbreaks. Food Environ Virol 2:136–145. doi: 10.1007/s12560-010-9043-4. [DOI] [Google Scholar]
- 30.Maalouf H, Zakhour M, Le Pendu J, Le Saux JC, Atmar RL, Le Guyader FS. 2010. Distribution in tissue and seasonal variation of norovirus genogroup I and II ligands in oysters. Appl Environ Microbiol 76:5621–5630. doi: 10.1128/AEM.00148-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroneman A, Vennema H, Deforche K, Avoort H, Penaranda S, Oberste M, Vinjé J, Koopmans M. 2011. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol 51:121–125. doi: 10.1016/j.jcv.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Eden J-S, Lim KL, White PA. 2012. Complete genome of the human norovirus GIV.1 strain Lake Macquarie virus. J Virol 86:10251–10252. doi: 10.1128/JVI.01604-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ao Y-Y, Yu J-M, Li L-L, Jin M, Duan Z-J. 2014. Detection of human norovirus GIV.1 in China: a case report. J Clin Virol 61:298–301. doi: 10.1016/j.jcv.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Martella V, Lorusso E, Decaro N, Elia G, Radogna A, D'Abramo M, Desario C, Cavalli A, Corrente M, Camero M. 2008. Detection and molecular characterization of a canine norovirus. Emerg Infect Dis 14:1306–1308. doi: 10.3201/eid1408.080062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martella V, Decaro N, Lorusso E, Radogna A, Moschidou P, Amorisco F, Lucente MS, Desario C, Mari V, Elia G. 2009. Genetic heterogeneity and recombination in canine noroviruses. J Virol 83:11391–11396. doi: 10.1128/JVI.01385-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mesquita JR, Barclay L, Nascimento MSJ, Vinjé J. 2010. Novel norovirus in dogs with diarrhea. Emerg Infect Dis 16:980–982. doi: 10.3201/eid1606.091861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinto P, Wang Q, Chen N, Dubovi EJ, Daniels JB, Millward LM, Buonavoglia C, Martella V, Saif LJ. 2012. Discovery and genomic characterization of noroviruses from a gastroenteritis outbreak in domestic cats in the US. PLoS One 7:e32739. doi: 10.1371/journal.pone.0032739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martella V, Campolo M, Lorusso E, Cavicchio P, Camero M, Bellacicco AL, Decaro N, Elia G, Greco G, Corrente M. 2007. Norovirus in captive lion cub (Panthera leo). Emerg Infect Dis 13:1071–1073. doi: 10.3201/eid1307.070268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benabbes L, Ollivier J, Schaeffer J, Parnaudeau S, Rhaissi H, Nourlil J, Le Guyader FS. 2013. Norovirus and other human enteric viruses in Moroccan shellfish. Food Environ Virol 5:35–40. doi: 10.1007/s12560-012-9095-8. [DOI] [PubMed] [Google Scholar]
- 40.Schaeffer J, Le Saux J-C, Lora M, Atmar RL, Le Guyader FS. 2013. Norovirus contamination on French marketed oysters. Int J Food Microbiol 166:244–248. doi: 10.1016/j.ijfoodmicro.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan M, Jiang X. 2011. Norovirus–host interaction: multi-selections by human histo-blood group antigens. Trends Microbiol 19:382–388. doi: 10.1016/j.tim.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rockx BH, Vennema H, Hoebe CJ, Duizer E, Koopmans MP. 2005. Association of histo-blood group antigens and susceptibility to norovirus infections. J Infect Dis 191:749–754. doi: 10.1086/427779. [DOI] [PubMed] [Google Scholar]
- 43.Tan M, Jiang X. 2010. Norovirus gastroenteritis, carbohydrate receptors, and animal models. PLoS Pathog 6:e1000983. doi: 10.1371/journal.ppat.1000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian P, Bates AH, Jensen HM, Mandrell R. 2006. Norovirus binds to blood group A-like antigens in oyster gastrointestinal cells. Lett Appl Microbiol 43:645–651. doi: 10.1111/j.1472-765X.2006.02010.x. [DOI] [PubMed] [Google Scholar]
- 45.Tian P, Engelbrektson AL, Jiang X, Zhong W, Mandrell RE. 2007. Norovirus recognizes histo-blood group antigens on gastrointestinal cells of clams, mussels, and oysters: a possible mechanism of bioaccumulation. J Food Prot 70:2140–2147. [DOI] [PubMed] [Google Scholar]
- 46.Tian P, Engelbrektson AL, Mandrell RE. 2008. Seasonal tracking of histo-blood group antigen expression and norovirus binding in oyster gastrointestinal cells. J Food Prot 71:1696–1700. [DOI] [PubMed] [Google Scholar]
- 47.Le Guyader FS, Loisy F, Atmar RL, Hutson AM, Estes MK, Ruvoën-Clouet N, Pommepuy M, Le Pendu J. 2006. Norwalk virus-specific binding to oyster digestive tissues. Emerg Infect Dis 12:931–936. doi: 10.3201/eid1206.051519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caddy S, Breiman A, le Pendu J, Goodfellow I. 2014. Genogroup IV and VI canine noroviruses interact with histo-blood group antigens. J Virol 88:10377–10391. doi: 10.1128/JVI.01008-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel MM, Hall AJ, Vinjé J, Parashar UD. 2009. Noroviruses: a comprehensive review. J Clin Virol 44:1–8. doi: 10.1016/j.jcv.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Zakhour M, Maalouf H, Di Bartolo I, Haugarreau L, Le Guyader FS, Ruvoën-Clouet N, Le Saux J-C, Ruggeri FM, Pommepuy M, Le Pendu J. 2010. Bovine norovirus: carbohydrate ligand, environmental contamination, and potential cross-species transmission via oysters. Appl Environ Microbiol 76:6404–6411. doi: 10.1128/AEM.00671-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brake F, Ross T, Holds G, Kiermeier A, McLeod C. 2014. A survey of Australian oysters for the presence of human noroviruses. Food Microbiol 44:264–270. doi: 10.1016/j.fm.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinjé J. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. 2004. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev 68:132–153. doi: 10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caroff M, Karibian D. 2003. Structure of bacterial lipopolysaccharides. Carbohydr Res 338:2431–2447. doi: 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Karst SM, Wobus CE. 2015. A working model of how noroviruses infect the intestine. PLoS Pathog 11:e1004626. doi: 10.1371/journal.ppat.1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flannery J, Keaveney S, Rajko-Nenow P, O'Flaherty V, Doré W. 2012. Concentration of norovirus during wastewater treatment and its impact on oyster contamination. Appl Environ Microbiol 78:3400–3406. doi: 10.1128/AEM.07569-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rajko-Nenow P, Waters A, Keaveney S, Flannery J, Tuite G, Coughlan S, O'Flaherty V, Doré W. 2013. Norovirus genotypes present in oysters and in effluent from a wastewater treatment plant during the seasonal peak of infections in Ireland in 2010. Appl Environ Microbiol 79:2578–2587. doi: 10.1128/AEM.03557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahmed SM, Lopman BA, Levy K. 2013. A systematic review and meta-analysis of the global seasonality of norovirus. PLoS One 8:e75922. doi: 10.1371/journal.pone.0075922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verhoef L, Hewitt J, Barclay L, Ahmed S, Lake R, Hall AJ, Lopman B, Kroneman A, Vennema H. 2015. Norovirus genotype profiles associated with foodborne transmission, 1999–2012. Emerg Infect Dis 21:592–599. doi: 10.3201/eid2104.141073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nenonen NP, Hannoun C, Olsson MB, Bergström T. 2009. Molecular analysis of an oyster-related norovirus outbreak. J Clin Virol 45:105–108. doi: 10.1016/j.jcv.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 61.Yu Y, Yan S, Li B, Pan Y, Wang Y. 2014. Genetic diversity and distribution of human norovirus in China (1999–2011). Biomed Res Int 2014:196169. doi: 10.1155/2014/196169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karst SM, Baric RS. 2015. What is the reservoir of emergent human norovirus strains? J Virol 89:5756–5759. doi: 10.1128/JVI.03063-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.