

Abstract
Flavobacterium columnare is an important bacterial pathogen of freshwater fish that causes high mortality of infected fish and heavy economic losses in aquaculture. The pathogenesis of this bacterium is poorly understood, in part due to the lack of efficient methods for genetic manipulation. In this study, a gene deletion strategy was developed and used to determine the relationship between the production of chondroitin lyases and virulence. The F. johnsoniae ompA promoter (PompA) was fused to sacB to construct a counterselectable marker for F. columnare. F. columnare carrying PompA-sacB failed to grow on media containing 10% sucrose. A suicide vector carrying PompA-sacB was constructed, and a gene deletion strategy was developed. Using this approach, the chondroitin lyase-encoding genes, cslA and cslB, were deleted. The ΔcslA and ΔcslB mutants were both partially deficient in digestion of chondroitin sulfate A, whereas a double mutant (ΔcslA ΔcslB) was completely deficient in chondroitin lyase activity. Cells of F. columnare wild-type strain G4 and of the chondroitin lyase-deficient ΔcslA ΔcslB mutant exhibited similar levels of virulence toward grass carp in single-strain infections. Coinfections, however, revealed a competitive advantage for the wild type over the chondroitin lyase mutant. The results indicate that chondroitin lyases are not essential virulence factors of F. columnare but may contribute to the ability of the pathogen to compete and cause disease in natural infections. The gene deletion method developed in this study may be employed to investigate the virulence factors of this bacterium and may have wide application in many other members of the phylum Bacteroidetes.
INTRODUCTION
Columnaris disease can affect almost all species of freshwater fish, including cultured and wild fish, often resulting in heavy economic losses in aquaculture worldwide (1, 2). Its causative agent, Flavobacterium columnare, has been the focus of microbiological as well as aquaculture research for decades, with respect to diagnosis, epidemiology, pathology, preventive measures, and possible virulence factors (2–4). The genetic diversity of F. columnare has been recognized by the presence of at least three distinct genomovars (5–7). However, the pathogenesis of this important pathogen is still rather unclear. Although a few molecules, such as chondroitin AC lyase, have been reported as possible virulence factors (8–11), the lack of genetic manipulation systems suitable for F. columnare has been an obstacle in understanding the mechanisms involved in the pathogenesis of this bacterium. Staroscik et al. (12) developed techniques to introduce replicative plasmids into F. columnare by conjugation and also to construct mutants by insertional mutagenesis and transposon-mediated mutagenesis. However, both processes remain inefficient, and few subsequent reports utilizing these techniques have been published (12, 13).
A technique to construct in-frame deletions was recently developed for the genetically tractable bacterium Flavobacterium johnsoniae (14). This involved the isolation of a streptomycin-resistant rpsL mutant of F. johnsoniae and the use of wild-type rpsL as a counterselectable marker (14). While this approach has been very successful, one disadvantage is that deletion mutants must be constructed in an rpsL mutant rather than in wild-type cells. In some other Gram-negative bacteria, sacB, which confers sensitivity to sucrose, has been used as a counterselectable marker to allow isolation of gene deletion mutants from wild-type cells (15–17). Previous attempts to adapt this approach to F. johnsoniae or to other members of the phylum Bacteroidetes were not successful (18, 19).
Here we demonstrate techniques to efficiently isolate targeted in-frame gene deletion mutants of F. columnare. Deletion mutants were isolated using an rpsL-based system similar to the one described for F. johnsoniae (14) and using a sacB-based system in which a Flavobacterium promoter drove expression of sacB. Gene deletion coupled with complementation analysis was used to characterize two genes encoding chondroitin lyases and to determine their roles in virulence.
MATERIALS AND METHODS
Bacterial strains and media.
Bacterial strains and plasmids used in this study are listed in Table 1. F. columnare strain G4 was grown at 28°C in Shieh medium or on Shieh agar (20), and its genomovar is currently being determined. Escherichia coli strains were cultured in Luria broth (LB) liquid medium or on LB agar at 37°C. When necessary, antibiotics (Sigma) were added at the following concentrations: 1 μg/ml tobramycin for F. columnare, 100 μg/ml or 10 μg/ml streptomycin for F. columnare, 15 μg/ml tetracycline for F. columnare, and 100 μg/ml ampicillin for E. coli.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics or genotypea | Reference or source |
|---|---|---|
| Strains | ||
| E. coli strain | ||
| S17-1 λpir | F− RP4-2-Tc::Mu aphA::Tn7 recA λpir; lysogen | 28 |
| F. columnare strains | ||
| G4 | Wild type; grass carp isolate | 29 |
| G4S | rpsL2 Smr | This study |
| G4S ΔcslA | rpsL2 mutant with chondroitin AC lyase gene null mutation; Smr | This study |
| ΔcslA | Chondroitin AC lyase gene null mutant; strain obtained from G4 | This study |
| ΔcslB | Chondroitin ABC lyase gene null mutant; strain obtained from G4 | This study |
| ΔcslA ΔcslB | Chondroitin AC and ABC lyase gene null mutant; strain obtained from ΔcslA | This study |
| Plasmids | ||
| pCP23 | E. coli-F. columnare shuttle plasmid; Apr (Tcr) | 30 |
| pCP29 | E. coli-F. columnare shuttle plasmid; Apr (Cfr Emr) | 31 |
| pCP23-rpsl | rpsL gene cloned into KpnI sites of pCP23; Apr (Tcr) | This study |
| pCS | Suicide plasmid derived from pCP23; Apr (Tcr) | This study |
| pCSR | rpsL-containing suicide vector; Apr (Tcr) | This study |
| pCSR-cslA | 2.0-kbp region upstream of cslA cloned into BamHI and SalI sites of pCSR; Apr (Tcr) | This study |
| pCSR-csl | Construct used to delete cslA in G4S; 2.2-kbp region downstream of cslA cloned into SalI and SphI sites of pCSR-cslA; Apr (Tcr) | This study |
| pMS75 | sacB-containing suicide vector; Apr (Tcr) | This study |
| pMS-cslA | Construct used to delete cslA in G4; 2.0-kbp region upstream of cslA fused to 2.2-kbp region downstream of cslA and cloned into BamHI and SphI sites of pMS75; Apr (Tcr) | This study |
| pMS-csl2A | 2.6-kbp region downstream of cslB cloned into KpnI and SalI sites of pMS75; Apr (Tcr) | This study |
| pMS-cslB | Construct used to delete cslB in G4 or ΔcslA mutant; 2.2-kbp region upstream of cslB cloned into SalI and SphI sites of pMS-csl2A; Apr (Tcr) | This study |
| pCP-A | 4.6-kbp fragment spanning cslA and its promoter region cloned into KpnI and PstI sites of pCP23; Apr (Tcr) | This study |
| pCP-B | 4.1-kbp fragment spanning cslB and its promoter region cloned into PstI and SphI sites of pCP23; Apr (Tcr) | This study |
| pCP-AB | 4.1-kbp fragment spanning cslB and its promoter region cloned into PstI and SphI sites of pCP-A; Apr (Tcr) | This study |
| pAS43 | gfp-containing shuttle plasmid; Apr (Emr) | 12 |
| pCP-gfp | gfp-containing shuttle plasmid originated from pCP23; Apr (Tcr) | This study |
| pDS-RED2-1 | rfp-containing shuttle plasmid; Kmr | Clontech Inc. |
| pCP-rfp | rfp-containing shuttle plasmid; Apr (Tcr) | This study |
Antibiotic resistance phenotypes: Apr, ampicillin resistant; Cfr, cefoxitin resistant; Emr, erythromycin resistant; Smr, streptomycin resistant; Tcr, tetracycline resistant. Unless otherwise indicated, the antibiotic resistance phenotypes are those expressed in E. coli. The antibiotic resistance phenotypes in parentheses are those expressed in Flavobacterium but not in E. coli.
Isolation of the streptomycin-resistant rpsL mutant G4S.
Streptomycin-resistant F. columnare cells were obtained by plating 109 wild-type cells on Shieh agar containing 100 μg/ml streptomycin. Streptomycin-resistant clones were streaked on fresh medium for isolation, and the rpsL gene from each clone was PCR amplified using primers rpslKpnF and rpslKpnR (Table 2) before sequencing. Point mutations in rpsL conferring streptomycin resistance were identified by comparison to the wild-type rpsL sequence.
TABLE 2.
Primers used in this study
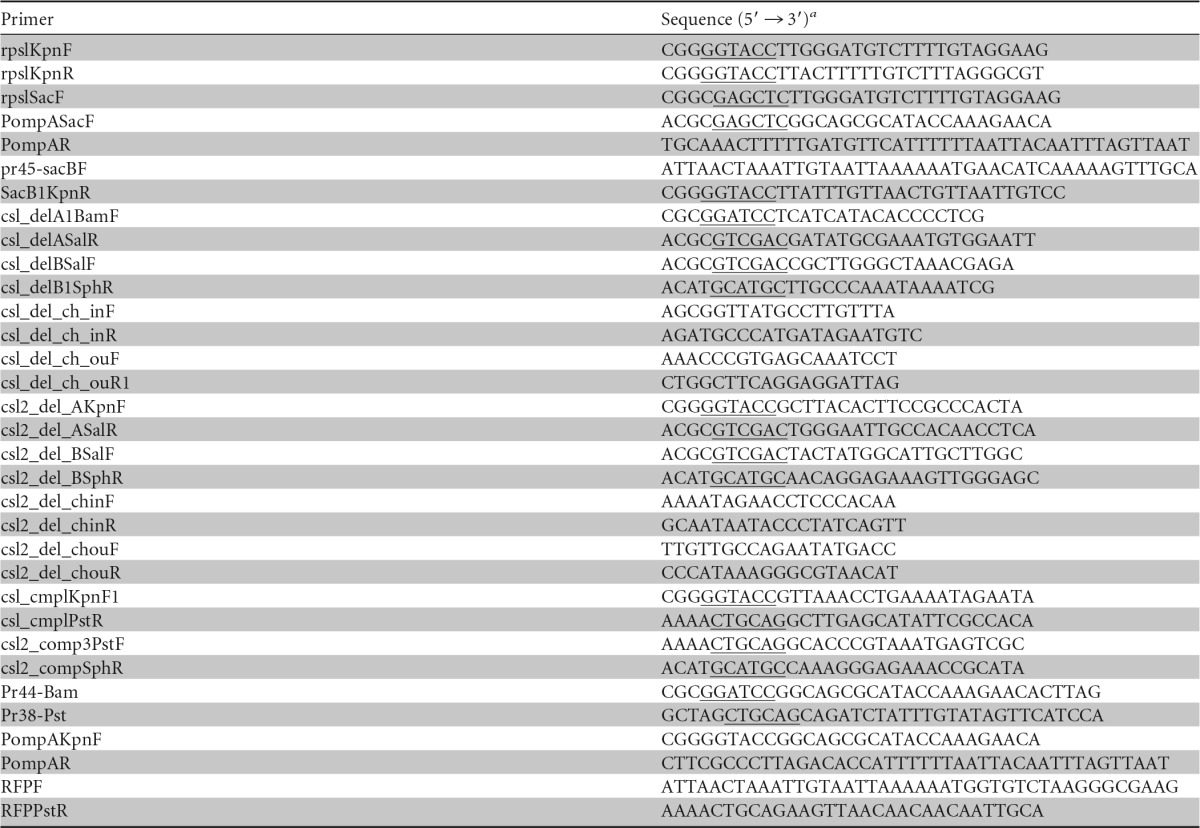
Restriction sites in the primer sequences are underlined.
To determine if wild-type rpsL was dominant to the mutant rpsL alleles, wild-type rpsL was cloned into the shuttle vector pCP23. Specifically, a fragment spanning wild-type rpsL and its presumed promoter region was amplified using primers rpslKpnF and rpslKpnR (Table 2), which were designed with engineered KpnI restriction sites. The fragment was digested with KpnI and cloned into the corresponding sites of pCP23 to generate pCP23-rpsl. pCP23-rpsl was introduced into streptomycin-resistant F. columnare by conjugation as previously described (12–14). All primers used in this study are listed in Table 2.
Construction of the rpsL-containing suicide vector pCSR.
To generate a suicide vector carrying rpsL as a counterselectable marker, wild-type rpsL was amplified using primers rpslSacF and rpslKpnR, which introduced SacI and KpnI restriction sites, respectively. Both the PCR product and the shuttle plasmid pCP23 were digested with SacI and KpnI. The wild-type rpsL gene was ligated with the 6.10-kb fragment of pCP23 lacking the replicase gene. The resultant plasmid, pCSR, is an rpsL-containing suicide vector (see Fig. S1 in the supplemental material).
Construction of the sacB-containing suicide vector pMS75.
In order to use sacB as a counterselectable marker in F. columnare, the F. johnsoniae ompA promoter was cloned in front of sacB by overlap PCR to replace its original promoter. Briefly, PompA was amplified using primers PompASacF and PompAR, with pAS43 as the template. sacB was amplified using primers pr45-sacBF and SacB1KpnR, with pRE112 as the template. To generate the PompA-sacB fusion, the PCR products were used as the templates for overlap PCR with primers PompASacF and SacB1KpnR, which were designed with engineered SacI and KpnI sites, respectively. The product was purified, digested with SacI and KpnI, and ligated into pCP23 that had been digested with the same enzymes. The resulting suicide plasmid, pMS75, lacked functions needed for replication in Flavobacterium (Fig. 1A).
FIG 1.
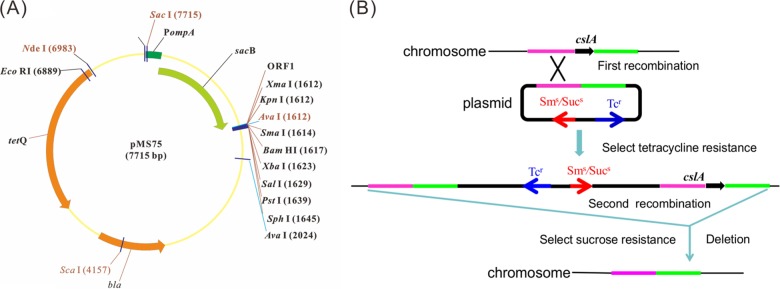
Map of the suicide plasmid pMS75 (A) and gene deletion strategy in Flavobacterium columnare (B). PompA, ompA promoter of F. johnsoniae; cslA, chondroitin AC lyase gene of F. columnare; Sms, Sucs, and Tcr, streptomycin sensitive, sucrose sensitive, and tetracycline resistant, respectively.
Construction of the cslA deletion mutant in the streptomycin-resistant F. columnare strain G4S.
The gene deletion strategy is indicated in Fig. 1B. First, a 2.0-kbp fragment including the final 153 bp of cslA and 1,841 bp of downstream sequence was amplified using primers csl_delA1BamF (introducing a BamHI site) and csl_delASalR (introducing a SalI site). The fragment was digested with BamHI and SalI and cloned into pCSR which had been digested with the same enzymes to generate pCSR-cslA. A 2.2-kbp fragment including the first 240 bp of cslA and 1,973 bp of upstream sequence was amplified with primers csl_delBSalF (introducing a SalI site) and csl_delB1SphR (introducing an SphI site). The PCR product was digested with SalI and SphI and cloned into pCSR-cslA which had been digested with the same enzymes to generate the deletion construct pCSR-csl.
Plasmid pCSR-csl was introduced into the streptomycin-resistant wild-type F. columnare strain G4S by conjugation (21). Briefly, 20 ml of E. coli S17-1 λpir with pCSR-cslA or G4S was cultured overnight at 37°C or 28°C, respectively. The cells were collected by centrifugation at 5,000 rpm for 10 min at room temperature. Pellets were washed twice with antibiotic-free Shieh medium and suspended in 100 μl Shieh medium. The cell suspensions were mixed, placed as spots on nitrocellulose membranes which had been placed on Shieh agar, and incubated at 28°C for 24 h. After incubation, selection for a positive clone whose genomic DNA was integrated with pCSR-csl after the first recombination was conducted by spreading the conjugation product on Shieh plates containing 15 μg/ml tetracycline. A tetracycline-resistant (streptomycin-sensitive) clone was streaked on Shieh agar with 15 μg/ml tetracycline. An isolated colony was inoculated into 1 ml antibiotic-free Shieh liquid medium and shaken at 28°C overnight. One milliliter of culture was spread on Shieh agar which contained 100 μg/ml streptomycin to screen for mutants that had lost the plasmid by a second recombination event. The colonies were streaked on Shieh agar with or without tetracycline, separately. Colonies which grew on Shieh agar but not on Shieh agar containing tetracycline were inoculated into liquid culture for further characterization. The mutants were confirmed by PCR amplification using primers csl_del_ch_inF and csl_del_ch_inR, which resulted in a 409-bp product from wild-type cells but produced no PCR product from cells of the deletion mutant. The mutant strain was further verified by PCR amplification using primers csl_del_ch_ouF and csl_del_ch_ouR1, which resulted in a 3.0-kb product from wild-type cells and a 1.1-kb product from cells of the deletion mutant.
Construction of a cslA deletion mutant in wild-type F. columnare strain G4.
A 4.2-kb fragment containing regions upstream and downstream of cslA was isolated from pCSR-csl by digestion with BamHI and SphI and was inserted into the same sites of pMS75 to generate the deletion construct pMS-cslA. This plasmid was introduced into wild-type F. columnare strain G4 by conjugation followed by growth on Shieh agar containing 15 μg/ml tetracycline to select colonies that contained the integrated plasmid after homologous recombination. The tetracycline-resistant colonies were inoculated into liquid Shieh medium without antibiotics and incubated with shaking overnight at 28°C to allow for loss of the plasmid by a second recombination event. The ΔcslA deletion mutant was selected by growth on Shieh agar medium containing 10% sucrose and was confirmed by PCR as described above.
Deletion of cslB in wild-type F. columnare strain G4 and in the ΔcslA mutant.
A 2.7-kb gene predicted to encode a chondroitin ABC lyase was identified and named cslB. To identify the function of this gene, a deletion mutant was constructed. A 2.6-kb fragment including the final 381 bp of cslB and 2,192 bp downstream of cslB was amplified using primers csl2_del_AKpnF (introducing a KpnI site) and csl2_del_ASalR (introducing a SalI site). The product was digested with KpnI and SalI and ligated into the corresponding sites of pMS75 to generate pMS-csl2A. A 2.2-kb fragment including the first 36 bp of cslB and 2,180 bp of upstream sequence was amplified by PCR with primers csl2_del_BSalF (introducing a SalI site) and csl2_del_BSphR (introducing an SphI site). The product was digested with SalI and SphI and cloned into the corresponding sites of pMS-csl2A to generate the deletion construct pMS-cslB.
Plasmid pMS-cslB was introduced into wild-type F. columnare strain G4 and into the ΔcslA mutant by conjugation, and deletion mutants were obtained as described above. Deletion of cslB in each strain was confirmed by PCR amplification using primers csl2_del_ch_inF and csl2_del_ch_inR, resulting in a 631-bp product from the parent cells but no PCR product from cells of the deletion mutants. The mutants were further confirmed by PCR amplification using primers csl2_del_ch_ouF and csl2_del_ch_ouR, which resulted in products of 5,008 bp from the parent cells and products of 2,663 bp from cells of the deletion mutants.
Construction of complementation strains.
Plasmids carrying cslA and cslB and their promoter regions were constructed using the shuttle vector pCP23. Primers csl_cmplKpnF1 (introducing a KpnI site) and csl_cmplPstR (introducing a PstI site) were used to amplify a 4,641-bp fragment spanning cslA and its putative promoter region. The product was digested with KpnI and PstI and cloned into the corresponding sites of pCP23 to generate pCP-A. Primers csl2_comp3PstF (introducing a PstI site) and csl2_compSphR (introducing an SphI site) were used to amplify a 4,075-bp fragment spanning cslB and its putative promoter region. The product was digested with PstI and SphI and ligated into the same sites of pCP23 to generate pCP-B. To construct a plasmid containing both cslA and cslB, the fragment spanning cslB was isolated from pCP-B by digestion with PstI and SphI and cloned into the corresponding sites of pCP-A to generate pCP-AB. The plasmids were transferred into the corresponding mutants of F. columnare by conjugation for complementation studies.
Construction of RFP-labeled wild-type and GFP-labeled mutant strains.
To construct a red fluorescent protein (RFP)-labeled wild-type G4 strain, a plasmid containing a fused fragment carrying PompA and rfp was constructed. Briefly, the promoter region of F. johnsoniae ompA was amplified from pAS43 with primers PompAKpnF and PompAR. The rfp gene was amplified by using pDS-RED2-1 as the template, with primers RFPF and RFPPstR. The fused fragment carrying PompA and rfp was obtained using overlap PCR with primers PompAKpnF and RFPPstR. The fused fragment was digested with KpnI and PstI and ligated into the corresponding sites of pCP23 to generate pCP-rfp. This plasmid was introduced into G4 by conjugation. The RFP-labeled strains were screened on Shieh agar containing tobramycin and tetracycline.
To construct green fluorescent protein (GFP)-labeled mutants, including ΔcslA, ΔcslB, and ΔcslA ΔcslB mutants, a fused fragment carrying PompA and gfp was obtained by PCR using the primers pr44-Bam and pr38-Pst, with pAS43 as the template. The fragment was cloned into pCP23 to generate plasmid pCP-gfp. The plasmid was introduced into mutants by conjugation. Colonies containing pCP-gfp were screened using Shieh agar with tobramycin and tetracycline.
In vitro growth rate determination.
In vitro growth rates of wild-type G4 and its mutants were determined by measuring the optical density at 600 nm (OD600) with a spectrophotometer (Eppendorf Biophotometer, Germany). Briefly, 100 μl of early-exponential-phase culture (OD600 = 0.3) was inoculated into 20 ml Shieh medium and incubated at 28°C with shaking at 150 rpm. The OD600 was determined every 30 min for 7.5 h.
ECP preparation and CslA and CslB identification.
The extracellular products (ECPs) of wild-type G4, deletion mutants, and complemented strains were prepared as described by Abbass et al. (22). Briefly, F. columnare cells were cultured to the early logarithmic growth phase in 30 ml Shieh medium at 28°C and 150 rpm. Cells were removed by centrifugation (3,000 × g for 10 min), and the supernatants were passed through 0.22-μm membrane filters to remove residual cells. The filtered supernatants (30 ml) were concentrated to 100 μl by using Amicon Ultra-15 concentrators (Millipore). The samples were diluted with 1 ml phosphate-buffered saline (PBS; pH 7.4) and concentrated again to 100 μl. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of the ECPs was performed according to standard procedures, and proteins were detected by staining with Coomassie blue. Proteins that were present in the culture fluid of wild-type G4 but not in the culture fluid of the mutants were characterized by matrix-assisted laser desorption ionization–tandem time of flight mass spectrometry (MALDI-TOF/TOF MS) by using an Applied Biosystems model 4800 Plus MALDI-TOF/TOF analyzer.
Generation of antibodies against CslA and CslB and Western blot analyses.
Affinity-purified polyclonal antibodies were prepared from whole rabbit serum by immunization with synthetic peptides (SEGKLKIESKDMRN for CslA and CNPDLGLPDKRSFDS for CslB) and were stored at −20°C (GenScript, Nanjing, China).
The ECPs were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Detection of CslA and CslB was carried out at room temperature as previously described (23). Briefly, the membranes were blocked with 5% skim milk in Tris-buffered saline (TBS; 25 mM Tris-Cl, 500 mM NaCl, pH 7.5) for 1 h at room temperature, followed by three washes in TBS containing 0.05% Tween 20 (TBST). The membranes were then incubated with antibody against CslA or CslB in 1% skim milk in TBS for 1.5 h, followed by three washes in TBST. The washed membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody for 0.5 h, followed by three washes with TBST, and bound antibodies were visualized using chemiluminescence detection (ChemiDoc MP imaging system).
Chondroitin lyase assay.
As previously described (23, 24), sterilized chondroitin sulfate A (Sigma-Aldrich) and bovine serum albumin (BSA) were added to Shieh agar, to final concentrations of 150 μg/ml and 4%, respectively. Two-milliliter mid-log-phase cultures of bacteria (OD600 = 0.4) were placed on the plates as spots and incubated at 28°C for 9 h. Degradation of chondroitin was visualized by flooding the plates with acetic acid followed by incubation for 5 min. Chondroitin lyase activity was identified as the clear zone around bacterial colonies.
The chondroitin sulfate degradation activity of each strain was also determined in 96-well flat-bottom microplates (10, 25). Briefly, bacterial cultures in the early logarithmic growth phase were collected by centrifugation at 1,000 × g for 10 min. Bacterial pellets were suspended and diluted with Shieh medium until the OD600 reached 0.3, and then the chondroitin sulfate A solution was added to the suspension, to a final concentration of 1.5 mg/ml. Next, 20 μl of bacterial suspension containing chondroitin sulfate A and 150 μl basal medium (0.2% tryptone, 0.05% yeast extract, pH 7.0) were added to each well of the 96-well microplate for 30 min of incubation at 28°C. Finally, 0.5% BSA (dissolved in 0.45 M acetate buffer, pH 4.0) was added to each well, and the turbidity of each well was measured at 405 nm, using an ELx800 microplate reader (BioTek Instruments, Inc.). The Student t test was used to evaluate the statistical significance of differences.
LD50 determination.
In order to ensure that the infection dose was optimal, 50% lethal dose (LD50) values were determined by using intraperitoneal infection. Healthy grass carp (Ctenopharyngodon idella), weighing 25 to 30 g each and with no disease symptoms, were obtained from a local fish farm, where columnaris disease has not been recorded, in Wuhan, China. Fish were kept at 25°C in an 8-m3 flowthrough tank with aeration and fed once a day with commercial fish feed pellets for more than 1 week before each experiment. The bacterial strains were cultured overnight in 5 ml Shieh medium with shaking at 28°C until the OD600 reached 0.4. The cultures were diluted in 10-fold series or 2-fold series in Shieh medium, and the number of cells per milliliter was determined by plating dilutions on Shieh agar and counting the colonies after 48 h of incubation at 28°C. For wild-type G4 and each mutant strain, bacterial suspensions used for injection were all adjusted to the following concentrations: 4 × 106, 6 × 106, 8 × 106, 1 × 107, 1.5 × 107, and 2 × 107 CFU/ml. The use of fish in the present study (i.e., the use of animals in the laboratory) was approved by the Institute of Hydrobiology, Chinese Academy of Sciences.
Fish were divided into seven groups, with 10 individuals in each group, and were further reared at 25°C for 1 week in a 10-liter tank with continuous aeration, water changes at 1-day intervals, and feeding with commercial pellets. Six groups of fish were injected correspondingly with bacterial suspensions at the six different concentrations indicated above, at a dose of 100 μl bacterial suspension per fish for the six experimental groups and 100 μl Shieh medium for the control. Mortality was recorded every 6 h for 7 days following infection. The LD50 values were calculated by using the method of Reed and Muench (26). The Student t test was used to evaluate the statistical significance.
CI analysis.
To compare infection abilities between each mutant and the wild type, competitive index (CI) analysis was used. GFP-labeled mutants and the RFP-labeled wild-type strain were grown in Shieh medium containing 1 μg/ml tobramycin and 15 μg/ml tetracycline at 28°C. When the OD600 value for the bacteria reached 0.4, the cultures were mixed at a 1:1 ratio, and 7 ml of the mixture was diluted in 4 liters of water in a plastic container that contained 10 fingerling grass carp at 26°C. After 30 min, an additional 10 liters of water was added, and the fish were maintained for 7 days at 26°C. For fish that died during this period, gills were collected and homogenized in Shieh medium. The homogenized liquid was serially diluted in buffer (0.3 g NaCl, 0.1 g KCl, 0.02 g CaCl2·2H2O, 0.04 g MgCl2·7H2O, pH 7.4) and spread on Shieh agar containing tobramycin and tetracycline, and the numbers of wild-type and mutant cells were determined using a stereo fluorescence microscope. The CI was defined as the output-to-input ratio for the mutant compared to the wild-type strain. A CI of <1 indicates a competitive virulence defect. The mean CI values were calculated for 12 fish in each group. The data were analyzed by the Student t test, with P values of <0.05 considered statistically significant.
Nucleotide sequence accession number.
Sequence data for cslB were deposited in GenBank under accession number KR083049.
RESULTS
Development of rpsL and sacB as counterselectable markers for F. columnare.
Ten spontaneously streptomycin-resistant mutants of wild-type F. columnare G4 were isolated, and their rpsL genes were amplified and sequenced. Two of the mutants had an adenine-to-guanine point mutation at bp 128 of the rpsL coding sequence that resulted in a K43R substitution in RpsL, which is similar to that in streptomycin-resistant rpsL mutants of F. johnsoniae (14). The wild-type F. columnare rpsL gene was inserted into the shuttle vector pCP23 and introduced into G4S. G4S with the plasmid containing wild-type rpsL failed to grow on Shieh agar with 10 μg/ml streptomycin, whereas G4S without the plasmid grew well in the presence of streptomycin. These results suggest that wild-type rpsL can function as a counterselectable marker in F. columnare.
The F. johnsoniae ompA promoter was used to express sacB in F. columnare. Wild-type cells carrying pCP23-sacB failed to grow on Shieh agar with 10% sucrose, whereas cells without the plasmid grew well, indicating that sacB can also function as a counterselectable marker in F. columnare.
Construction of cslA deletion mutants of F. columnare strains G4S and G4.
To investigate the feasibility of constructing in-frame deletion mutants of F. columnare by using rpsL or sacB as a counterselectable marker, the suicide vectors pCSR, carrying rpsL (see Fig. S1 in the supplemental material), and pMS75, carrying PompA-sacB (Fig. 1), were used. Fused fragments of upstream and downstream regions of cslA (GenBank accession no. AY912281) were cloned into pCSR and pMS75 and introduced by conjugation into G4S and G4, respectively. After two rounds of recombination events, hundreds of streptomycin-resistant colonies were obtained on the Shieh plate containing 100 μg/ml streptomycin (pCSR-mediated deletion), and dozens of sucrose-resistant colonies were obtained on the Shieh plate containing 10% sucrose (pMS75-mediated deletion). About 50% of the streptomycin-resistant colonies and about 20% of the sucrose-resistant colonies were identified as cslA deletion mutants by PCR analysis. These results demonstrate that both rpsL and sacB can function as counterselectable markers to isolate deletion mutants of F. columnare. Although sacB was less effective as a counterselectable marker, it has a significant advantage over rpsL in that mutations are constructed in the wild-type strain rather than in a streptomycin-resistant rpsL mutant. For this reason, sacB-mediated gene deletion was used to construct most of the in-frame deletion mutants analyzed in this study.
Deletion of cslA resulted in a partial reduction in chondroitin lyase activity, which was restored following the introduction of the wild-type gene on pCP-A (Fig. 2). The residual chondroitin lyase activity exhibited by the mutant suggested the presence of an additional chondroitin lyase that had not previously been recognized in wild-type F. columnare cells.
FIG 2.

Comparison of chondroitin sulfate A digestion activities among Flavobacterium columnare wild-type, mutant, and complemented strains. (A) Chondroitin lyase activity can be seen as a clear zone around bacterial colonies against a cloudy background. (B) The chondroitin lyase activity of each strain was determined by measuring the amount of residual chondroitin sulfate A after incubation of this compound with the strains for 30 min in a 96-well flat-bottom microplate. The values are means and standard deviations (SD) (n = 3). Asterisks indicate significant differences (P < 0.05).
F. columnare cslB encodes a second chondroitin lyase.
Chondroitin lyases typically have catalytic polysaccharide lyase family 8 (PL8) domains corresponding to Pfam 02278 (27). Analysis of the F. columnare G4 genome (unpublished data) revealed two genes encoding proteins with PL8 domains: cslA and a gene that we labeled cslB (GenBank accession no. KR083049). To verify the function of F. columnare cslB, in-frame deletions were made in G4 and in the ΔcslA mutant. The ΔcslB deletion mutant had little change in chondroitin sulfate digestion compared to the wild type (Fig. 2A). However, the mutant lacking both cslA and cslB failed to digest chondroitin sulfate. The ability to digest chondroitin sulfate was restored to the ΔcslA mutant, the ΔcslB mutant, and the double deletion mutant by complementation with plasmids expressing CslA, CslB, and both CslA and CslB, respectively (Fig. 2A).
To quantify the decrease of chondroitin sulfate degradation activity, chondroitin sulfate was incubated with bacterial cells for 30 min, and the turbidity of the mixture was measured. As shown in Fig. 2B, deletion of cslB resulted in a slight decrease in chondroitin lyase activity, whereas deletion of cslA resulted in a much larger decrease in chondroitin lyase activity. Deletion of both cslB and cslA had a cumulative effect, essentially eliminating chondroitin lyase activity. Complementation of the mutants with plasmids expressing the appropriate chondroitin lyase genes restored the ability to digest chondroitin sulfate (Fig. 2).
Extracellular products (ECPs) of wild-type, mutant, and complemented cells were collected and separated by SDS-PAGE. A band corresponding to approximately 82 kDa was missing in the ECP fraction from the ΔcslA mutant, and a band corresponding to approximately 96 kDa was missing in the ECP fraction from the ΔcslB mutant (Fig. 3). Both bands were missing from the double deletion mutant (data not shown). The protein bands at 82 and 96 kDa for wild-type cells were identified by MALDI-TOF/TOF MS as CslA and CslB, respectively.
FIG 3.
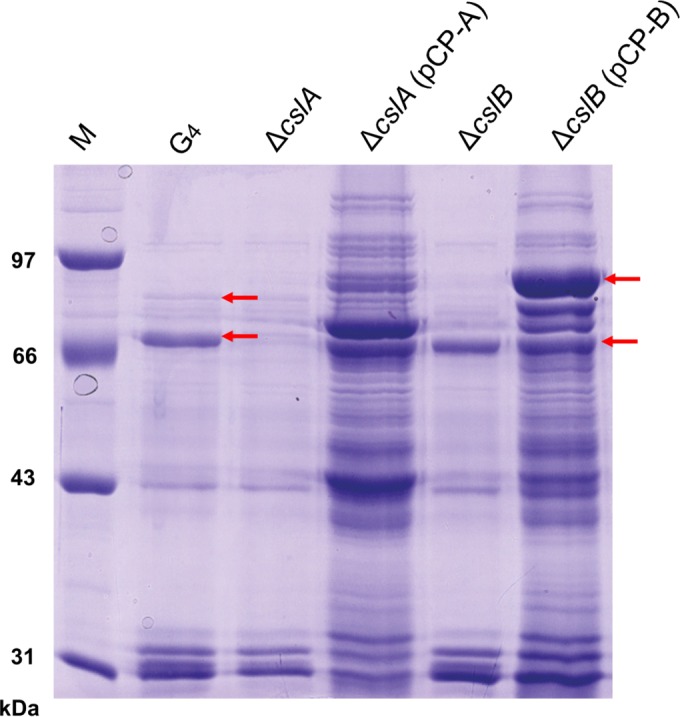
Extracellular products of wild-type G4 and the ΔcslA, ΔcslA(pCP-A), ΔcslB, and ΔcslB(pCP-B) mutants as determined by SDS-PAGE. The upper arrows indicate CslB, and the lower arrows indicate CslA.
Antibodies raised against peptides corresponding to CslA and CslB were used for Western blot analyses of the ECPs. The results confirmed that CslA was missing from the cslA deletion mutants and that CslB was missing from the cslB deletion mutants (Fig. 4). They also demonstrated a restoration of production of each protein by complementation with the appropriate plasmid (Fig. 4).
FIG 4.
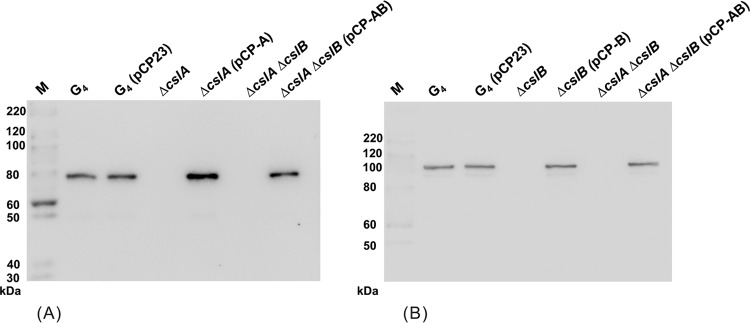
Extracellular products of wild-type G4 and the ΔcslA, ΔcslA(pCP-A), ΔcslB, ΔcslB(pCP-B), ΔcslA ΔcslB, and ΔcslA ΔcslB(pCP-AB) mutants, separated by SDS-PAGE and recognized by rabbit anti-CslA (A) or rabbit anti-CslB (B) polyclonal antiserum. M, molecular size marker.
The wild-type and mutant strains grew at similar rates in Shieh medium (Fig. 5), indicating that deletion of cslA or cslB was not detrimental to the growth of cells in vitro.
FIG 5.
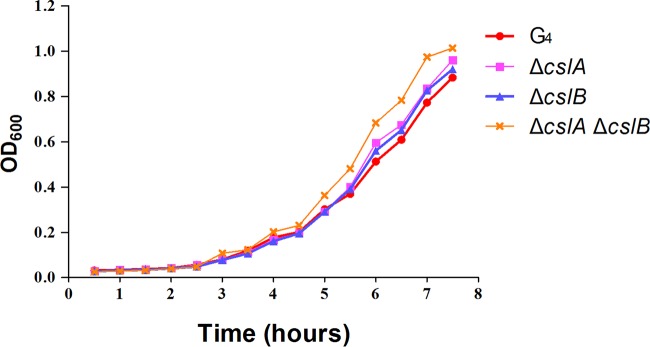
Growth curves for wild-type G4 and the ΔcslA, ΔcslB, and ΔcslA ΔcslB mutants. Cells were grown in Shieh medium at 28°C.
LD50 and CI of G4 and mutants.
The LD50s of wild-type G4 and its mutants were determined by injection into grass carp. The results showed that the LD50 values for G4 and the ΔcslA, ΔcslB, and double mutants were 5.1 × 104, 6.3 × 104, 3.2 × 104, and 4.4 × 104 CFU/fish, respectively (Fig. 6A). Compared with wild-type G4, the ΔcslA mutant had a higher LD50. Unexpectedly, the LD50 of the ΔcslB mutant was reduced, and the LD50 of the double mutant was similar to the LD50 exhibited by wild-type cells (Fig. 6A).
FIG 6.
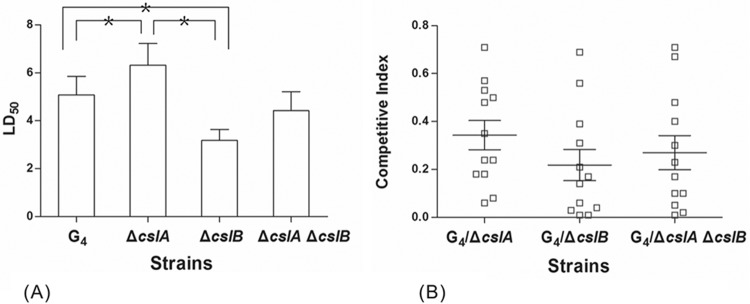
LD50s (A) and CIs (B) for F. columnare wild-type G4 and its mutants, determined in grass carp by using intraperitoneal infection. LD50 values are expressed as means and SD for three replicate experiments. Asterisks indicate significant differences (P < 0.05). The CI values were obtained 36 h after infection with wild-type G4 and its mutants. A CI of <1 indicates a virulence defect. Each data point represents the result for an individual animal, and the mean ± SD of CIs for all fish in each competitive experiment is shown as a horizontal line and error bars.
The CI for the wild type versus each mutant strain was obtained through coinfection with the wild type and each mutant in fingerling grass carp by immersion. The ΔcslA, ΔcslB, and ΔcslA ΔcslB mutants each exhibited a decreased ability to compete with the wild type, with CI values of 0.34 ± 0.21, 0.22 ± 0.22, and 0.27 ± 0.25, respectively (Fig. 6B). These values indicate that the ΔcslA, ΔcslB, and ΔcslA ΔcslB mutants had 2.94-, 4.55-, and 3.70-fold decreases, respectively, in the number of CFU obtained from grass carp gills compared to the number of CFU of the wild type. The results suggest that chondroitin lyases may have roles in competition with other strains and in the ability to cause disease in nature.
DISCUSSION
Although F. columnare is an important pathogen of almost all species of freshwater fish, many details regarding its pathogenesis have not been elucidated, in part because of the lack of suitable genetic techniques. We constructed two suicide vectors, with rpsL and sacB as counterselectable markers, and used these to construct targeted gene deletions. Both rpsL and sacB were effective counterselectable markers that allowed efficient isolation of deletion mutants. Because rpsL-based gene deletion only works in a streptomycin-resistant mutant strain, sacB was established as the preferred counterselectable marker for construction of in-frame deletion mutants. Previous attempts to use sacB as a counterselectable marker in members of the phylum Bacteroidetes failed, perhaps because of a lack of expression of the sacB gene (18, 19). We eliminated this problem by introducing a Bacteroidetes promoter that is known to function in many members of the phylum. It is likely that this approach will allow sacB to function as a counterselectable marker in many other members of the large and diverse phylum Bacteroidetes and will thus facilitate genetic analysis of these important bacteria.
In this study, two genes encoding chondroitin lyases were deleted: cslA, which was identified previously (4), and cslB. Similar lyases are also present in many other members of the Bacteroidetes phylum (see Fig. S2 in the supplemental material). With the deletion of both F. columnare cslA and cslB, the degradation of chondroitin was completely eliminated. CslA may be more important than CslB for degradation of chondroitin, because CslA was more abundant than CslB and deletion of cslA caused a more dramatic reduction of chondroitin lyase activity than did deletion of cslB.
Chondroitin lyases have been suggested to be important virulence factors of F. columnare (9–11). However, in this study, deletion of cslA and cslB resulted in little change in virulence as measured by the LD50 following infection by injection. This indicates that chondroitin lyases are not critical virulence factors for bacterial cells introduced by injection. In contrast, deletion of the chondroitin lyase genes resulted in a decreased competitive index with respect to the wild type during coinfection by immersion, a very different and perhaps more natural challenge method. This suggests that chondroitin lyases may play a role in the process of natural infection. The exact step at which these enzymes confer their competitive advantage remains to be determined.
The genetic techniques developed in this study can be applied to examine other predicted F. columnare virulence factors and should help to elucidate their roles in infection and pathogenesis in fish. The techniques may also be used to construct rationally designed avirulent mutants as candidate vaccine strains to protect freshwater aquaculture fish from columnaris disease.
Supplementary Material
ACKNOWLEDGMENTS
This work has been a rather long-term effort in the laboratory, and financial support from the National Basic Research Program of China (973 Program; grant 2009CB118703) and from the State Key Laboratory of Freshwater Ecology and Biotechnology is gratefully acknowledged, as is partial support from the National Natural Science Foundation of China (grant 31001135).
We thank J.-F. Bernardet in France for his tremendous support and help.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01586-15.
REFERENCES
- 1.Hawke JP, Thune RL. 1992. Systemic isolation and antimicrobial susceptibility of Cytophaga columnaris from commercially reared channel catfish. J Aquat Anim Health 4:109–113. doi:. [DOI] [Google Scholar]
- 2.Declercq AM, Haesebrouck F, Van den Broeck W, Bossier P, Decostere A. 2013. Columnaris disease in fish: a review with emphasis on bacterium-host interactions. Vet Res 44:27. doi: 10.1186/1297-9716-44-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li N, Zhang J, Zhang LQ, Nie P. 2010. Difference in genes between a high virulence strain G4 and a low virulence strain G18 of Flavobacterium columnare by using suppression subtractive hybridization. J Fish Dis 33:403–412. doi: 10.1111/j.1365-2761.2009.01132.x. [DOI] [PubMed] [Google Scholar]
- 4.Xie HX, Nie P, Sun BJ. 2004. Characterization of two membrane-associated protease genes obtained from screening outer-membrane protein genes of Flavobacterium columnare G4. J Fish Dis 27:719–729. doi: 10.1111/j.1365-2761.2004.00596.x. [DOI] [PubMed] [Google Scholar]
- 5.LaFrentz BR, Waldbieser GC, Welch TJ, Shoemaker CA. 2014. Intragenomic heterogeneity in the 16S rRNA genes of Flavobacterium columnare and standard protocol for genomovar assignment. J Fish Dis 37:657–669. doi: 10.1111/jfd.12166. [DOI] [PubMed] [Google Scholar]
- 6.Triyanto Wakabayashi H. 1999. Genotypic diversity of strains of Flavobacterium columnare from diseased fishes. Fish Pathol 34:65–71. doi: 10.3147/jsfp.34.65. [DOI] [Google Scholar]
- 7.Wang LF, Xie HX, Zhang J, Li N, Yao WJ, Xiong CX, Nie P. 2010. Columnaris disease and genetic diversity of its bacterial pathogen Flavobacterium columnare in freshwater fish in China. Acta Hydrobiol Sin 34:1–11. (In Chinese with English abstract.). [Google Scholar]
- 8.Beck BH, Li C, Farmer BD, Barnett LM, Lange MD, Peatman E. 23 February 2015. A comparison of high- and low-virulence Flavobacterium columnare strains reveals differences in iron acquisition components and responses to iron restriction. J Fish Dis doi: 10.1111/jfd.12343. [DOI] [PubMed] [Google Scholar]
- 9.Stringer-Roth KM, Yunghans W, Caslake LF. 2002. Differences in chondroitin AC lyase activity of Flavobacterium columnare isolates. J Fish Dis 25:687–691. doi: 10.1046/j.1365-2761.2002.00421.x. [DOI] [Google Scholar]
- 10.Suomalainen L-R, Tiirola M, Valtonen ET. 2006. Chondroitin AC lyase activity is related to virulence of fish pathogenic Flavobacterium columnare. J Fish Dis 29:757–763. doi: 10.1111/j.1365-2761.2006.00771.x. [DOI] [PubMed] [Google Scholar]
- 11.Xie HX, Nie P, Chang MX, Liu Y, Yao WJ. 2005. Gene cloning and functional analysis of glycosaminoglycan-degrading enzyme chondroitin AC lyase from Flavobacterium columnare G4. Arch Microbiol 184:49–55. doi: 10.1007/s00203-005-0009-0. [DOI] [PubMed] [Google Scholar]
- 12.Staroscik AM, Hunnicutt DW, Archibald KE, Nelson DR. 2008. Development of methods for the genetic manipulation of Flavobacterium columnare. BMC Microbiol 8:115. doi: 10.1186/1471-2180-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Zou H, Wang LF, Li N, Wang GT, Huang B, Nie P. 2012. Construction of two selective markers for integrative/conjugative plasmids in Flavobacterium columnare. Chin J Oceanol Limnol 30:269–278. doi: 10.1007/s00343-012-1077-z. [DOI] [Google Scholar]
- 14.Rhodes RG, Pucker HG, McBride MJ. 2011. Development and use of a gene deletion strategy for Flavobacterium johnsoniae to identify the redundant gliding motility genes remF, remG, remH, and remI. J Bacteriol 193:2418–2428. doi: 10.1128/JB.00117-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Link A, Phillips D, Church G. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol 179:6228–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelicic V, Jackson M, Reyrat J-M, Jacobs WR Jr, Gicquel B, Guilhot C. 1997. Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 94:10955–10960. doi: 10.1073/pnas.94.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng J, Tung SL, Leung KY. 2005. Regulation of a type III and a putative secretion system in Edwardsiella tarda by EsrC is under the control of a two-component system, EsrA-EsrB. Infect Immun 73:4127–4137. doi: 10.1128/IAI.73.7.4127-4137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjornsdottir SH, Fridjonsson OH, Hreggvidsson GO, Eggertsson G. 2011. Generation of targeted deletions in the genome of Rhodothermus marinus. Appl Environ Microbiol 77:5505–5512. doi: 10.1128/AEM.02070-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McBride MJ, Kemp PF. 1996. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J Bacteriol 178:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Decostere A, Haesebrouck F, Devriese LA. 1997. Shieh medium supplemented with tobramycin for selective isolation of Flavobacterium columnare (Flexibacter columnaris) from diseased fish. J Clin Microbiol 35:322–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alvarez B, Secades P, McBride MJ, Guijarro JA. 2004. Development of genetic techniques for the psychrotrophic fish pathogen Flavobacterium psychrophilum. Appl Environ Microbiol 70:581–587. doi: 10.1128/AEM.70.1.581-587.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abbass A, Sharifuzzaman SM, Austin B. 2010. Cellular components of probiotics control Yersinia ruckeri infection in rainbow trout, Oncorhynchus mykiss (Walbaum). J Fish Dis 33:31–37. doi: 10.1111/j.1365-2761.2009.01086.x. [DOI] [PubMed] [Google Scholar]
- 23.Guthrie EP, Salyers AA. 1986. Use of targeted insertional mutagenesis to determine whether chondroitin lyase II is essential for chondroitin sulfate utilization by Bacteroides thetaiotaomicron. J Bacteriol 166:966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shotts EB, Cooper RK. July 1996. Chondroitinase attenuated Edwardsiella ictaluri and a vaccine for prevention of enteric septicemia (ES) in fish. US patent 5536658.
- 25.Teska JD. 1993. Assay to evaluate the reaction kinetics of chondroitin AC lyase produced by Cytophaga columnaris. J Aquat Anim Health 5:259–264. doi:. [DOI] [Google Scholar]
- 26.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Hyg (Lond) 27:493–497. [Google Scholar]
- 27.Féthière J, Eggimann B, Cygler M. 1999. Crystal structure of chondroitin AC lyase, a representative of a family of glycosaminoglycan degrading enzymes. J Mol Biol 288:635–647. doi: 10.1006/jmbi.1999.2698. [DOI] [PubMed] [Google Scholar]
- 28.de Lorenzo V, Timmis KN. 1994. Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol 235:386–405. doi: 10.1016/0076-6879(94)35157-0. [DOI] [PubMed] [Google Scholar]
- 29.Lu QZ, Ni DS, Ge RF. 1975. Studies on the gill diseases of the grass carp (Ctenopharyngodon idellus). I. Isolation of a myxobacterial pathogen. Acta Hydrobiol Sin 5:315–334. (In Chinese with English abstract.). [Google Scholar]
- 30.Agarwal S, Hunnicutt DW, McBride MJ. 1997. Cloning and characterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) gliding motility gene, gldA. Proc Natl Acad Sci U S A 94:12139–12144. doi: 10.1073/pnas.94.22.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kempf MJ, McBride MJ. 2000. Transposon insertions in the Flavobacterium johnsoniae ftxX gene disrupt gliding motility and cell division. J Bacteriol 182:1671–1679. doi: 10.1128/JB.182.6.1671-1679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.