

Abstract
Microtubules are dynamic and structural cellular components involved in several cell functions, including cell shape, motility, and intracellular trafficking. In proliferating cells, they are essential components in the division process through the formation of the mitotic spindle. As a result of these functions, tubulin and microtubules are targets for anticancer agents. Microtubule-targeting agents can be divided into two groups: microtubule-stabilizing, and microtubule-destabilizing agents. The former bind to the tubulin polymer and stabilize microtubules, while the latter bind to the tubulin dimers and destabilize microtubules. Alteration of tubulin-microtubule equilibrium determines the disruption of the mitotic spindle, halting the cell cycle at the metaphase-anaphase transition and, eventually, resulting in cell death. Clinical application of earlier microtubule inhibitors, however, unfortunately showed several limits, such as neurological and bone marrow toxicity and the emergence of drug-resistant tumor cells. Here we review several natural and synthetic microtubule-targeting agents, which showed antitumor activity and increased efficacy in comparison to traditional drugs in various preclinical and clinical studies. Cryptophycins, combretastatins, ombrabulin, soblidotin, D-24851, epothilones and discodermolide were used in clinical trials. Some of them showed antiangiogenic and antivascular activity and others showed the ability to overcome multidrug resistance, supporting their possible use in chemotherapy.
1. Introduction
Microtubules are dynamic and structural cellular components, typically formed by 13 protofilaments, which constitute the wall of a tube; each of the protofilaments consists of a head-to-tail arrangement of α/β tubulin heterodimers [1]. They are involved in several cell functions, including cell shape, motility, and intracellular trafficking. In proliferating cells, they are one of the essential components in the division process through the formation of the mitotic spindle. This event can take place because of the dynamic nature of microtubules through polymerization and depolymerization cycles [2]. As a result of these functions, tubulin and microtubules are targets for anticancer agents [3, 4]. Microtubule-targeting agents can be divided into two groups: microtubule-stabilizing and microtubule-destabilizing agents. The former bind to the tubulin polymer and stabilize microtubules, while the latter bind to the tubulin dimers and destabilize microtubules [5, 6].
Despite these differences, alteration of tubulin-microtubule equilibrium leads to the same final result: it disrupts the mitotic spindle, halting the cell cycle at the metaphase-anaphase transition and eventually resulting in cell death [7] (Figure 1).
Figure 1.
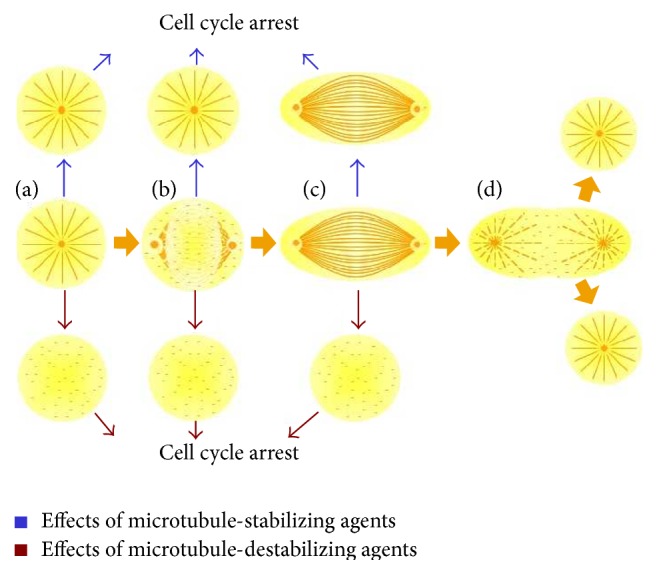
The dynamic nature of cytoskeleton is due to cycles of microtubule catastrophes. (a) Model structure of assembled cytoskeleton. The variety of shapes and sizes of the microtubule cytoskeleton is as great as the number of different cell types. In interphase, microtubules are long and stable because there are almost no catastrophes. (b) In mitosis, catastrophes are relatively frequent, resulting in highly dynamic microtubules that reach a steady-state length after a few minutes of growth (c). (d) After the segregation of chromatids, a new cycle of depolymerization and polymerization begins, resulting in a new stable microtubule cytoskeleton in daughter's cells (d). Blue and red arrows indicate effects of stabilizing and destabilizing agents, all resulting in cell cycle arrest.
Clinical application, however, has unfortunately shown several limits, such as a high level of neurological and bone marrow toxicity and the emergence of drug-resistant tumor cells due to the overproduction of P-glycoprotein (Pgp), an ATP-binding cassette (ABC) transmembrane transporter [8], the overexpression of different beta-tubulin isotypes, including βIII-tubulin [9, 10], or tubulin mutations [11].
Several natural and synthetic microtubule-targeting agents, exhibiting antitumor activity and increased efficacy in comparison to traditional drugs in various preclinical and clinical studies, have been discovered and their mechanisms have been elucidated [12, 13]. Apart from the well-known antimitotic function, for some of these drugs antiangiogenic and antivascular activity were demonstrated; for others the ability to overcome multidrug resistance was found. Many of these new generation microtubule-targeting agents are still under evaluation for clinical use. Some of them showed good tolerability and antitumor activity in particular cancers.
This review provides an overview of those microtubule-targeting drugs which are to date under clinical evaluation. A particular attention will be paid to the translation of preclinical data into the design of clinical trials.
2. Microtubule-Destabilizing Agents
Colchicine and Vinca alkaloids are two of the first microtubule-destabilizing agents to be discovered. These two compounds depolymerize microtubules by interacting with various β-tubulin sites. In particular, Vinca alkaloids interact with tubulin at specific binding sites which differ from those of other agents, including colchicine or taxanes, interfering with microtubule dynamics, blocking polymerization at the end of the mitotic spindle, and leading to metaphase arrest. Thanks to their peculiar mechanism of action, Vinca alkaloids have been widely used in anticancer therapy, usually in combination with other chemotherapeutic agents which do not have cross-resistance with them. First-generation Vinca alkaloids such as vinblastine have been included in the treatment protocol of both Hodgkin and non-Hodgkin lymphomas and testicular carcinoma, while vincristine has been approved for several years in the treatment of hematological tumors such as acute leukemia and multiple myeloma but also of rare tumors such as rhabdomyosarcoma and neuroblastoma. However, vincristine treatment was associated with a severe neurotoxicity, while the suppression of the bone marrow was more frequently reported during vinblastine therapy [14]. Second-generation semisynthetic Vinca alkaloids, vinorelbine and vindesine, have shown a broader spectrum of antitumor activity in vitro, along with a decreased neurotoxicity. Vinorelbine was approved as single agent and in combination therapy for the treatment of both hematological and solid tumors, including lung cancer, breast cancer, and gynecological tumors [15]. Recently, another synthetic Vinca alkaloid, vinflunine, has been approved in Europe for the second-line treatment of metastatic urothelial carcinoma. It is the first fluorinated microtubule inhibitor, which was associated with a higher antitumor activity than other Vinca alkaloids, showing also an excellent safety profile [16].
In order to overcome the clinical limits of these agents, in the last years attention has been focused on natural and synthetic compounds with a different structure but which act in a similar way [7, 17] (Table 1).
Table 1.
Microtubule-destabilizing agents.
| Chemical lead | Properties and effects | Clinical trial/status | References |
|---|---|---|---|
| Cryptophycins | Apoptosis induction. Synergistic with chemotherapy and radiation. | Phase II clinical trials in platinum-resistant ovarian cancer and in NSCLC (C-52) but withdrawn due to peripheral neuropathy. | [26, 28, 31, 32, 36] |
|
| |||
| Combretastatin A-4-P | Antivascular and antiangiogenic activity. Synergistic with radiation, hyperthermia, chemotherapy, and immunoradiotherapy. | Phases II and III clinical trials in advanced solid tumors (lung and thyroid cancer) and in combination with carboplatin. | [63, 64, 66–70, 72, 73] |
|
| |||
| Combretastatin A-1-P | Antivascular and antitumoral activity superior to CA-4-P. Synergistic with chemotherapy. | Phase I clinical trials in solid tumors and in acute myelogenous leukaemia and myelodysplastic syndromes. | [78, 79] |
|
| |||
| Ombrabulin | Antivascular and antitumoral activity superior to CA-4-P. Synergistic with chemotherapy. | Phase I clinical trials as a single agent or in combination; phase III clinical trial in advanced soft-tissue sarcoma. | [86] |
|
| |||
| Soblidotin | Apoptosis induction. Antivascular activity. Antitumoral activity in tumors resistant to vincristine, docetaxel, and paclitaxel. | Phase II clinical trials in advanced solid tumors (soft-tissue sarcoma, NSCLC). | [99–105] |
|
| |||
| D-24851 | Curative at nontoxic doses in rat tumor. No neurotoxic effects. Oral applicability. Activity versus MDR cell lines. | Phase I/II clinical trials in advanced solid tumors. | [140, 141] |
|
| |||
| Pseudolaric acid B | Antiangiogenic activity. No neurotoxic effects in tested animal. Activity versus MDR cell lines. | Preclinical phase. | [148, 149] |
|
| |||
| Embellistatin | Antiangiogenic activity. | Preclinical phase. | [150] |
2.1. Cryptophycins
Cryptophycins are synthetic derivatives of macrocyclic depsipeptides, isolated by Nostoc sp. [18]. They block cell division and prevent the correct formation of the mitotic spindle, by inhibiting tubulin polymerization, probably at the binding site of the Vinca alkaloids [19]. In particular, C-52 and C-55 induce apoptosis by means of Bcl-2 hyperphosphorylation and inactivation [20–22] (Figure 2). These compounds are able to induce this phosphorylation at a greater extent than other microtubule inhibitors [23]. The first form discovered was epoxide cryptophycin 1, which showed antitumoral activity both in preclinical in vitro (colon, breast, ovarian, lung, and nasopharyngeal carcinomas) and in vivo (lung, breast, and prostate tumors) models. This has led to isolation and synthesis of cryptophycin analogs, divided into epoxides, chlorohydrins, and glycinate chlorohydrins [24] (Figure 3).
Figure 2.
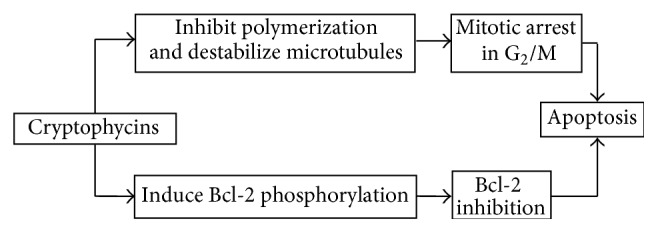
Mechanism of action of cryptophycins.
Figure 3.
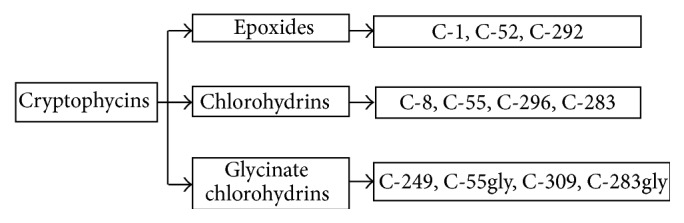
Classification of cryptophycins.
Cryptophycin 8 is the first C-1 analog synthesized in order to improve its antitumoral efficacy by means of conversion of the epoxide group into chlorohydrin. Its activity has been shown both in murine and human tumors. Although it is not as powerful as C-1, it is more soluble in water and has a stronger therapeutic effect. Nevertheless, it is still too unstable in solution to be considered clinically relevant [25].
2.1.1. Cryptophycins 52 and 55
Cryptophycin 52 (LY355703) is a synthetic epoxide, used in phase II clinical trials, which presents a cytotoxic effect 400 times stronger than paclitaxel and Vinca alkaloids [26, 27]. It shows in vitro antitubulin, antimitotic, and cytotoxic activity which is dose-dependent against tumor cells. Furthermore, its activity has been evaluated both in murine tumor models and in human tumor xenografts [23]. C-52 resulted to be also effective against multidrug-resistant tumors [26, 28, 29].
Paclitaxel and the Vinca alkaloids are sensitive to the multidrug resistance (MDR) transporters P-glycoprotein (P-gp, MDR-1) and/or MDR-associated protein (MRP-1). Cryptophycin 52 was tested for its sensitivity to multidrug resistance in several paired cell lines in which a sensitive parental line was matched with a multidrug-resistant derivative line. Compared to other antimitotic agents (paclitaxel, vinblastine, and vincristine), the potency of cryptophycin 52 was shown to be minimally affected in multidrug-resistant cells compared to their sensitive parental lines [30]. Cryptophycin 52 fragment A analogues was synthesized to improve the potency and the aqueous solubility of the molecule allowing for the modification of its formulation. However, the same functional groups that rendered these analogues more potent and more water soluble also contributed to making them better substrates of the Pgp efflux pump. It is an unacceptable feature in the development of a clinically relevant antitumor agent [29].
Preclinical toxicological studies on animals (rats and dogs) have shown that above a certain concentration level C-52 causes secondary effects such as neutropenia and gastrointestinal problems but not neurotoxicity. These studies have allowed evaluating the optimum phase II dosage and tracing the plasma pharmacokinetic profile [26]. Furthermore, phase I clinical trials identified 1.5 mg/m2 as a well-tolerated dose level of C-52. It was delivered as a 2-hour i.v. infusion on day 1 and day 8 repeated every 3 weeks [31]. This schedule was employed in a phase II study to determine the activity of C-52 in non-small cell lung cancer (NSCLC) patients previously treated with platinum-based chemotherapy and to characterize its toxicity profile. A good rate of disease stabilization and an unacceptable toxicity was found in this setting [32]. Also, a multicenter trial was performed to evaluate the same schedule of the drug in patients with platinum-resistant advanced ovarian cancer. A considerable clinical benefit without serious adverse events was achieved [28]. Afterwards, these phase II clinical trials were terminated due to significant neurological toxicity [12].
Cryptophycin 55, a C-52 chlorohydrin, shows higher cytotoxic activity and therapeutic efficacy than its epoxide precursor, but its low stability in solution has delayed its clinical application [33]. This problem has been overcome, however, by means of the synthesis of glycinate esters (C-55gly, C-283gly, and C-309) which show not only an in vivo activity similar to their precursors but also a high level of stability [34].
Treatment with C-52 and C-55 combined with other chemotherapy agents has produced synergic effects without increased toxicity, bringing about a greater survival rate in ovarian carcinoma murine models [23, 28]. The use of human tumor xenografts has made it possible to evaluate C-52 and C-55 activity combined with cisplatin, carboplatin, and oxaliplatin in different tumors. C-52 showed a synergic effect only when associated with cisplatin, whereas C-55 showed increased activity with all the platinum compounds [35]. In vivo antitumoral activity of C-52 and C-55 has been assessed in combination with radiotherapy (2γ) or with 5-FU in tumor xenografts, showing an increased effect. Pharmacokinetic analyses performed in murine models have demonstrated that C-52 concentration in the tumor increased after administration and remained high for 24 hours. The mean life of C-55 was the longest in the liver, intermediate in tumor tissue, and less in plasma. After C-55 administration, the mean life of C-52 was the longest in tumor tissue, less in plasma, and even less in the liver, suggesting almost total conversion of C-55 into C-52 in the tumor. The greater C-52 accumulation in tumor tissue depends on the bioconversion of C-55 in C-52 and different binding affinities towards different tissue proteins. The use of C-55 to deliver C-52 increased the retention of C-52 in tumor tissue and reduced its presence in all studied normal tissues. Furthermore, extracellular acid pH of the tumor increased C-55 stability, whereas intracellular basic pH encouraged bioconversion by stimulating its pharmacological activity [36].
The obtained results indicated that C-52 and C-55 fulfilled all the criteria required by ideal chemotherapy agents, since they showed an action mechanism against a specific target and considerable activity against drug-resistant cells. However, the lack of response observed in some tumors and peripheral neuropathy have been limiting factors in the development of these agents leading to termination of their study.
2.1.2. Second-Generation Cryptophycins
C-309, C-249, and C-283 are second-generation candidates for clinical use. The first two are glycinate esters, synthesized in order to provide a higher chemical stability and more solubility in water. C-309 is a derivative of C-296 which has proved to have more therapeutic activity than C-55, C-283, C-249, and C-296; it is able to bring about a complete or partial regression of murine tumors at lower doses than those of other glycinate analogs. C-249 derives from C-8 and is active against MDR tumors. Moreover, it has the advantage of being easier to synthesize.
These second-generation analogs have proved to be up to 1000 times as active as those of the first clinical candidates (C-52) but with the same or even less toxicity [34].
2.2. Combretastatins
The combretastatins, isolated from Combretum caffrum, are molecules structurally related to colchicine which have been extensively developed since the late 1990s as vascular-disrupting agents (VDAs) [37]. The vascular-disrupting effect of these compounds is present well below the maximum tolerated dose, with a wide therapeutic window [38]. A number of combretastatins are currently in clinical trials: combretastatins A4- and A1-phosphate, verubulin, crolibulin, plinabulin, and ombrabulin [12].
2.2.1. CA-4-P
Combretastatin A-4 interacts with tubulin at the colchicine binding site but not in the same pseudoirreversible manner. It is used as a combretastatin A-4 3-O-phosphate (CA-4-P), a prodrug which is soluble in water and transformed into its active form by endogenous phosphatases [39]. It showed cytotoxicity in tumor cell lines and in human endothelial cells, HUVEC, which are sensitive to the drug only if they are actively proliferating, suggesting a potential use as an antiangiogenic agent [38]. By interfering with microtubule polymerization and with mitotic spindle assembly, CA-4-P induces G2/M arrest, thus bringing about cell death by either mitotic catastrophe or apoptosis [38, 40, 41].
Recent computational studies, using fluorescence spectroscopy, identified a potential binding site on γ-tubulin for both CA-4-P and colchicines [42]. Since high levels of γ-tubulin have been reported in poorly differentiated and aggressive brain tumors, such as human glioblastoma and medulloblastoma [43, 44] and lung [45] and breast cancer [46], the discovery of a potential site interaction on this molecule would offer the possibility of targeting inhibition with a new class of chemotherapeutic agents. However, the experimental validation of such interesting observation is underway.
CA-4-P (also known as Zybrestat or fosbretabulin) shows a potent in vivo antivascular activity since it causes a rapid and widespread reduction of the tumoral blood flow and an increased vascular resistance, effects which are extremely reduced in the normal tissues [47]. At a dose of 1/5–1/10 of the maximum tolerated dose (MTD), the central area of the tumor undergoes hemorrhagic necrosis, while a thin peripheral ring of live cells remains [38, 48, 49]. On the contrary, colchicine and other drugs act only at approximately MTD [50]. This constitutes an important advantage for the therapeutic application of CA-4-P. An immediate effect of CA-4-P treatment is an increased vascular permeability, which is important for the reduction of blood flow through vascular collapse, and an increased viscosity consequent to fluid loss from the vasculature. However, endothelial barrier function alterations and increased vascular permeability might contribute to hastening tumor cell extravasation, causing progression to stages of greater malignancy, with heightened invasiveness and, in some cases, increased distant metastasis. It is no coincidence that susceptibility of tumors to CA-4-P showed a positive correlation with tumor vascular permeability [51]. Experiments conducted on HUVEC cells have shown that the CA-4-P-inducted microtubule depolymerization triggers off the actin reorganization through Rho activation and MLC (Myosin Light Chain) phosphorylation, thus causing rounding and retraction of cells and membrane blebbing. These events are associated with increased permeability, while the morphological cell change might contribute to determining the effects observed in vivo by means of vascular constriction [52, 53]. Furthermore, since CA-4-P interferes with the formation of stress fibers, it inhibits the VE-cadherin/β-catenin complex, thus leading to the destabilization of cell-cell junctions and increasing endothelial permeability [54].
The ex vivo perfusion of animal tumors highlights a lower increase in vascular resistance to that found in vivo, suggesting that the blood might also contribute towards the antivascular action of the drug [48]. It has been demonstrated that CA-4-P induces increased expression of endothelial CAM, responsible for the observed neutrophil recruitment which in vivo probably contributes both to vascular damage and to tumor cell death [55].
Apart from being an antivascular agent, CA-4-P inhibits the formation of new blood vessels, both in vitro and in vivo, presumably through inactivation of the VE-cadherin/β-catenin complex and Akt, all proteins required for cell adhesion, survival, and proliferation during neoangiogenesis. The same study has shown that smooth muscle cells, which are resistant to the drug, interfere with its antiangiogenic activity in vitro, suggesting that they may confer resistance to the endothelium by stabilizing cell-cell junctions [54]. CA-4-P selectivity towards the neoplastic tissue might therefore depend on the immaturity of tumor vessels, together with the proliferative status of tumor endothelial cells. Moreover, CA-4-P reduces in vitro HIF-1 expression (Hypoxia Inducible Factor-1) under hypoxia mainly in endothelial cells compared to that in cancer cell lines, suggesting a further possible mechanism of action for the drug [56] (Figure 4).
Figure 4.

Combretastatin A-4-P: mechanisms of action at tumor level.
However, the effects of CA-4-P on tumor growth are not particularly significant, probably because of the persistent presence of vital peripheral cells [50], although the administration of several doses compared to the same total dose of the drug does increase its antitumoral effect [57, 58]. Furthermore, CA-4-P activity is directly proportional to tumor size [49]. This aspect, together with its capacity to act on the tumor core, differentiates this drug from more common therapeutic approaches, which target the peripheral tumor area. These complementary properties, together with the limited action of CA-4-P as a single agent, have led to experimentation involving combined treatments. It has been demonstrated that CA-4-P increases the response to radiotherapy and hyperthermia in treated tumors [57, 59] and, what is more, leads to a 90% increase in the retention of the anti-CEA antibody marked with I131 in the tumor, which is eradicated in 83% of the cases [60]. Similarly, CA-4-P increases the effect of chemotherapy drugs such as cisplatin, vinblastine, 5-fluorouracil, and irinotecan [57, 61].
The overall in vivo results obtained with CA-4-P have led to its introduction in phase I clinical trials [12, 62–66]. A phase I trial was performed to determine the MTD, safety, and pharmacokinetic profile of CA-4-P. This study showed absence of traditional cytotoxic side effects, with a toxicity profile which seems consistent with a “vascularly active” drug [67, 68]. The effects on tumor blood flow were assessed using dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) techniques. Dosages < or = 60 mg/m2, as a 10 min infusion at 3-week intervals, define the upper boundary of the MTD. Similar effects were seen in other phase I clinical trials using a weekly and daily schedule [69, 70]. Afterwards, a further phase I trial investigated the combination of CA-4-P with carboplatin. A greater thrombocytopenia was observed as a consequence of altered carboplatin pharmacokinetics [63].
In order to improve its efficiency and reduce its side effects, a specific therapeutic system has been realized, based on the use of liposomes containing CA-4-P, carrying superficial RGD-peptides able to bind with the α v β3 integrins overexpressed on proliferating tumor endothelium. In vitro tests have demonstrated the specificity and stability of the system, essential properties for its in vivo application [71]. To date, phase II/III clinical trials in lung and thyroid cancer are currently being evaluated [12]. These studies showed that CA-4-P with or without carboplatin and paclitaxel combination therapy was well tolerated in thyroid cancer patients, although it did not meet statistical significance in OS improvement [72]. Instead, preliminary data suggests survival benefits and increased responses without significant additional toxicity in NSCLC patients treated with CA-4-P in combination with carboplatin, paclitaxel, and bevacizumab compared to patients treated with carboplatin, paclitaxel, and bevacizumab only [73].
2.2.2. CA-1-P
Combretastatin A1 phosphate (also known as Oxi4503), a CA-1 water-soluble prodrug, shows a powerful antivascular activity. When used in murine and human tumor xenografts at much lower doses than those required by CA-4-P, CA-1-P brings about a drastic reduction of blood flow, with resulting necrosis [74]. CA-1-P causes an increase in vessel permeability, in VEGF production and apoptosis induction in endothelial cells [75]. At high doses it is more easily tolerated than CA-4-P and shows a much higher antitumoral activity, leading to complete regression of human tumors even at extremely low doses [74]. Excellent results have been obtained with combined treatments involving several chemotherapy agents [76].
In vitro pharmacokinetic studies have suggested that CA-1-P is transformed into a more reactive metabolite than CA-4-P, which is responsible for most of the antitumoral activity; this has formed the basis for further clinical developments of the drug as an antivascular and antitumor agent [77]. The drug has completed the phase I evaluation as a potential anticancer drug at three different centres in the United Kingdom, and it was studied in other phase I clinical trials [78, 79]. Recently, a new series of combretastatin derivatives have been synthesized and evaluated in seven cancer cell lines, exhibiting good anticancer activity [80, 81].
2.2.3. Ombrabulin
Ombrabulin (also known as AC-7700) is a serinamide hydrochloride, synthetic derivative of CA-4-P, which inhibits growth in a large number of drug-resistant animal tumors and carcinogen-induced tumors [39, 82]. Differently from CA-4-P, it does not act directly on the tumor vessels but instead causes constriction of the arterioles, resulting in complete downstream arrest of the blood flow and tumor growth [83]. These effects are obtained at doses half of MTD and 100 times less than that of CA-4-P [84]. Finally, the combination of AC-770 with cisplatin increases the effect of both drugs in murine tumors, with curative effects, and in human tumor xenografts [85]. In 2002, AC-7700 was introduced into phase I clinical trials in the United States and in Europe (AVE8062, Aventis Pharma). Recently, ombrabulin in combination with cisplatin was used in a phase III clinical trial for patients with advanced soft-tissue sarcomas after failure of anthracycline and ifosfamide chemotherapy, significantly improving progression-free survival. However, this improvement was not clinically relevant, despite being statistically significant [86].
2.3. Dolastatins
Dolastatins are pseudopeptides isolated from the sea hare Dolabella auricularia [50]. Dolastatins 10 and 15 showed antiproliferative activity. These agents induce apoptosis through interaction with tubulin [87]. Dolastatin 10 is a natural peptide able to interfere in microtubule assembly by means of the noncompetitive binding to the Vinca alkaloid site [88]. A phase II trial investigated dolastatin 10 in NSCLC patients. A low response rate was observed, even though a good tolerability was achieved. Myelosuppression was confirmed as the only noteworthy toxicity [89].
Other phase II clinical trials of dolastatin 10 were carried out in patients with metastatic melanoma, advanced colorectal and breast cancers, recurrent platinum-sensitive ovarian carcinoma, and hormone-refractory metastatic prostate adenocarcinoma [90–94]. These studies confirmed the same results previously obtained in terms of tumor response and toxicity. No activity was found in advanced pancreaticobiliary cancers and metastatic soft-tissue sarcomas [95, 96]. For this reason, it was suggested to not pursue the clinical development of this drug in further studies, not only because of its side effects [97] but also because of the low mean survival rate of the treated patients [95].
2.3.1. Soblidotin
Soblidotin (TZT-1027) is a synthetic analog of dolastatin 10 which inhibits the growth of several tumoral cell lines and induces caspase-3-dependent apoptosis. It shows in vivo antivascular effects in tumoral models overexpressing VEGF and in murine colon tumors, with an increase in vascular permeability, vessel closure, and widespread hemorrhage. Soblidotin also shows antitumoral activity in vincristine-, docetaxel-, and paclitaxel-resistant tumors, which makes it a potential chemotherapy drug for use in tumors which do not respond to other microtubule inhibitors [98].
The first two European phase I clinical trials identified a recommended dose of soblidotin between 2.4 and 2.7 mg/m2 for a 3-weekly administration with neutropenia, fatigue, and a reversible peripheral neuropathy as the DLT. Moreover neurological side effects seemed to correlate with previous exposure to other neurotoxic agents such as platinum compounds. No correlation was found with body surface area suggesting possible use of flat dose regimen for next trials [99, 100]. In a Japanese phase I clinical trial MTD of 1.5 mg/m2 administered on days 1 and 8 in 3-week courses was found [101]. A combination of this drug with carboplatin was also tested. The recommended dose was 1.5 mg/m2 for soblidotin and AUC 5 for carboplatin and no pharmacokinetics interaction was observed [102]. In NSCLC patients a phase I trial indicated a recommended dose of 4.8 mg/m2, administered every 3-4 weeks as recommended dose [103].
Phase II clinical investigations suggested activity in advanced or metastatic soft-tissue sarcomas with prior treatment with anthracycline-based chemotherapy. This study confirmed tolerability profile, but objective response was demonstrated in none of the patients [104]. Another phase II trial showed no anticancer activity for soblidotin in NSCLC patients previously treated with platinum-based chemotherapy [105].
2.3.2. Dolastatin 15
Dolastatin 15 is very similar to dolastatin 10. It was demonstrated by chromatography that the binding domain is the same as Vinca alkaloids and antimicrotubule peptides. The site of the binding is not a well-defined locus but a series of overlapping domains [106]. This drug showed an effect on growth and differentiation in leukaemia cell lines [107], induction of apoptosis through Bcl-2 phosphorylation in small cell lung cancer cell lines [108], and G2/M cell cycle arrest in human myeloma cell lines [109]. Romidepsin (Istodax), a dolastatin 15 analog, which also possesses activity as a histone deacetylase inhibitor, was found to be active in cutaneous T-cell lymphoma with a 34% objective response rate and for this it was approved in 2009 [110, 111]. Other two analogues of dolastatin 15 are used in clinical trials: cemadotin and tasidotin.
2.3.3. Cemadotin
Cemadotin (LU103793) exerts its effect by inhibition of microtubule polymerization [112]. This drug is not able to inhibit the binding of vinblastine to tubulin and it can suppress microtubule growth without a significant microtubule depolymerization [113]. This agent was first evaluated in three phase I clinical trials for advanced refractory solid tumors with different schedules. Daily 5-day every 3 weeks schedule identified a recommended dose of 2.5 mg/m2 per day. It was associated with neutropenia, peripheral edema, and liver function test abnormalities as DLTs. This dose showed lack of prohibitive cardiovascular effects. Acceptable general toxicity profile has allowed prompting phase II trials [114]. Meanwhile, cemadotin was studied for 24-hour intravenous (i.v.) continuous infusion every three weeks. 15 mg/m2 was the recommended dose for this schedule. Hypertension was highlighted as the DLT, even if its nature remained unclear [115]. Even 5-day continuous intravenous (CIV) infusion was investigated. MTD was 12.5 mg/m2. There were moderate nonhematologic toxicities and no evidence of the cardiovascular toxicity [116]. Pharmacokinetic analysis in these phase I trials suggested that cardiovascular toxicity may be associated with the magnitude of the peak blood levels of cemadotin or its metabolites, whereas myelotoxicity depends on the duration of time that blood levels exceed a threshold concentration.
The first phase II clinical trial which used this drug at 2.5 mg/m2 daily 5-day schedule repeated every three weeks obtained clinical activity with durable response in chemotherapy-naïve patients with metastatic melanoma. Toxicity profile previously determined for this schedule was confirmed [117]. In contrast, no activity was observed with the same schedule in metastatic breast cancer patients previously treated with two lines of chemotherapy and in untreated non-small cell cancer patients [118, 119].
2.3.4. Tasidotin
Tasidotin (ILX651) is a third-generation dolastatin 15 analogue that is metabolically stable through its resistance to hydrolysis [120]. It demonstrated in vitro cell cycle arrest in the G2 and M phases and inhibition of tubulin polymerization similar to cemadotin and the Vinca alkaloids. It can inhibit the extent of microtubule assembly even at low concentrations [121]. In vitro study with MCF7/GFP breast cancer cells and in vivo pharmacokinetic analysis through LOX tumors xenografts proposed that tasidotin is converted in tasidotin C-carboxylate, a functionally active intracellular metabolite, 10 to 30 times more potent [122]. Capability of inducing apoptosis was observed in Ewing's sarcoma, rhabdomyosarcoma, synovial sarcoma, and osteosarcoma cell lines. Preclinical xenograft models of pediatric sarcomas showed antitumor activity [123].
Like cemadotin the schedule indicated for clinical use is daily administration for 5 days every 3 weeks. The recommended dose for investigation in phase II trial was 27.3 mg/m2/day. The toxicity profile was favourable and antitumor activity was found in melanoma patients [124]. The other two schedules were evaluated in phase I trial: 34.4 mg/m2 d1,3,5 q3 wk and 46.8 mg/m2 d1,8,15 q4 wk [125, 126]. Tolerability was similar with these schedules.
2.4. Rhizoxin
Rhizoxin (NSC 332598) is a macrolide antitumor antibiotic extracted from a pathogenic fungus, Rhizopus chinensis. It is known for its antifungal activity, but it is also studied for cytotoxic activity in a variety of human tumor cell lines, including melanoma, leukaemia, sarcoma, and some human tumor xenografts of melanoma, lung, and breast cancer [127]. The drug can bind to tubulin and inhibits microtubule assembly, blocking the cell cycle at the G2-M phase [128]. It is a more potent cytotoxic compound than vincristine in vitro, and, in addition, it showed activity in vincristine-resistant cells [129].
A recommended dose of 2.0 mg/m2 administered by i.v. bolus injection at 3-week intervals was identified through phase I trial because of its good tolerability with mucositis and neutropenia as the main toxicities [130]. Minimal or absent antitumor activity was found in phase II studies for patients with various advanced solid tumors [131–134]. A pharmacological study demonstrated the rapid and variable elimination of rhizoxin. These data could explain the low levels of systemic toxicity and the little response rates [135]. For this reason, alternative dosage and schedule were studied in phase I trial. A 72-hour continuous i.v. infusion indicated the dose of 1.2 mg/m2/72 hours as the MTD. The toxicity profile was similar to that obtained with brief infusion, but yet no antitumor responses were found [136].
2.5. D-24851
D-24851 (N-(pyridin-4-yl)-[1-(4-chlorbenzyl)-indol-3-yl]-glyoxyl-amid) is a synthetic compound which has been selected by a cell-based screening assay by ASTA Medica AG, Germany. This drug destabilizes microtubules by interacting with a binding site that does not overlap with those of known microtubule-destabilizing agents like vincristine or colchicine [137, 138].
D-24851 (also known as indibulin) induces Bcl-2 and Bax-mediated apoptosis in both p53wt and p53−/− cell lines [137, 139]. It produces in vivo curative effects in rat sarcomas at nontoxic doses, is suitable for oral use, does not give rise to neurotoxic effects at curative doses, unlike vincristine and paclitaxel, and is effective in MDR tumor cells, so that it is an excellent candidate as a chemotherapy agent [137]. In 2004, an LC/MS/MS (liquid chromatography/tandem mass spectrometry) system was proposed for quantitative analysis of D-24851 in human plasma and urine in phase I clinical trials. Indibulin was used in phase I/II clinical trials of patients with advanced solid tumors (metastatic breast cancer) [27, 140, 141]. In a phase I clinical trial indibulin was studied for oral administration once daily for 14 days every 3 weeks in patients with various solid tumors. Pharmacokinetic analysis showed a better tolerability under feeding condition. The recommended dose identified for further studies was 60 mg daily for 14 days. Dose-limiting toxicities were nausea and vomiting, which seemed to be related to solvent lactic acid [141].
Furthermore, the effects of two N-heterocyclic indolyl glyoxylamides derivatives of D-24851, BPR0C259, and BPR0C123 were investigated in NSCLC cells. The obtained results showed that these compounds can suppress the cell proliferation, by inducing p53-independent apoptosis and G2/M phase arrest, and potentially increase radiosensitivity of human lung cancer cells in a p53-independent manner [142].
2.6. Pseudolaric Acid B
Pseudolaric acid B (PAB) is a diterpene isolated from Pseudolarix kaempferi Gordon which is able to selectively inhibit the growth of actively proliferating cancer cells. It induces apoptosis through the intrinsic pathway, involving JNK/SAPK and p53. Nevertheless, its cytotoxic effects were found also in p53−/− cell lines, which is interesting for its therapeutic use [143, 144].
It interacts with a different binding site on tubulin compared with those of colchicine and vinblastine [143] and, both in vitro and in vivo, inhibits endothelial cells proliferation and VEGF-dependent formation of blood vessels. In fact, PAB antagonizes VEGF-mediated antiapoptotic activity by inhibiting the phosphorylation/activation of KDR, the VEGF receptor implicated in mediating this effect [145]. Furthermore, at nontoxic doses, PAB inhibits VEGF secretion from tumor cells by reducing its HIF-1-dependent transcription. PAB, in fact, acts by accelerating the proteasome-mediated degradation of HIF-1α, by means of a mechanism so far unknown [146]. PAB also induces endothelial cell retraction, intercellular gap formation, and actin stress fiber formation, effects which can be attributed to disruption of tubulin cytoskeleton and which contribute to its antiangiogenic action [147]. Moreover, PAB circumvents P-glycoprotein overexpression-induced drug resistance and the doses used are well tolerated and nontoxic and have not proved lethal on tested animals [143]. PAB showed significant inhibitory effect and an additive inhibitory effect in combination with adriamycin on the growth of gastric cancer in vivo [148, 149].
2.7. Embellistatin
Embellistatin is a ketone isolated from Embellisia chlamydospora which inhibits microtubule polymerization and shows a strong antiangiogenic activity. It inhibits in vitro bovine endothelial cells (BAEC) proliferation through p53 and p21 activation, thus inhibiting bFGF-induced formation of vessels. This antiangiogenic activity has been confirmed in vivo on murine models. Similar effects have been found in human tumor cell lines, suggesting that it could be suitable for use in the development of new anticancer drugs [150].
2.8. CI-980
CI-980 (mivobulin) acts at the colchicine binding site and it appears to have significantly less vesicant activity than vinblastine [151]. It is a mitotic inhibitor with in vivo and in vitro activity against murine multidrug-resistant sublines. Its interactions with microtubules in vitro are similar to other drugs, but cellular microtubule and mitotic inhibition is more potent [152]. The uptake of CI-980 is not temperature or energy dependent, and its passive diffusion is followed by a significant but largely reversible binding to intracellular or membrane components [153].
Mivobulin was tested in a phase I trial using 24-hour infusion repeated every 3 weeks. MTD was 14.4 mg/m2. The main toxicities were neutropenia, dose-dependent but not dose-limiting, and early and reversible neurotoxicity characterized by dizziness, headache, loss of coordination, loss of consciousness, nervousness, and other symptoms. Tumor responses and tumor marker reductions were observed in a colon cancer patient and two ovarian cancer patients, respectively [154]. The same toxicity profile was confirmed in other studies [155, 156]. A continuous 72-hour infusion of MTD 4.5 mg/m2/day every 21 days was associated with reduced neurotoxicity but dose-limiting neutropenia [157]. For this reason, it was used in phase II clinical trials. A similar tolerability profile was found. CI-980 seems inactive in metastatic colorectal carcinoma, advanced soft-tissue sarcomas, treated and untreated melanoma, hormone-refractory prostate cancer, and malignant gliomas [158, 159]. Minimal activity was observed in platinum-refractory advanced epithelial ovarian carcinoma [160].
2.9. T138067
T138067 is a synthetic compound which irreversibly disrupts microtubule assembly by a selective and covalent binding to beta1, beta2, and beta4 isotypes of beta-tubulin at a conserved cysteine residue (Cys-239). Its action results in cell cycle arrest at G2/M and induction of apoptosis. It exhibits cytotoxic activity in tumor cell lines resistant to various antimicrotubule agents (vinblastine, paclitaxel, etc.) and in multidrug-resistant human tumor xenografts [161]. The covalent interaction of T138067 with β-tubulin may be proposed as a new way to overcome MDR. In vivo studies showed that this drug can cross the blood-brain barrier in mice, suggesting a possible use for brain tumors [162].
Phase I trials of T138067 were conducted by using a 3-hour infusion of drug given weekly or every 21 days with a recommended dose of 330 mg/m2 per week. DLTs were neutropenia and neurological effects, consisting of encephalopathy, headache, hearing loss, and ataxia [163, 164]. This weekly dosage was used in two phase II clinical trials for patients with malignant glioma and metastatic colorectal cancer previously treated with irinotecan and 5-fluorouracil, respectively. The good toxicity profile was confirmed in both studies. No clinical activity in terms of antitumor responses was observed in both cases [165, 166].
2.9.1. T900607
T900607 is similar to T138067 for the kind of binding to tubulin in Cys-239 residue, but it is distinguished for a reduced ability to cross the blood-brain barrier.
A phase I trial indicated a recommended dose of 130 mg/m2 delivered in i.v. infusion over 60 minutes on a 21-day cycle. No objective responses were observed but stable disease was reported in 7/20. Cardiac toxicity is the main drug-related side effect with this schedule. A different schedule consisting of weekly administration of T900607 identified MTD of 100 mg/m2. This schedule was used in a phase II clinical trial for untreated patients with unresectable hepatocellular carcinoma. It showed good tolerability and moderate activity in some of these patients [167].
2.10. ABT-751
ABT-751, also known as E7010, is a sulfonamide able to impair microtubule formation and inhibit cell growth. Its binding characteristics seem to be different from that of colchicine and Vinca alkaloids. This agent has antiproliferative effects in many tumor cell lines which are drug-resistant due to the P-glycoprotein overexpression [168]. It showed a broad spectrum of activity against a variety of tumors in mice and human tumor xenografts, when administered orally [169]. Beta3 isotype is the preferential binding target. ABT-751-resistant cells were characterized by decreased expression of this tubulin isotype [170]. A warning derived from an in vivo study, which shed light on a possible testicular toxicity related to this drug administration in mice. It consisted of loss of seminiferous epithelial cells due to apoptosis of meiotic spermatocytes [171]. This drug selectively reduces tumor blood flow through tumor necrosis, regardless of a direct cytotoxic effect on tumor cells. Negligible is the effect on normal vascular function [172].
In a phase I clinical trial ABT-751 was administered as oral single or 5-day doses. The recommended dose for phase II trials is identified at 320 mg/m2 for single dose and 200 mg/m2/day for 5-day repeated dose. Peripheral neuropathy and intestinal paralysis were the DLTs. Gastrointestinal toxicity was dose-dependent but hematological toxicity was not dose-dependent [173]. Pharmacokinetic analysis of this study suggested that activity of ABT-751 may be time-dependent. For this reason a new schedule using a divided dose in order to maintain the blood level of ABT-751 has been formulated. The recommended dose in hematologic malignancies is 175 mg/m2/day orally for 21 days every 4 weeks [174]. In a phase I trial for a pediatric population of patients with solid tumors ABT-751 was administered orally once daily for 21 days, repeated every 28 days. The MTD obtained for this schedule was 100 mg/m2/day. DLTs included fatigue, sensory neuropathy, transient hypertension, neutropenia, thrombocytopenia, nausea, vomiting, dehydration, abdominal pain, and constipation [175]. In a phase II clinical trial, 21-day every 28 days schedule at the dose of 200 mg daily was studied in taxane-refractory NSCLC patients. Toxicity was acceptable. Median time to tumor progression and overall survival was 2.1 and 8.4 months, respectively. The objective response rate was 2.9% [176]. The combination of this agent with other cytotoxic drugs was proposed for future clinical studies. A phase IB study investigated clinical antitumor activity of ABT-751 in combination with docetaxel in patients with castration-resistant prostate cancer. Based on the cumulative safety analysis, the recommended phase II dose of ABT-751 is 200 mg daily with docetaxel 60 mg/m2 for this patient population [177]. Further phases I and II clinical trials were carried out to evaluate activity of ABT-751 in combination with other drugs in advanced or metastatic NSCLC patients [178, 179]. ABT-751 showed adverse effects, although it has the advantage of being orally bioavailable.
3. Microtubule-Stabilizing Agents
Unlike the microtubule-destabilizing agents, there are other compounds that enhance microtubule polymerization. One of the most important classes of microtubule-stabilizing chemotherapy agents is that of taxanes, which target the cytoskeleton and spindle apparatus of tumor cells by binding to the microtubules, thereby disrupting key cellular mechanisms, including mitosis. The first microtubule-stabilizing agent used in anticancer chemotherapy [180] was paclitaxel. Thanks to their peculiar mechanism of action, taxanes are among the most effective chemotherapeutic agents used in the treatment of multiple solid tumors, such as breast, ovarian, lung, and prostate cancers. However, the occurrence of resistance limits treatment options and creates a major challenge for clinicians. Several potential mechanisms of resistance to these drugs have been identified, occurring at different pharmacodynamics levels. Besides the well-known overexpression of Pgp, an ABC transmembrane transporter which pumps the drugs out of the tumor cells [8], the alterated expression of specific beta-tubulin isotypes, seems to play an important role. Most notably, the increased expression of βIII-tubulin isotype has been associated with resistance to taxanes in several cancers, including ovarian, breast, and lung cancer [9, 181, 182]. It was originally correlated to the qualitative or quantitative modifications of the microtubule complex, which represents the target of such agents, definitively reducing the drug binding affinity [27]. However, the aberrant expression of βIII-tubulin can also interfere with microtubule dynamics, increasing the dynamic instability and counteracting the stabilizing effect of taxanes, with consequences for drug sensitivity/resistance [183]. Recent studies have suggested βIII-tubulin as a prosurvival factor adaptively expressed by cancer cells exposed to microenvironmental stressors, such as hypoxia or deficient nutrient supply [184, 185]. The activation of the βIII-tubulin-dependent pathway in partnership with GTPases, such as guanylate-binding protein 1 (GBP1), is associated with the incorporation of PIM1 into the cytoskeleton of tumor cells, conferring a survival advantage in a hostile microenvironment and ultimately leading to the development of drug resistance [186]. Finally, a multitude of alterations involving the apoptotic signaling pathways downstream the microtubule complex, as well as aberrant expression of microRNA, have been also found in resistant tumors. A better understanding of the mechanism underlying the occurrence of acquired resistance has led to the development of a new class of microtubule-stabilizing agents, including epothilones, discodermolide, sarcodictyins, eleutherobin, and laulimalide, which are more readily modifiable, with different structures but a similar mechanism of action [187] (Table 2). Epothilones, discodermolide, eleutherobins, and sarcodictyins compete with paclitaxel for binding to microtubules and bind at or near the taxane site, whereas laulimalide seems to bind to unique sites on microtubules (Figure 5). Recently, a novel generation of paclitaxel derivatives have been designed, targeting a specific intermediate binding site in the microtubule with differential affinity, depending on the β-tubulin isotype expressed in the tumor. Since βIII-tubulin is overexpressed in the majority of aggressive, resistant tumors, the design of a βIII-tubulin targeted agent was expected to enhance the drug activity, reducing common toxicities. However, none of the new molecules tested in breast cancer cell lines was superior to the currently used taxanes [188].
Table 2.
Microtube-stabilizing agents.
| Chemical lead | Properties and effects | Clinical trial/status | References |
|---|---|---|---|
| Epothilones | Elevated water solubility, activity versus MDR cell lines, and chemical malleability. | Phase II/III clinical trials in taxane-sensitive solid tumors (breast, lung, and prostate). | [195, 196] |
|
| |||
| Ixabepilone | Epothilone B analog, superior metabolic stability, and activity versus MDR cell lines. | Approved in 2007 for metastatic breast cancer; several ongoing trials in solid tumors. | [194, 197] |
|
| |||
| Laulimalide | Activity versus MDR cell lines and angiogenic activity, synergistic with docetaxel. | Preclinical phase. | [199, 201] |
|
| |||
| Dictyostatin | Activity against MDR cell lines, synergistic with taxol. | Preclinical phase. | [205, 206] |
Figure 5.

Similarities and differences between mechanisms of action and activity of microtubule-stabilizing agents.
3.1. Epothilones
Among several classes of microtubule-targeting chemotherapy agents that may maintain activity despite clinical resistance to taxanes, there are the epothilones which have been isolated from the soil bacterium Solangium cellulosum and have been studied most extensively in the clinical setting [189]. They induce the formation of an aberrant mitotic spindle, mitotic arrest, and apoptosis [190]. Their greater solubility in water and their activity in MDR cells have made them an alternative to paclitaxel in anticancer treatment [191, 192]. Moreover, their simple structure makes it easy to produce synthetic analogs during the clinical experimentation phase [190]. There are 4 classes of natural epothilones (A, B, C, and D). By means of the selection of resistant or taxane-dependent cells, it has been observed that tubulin β1 plays an important role in epothilone B functionality [193].
Ixabepilone (Ixempra) is a semisynthetic analog of epothilone B, selected because of its greater metabolic stability and its simple preparation. It is more powerful in vitro than paclitaxel and also presents cytotoxicity against MDR cells. It causes regression of MDR tumors and is more effective than paclitaxel in a wide spectrum of pediatric tumors [194]. Ixabepilone is currently the only approved epothilone derivative and the most clinically advanced (phases II and III clinical trials), showing efficacy in several patient subgroups and in various stages of breast cancer. This analog is used for the treatment of locally advanced or metastatic breast cancer as monotherapy after failure of a taxane, an anthracycline, and capecitabine, or in combination with capecitabine after failure of a taxane and an anthracycline [195].
A great number of phase II clinical studies of epothilones in cancer treatment have been reported, and significant activity in taxane-sensitive tumor types (such as breast, lung, and prostate cancers) has been observed [12, 17]. Response rates in taxane-refractory metastatic breast cancer are relatively modest, but ixabepilone and patupilone have shown promising efficacy in hormone-refractory metastatic prostate cancer and in taxane-refractory ovarian cancer [196, 197].
3.2. Laulimalide
Laulimalide is a macrolide isolated from marine sponge (Cacospongia mycofijiensis) which inhibits cell proliferation, promoting assembly of the microtubules and stabilizing them in a taxol-like way, but at a different binding site located on two adjacent β-tubulin units between tubulin protofilaments of a microtubule [198–200]. This agent is also active in MDR cells which overexpress glycoprotein-P. When administered below cytotoxic doses, the drug prevents blood vessel formation and the VEGF-induced endothelial cell migration [201]. Docetaxel and laulimalide possess a synergic effect in these two processes, whereas they have antagonistic effects towards cell proliferation.
Used at low doses, laulimalide inhibits events downstream of VEGFR-2 activation, such as FAK and paxillin phosphorylation, VEGFR-2/FAK/Hsp90 interaction, and integrin activation. Compared with docetaxel, laulimalide has less effect on the VEGF-induced VEGFR-2/integrin α5β1 interaction and is more effective with regard to phosphorylated paxillin levels. Furthermore, it inhibits RhoA/integrin α5β1 association, suggesting that synergic effects of the two drugs might be explained by two different modalities of action.
The low quantities of the drug found in nature, together with its instability caused by its transformation into isolaulimalide, have led to the synthesis of the drug itself and of several analogs. The removal of a electrophilic and/or nucleophilic group, which prevents the substitution process, leads to major functional stability of the drug [202]. In preclinical phase, laulimalide so far showed poor efficacy and systematic toxicity [12]. The macrolide peloruside A shared many of the same properties of laulimalide, including its binding site and synergistic effects with the taxanes [203].
3.3. Dictyostatin
Dictyostatin is a macrolactone produced from sponges which induces in vitro tubulin assembly in the same way of paclitaxel but more rapidly. Like discodermolide, this drug possesses an antiproliferative action against paclitaxel-resistant human tumor cells as a result of β-tubulin mutations [204]. Dictyostatin inhibits the binding of discodermolide with microtubules and both drugs are able to inhibit the binding of epothilone B and paclitaxel with microtubules [204]. Several discodermolide/dictyostatin hybrids have been designed and have been found to maintain antiproliferative activities against several taxane-resistant cell lines [205, 206].
3.4. Eleutherobin
Eleutherobin is a glycosylate diterpene isolated from Eleutherobia sp. [207], which inhibits cell proliferation stabilizing microtubules. It binds at a site overlapping that of paclitaxel [208]. There is another group of cytotoxic agents, called sarcodictyins, which are structurally and functionally correlated to eleutherobins but not so toxic [209]. A form of cytotoxic diterpene, known as (Z)-sarcodictyine A, has been isolated from Bellonia albiflora; this exhibits a high level of toxicity towards human HeLa cells of the cervix [210]. Eleutherobin and the sarcodictyins have not been pursued clinically likely due to their susceptibility to Pgp-mediated transport [211].
4. Clinical Implications
In the last few years, a great amount of efforts has been put into the identification of new microtubule-targeting agents for use in anticancer therapy [212]. These last generation agents are also active in MDR tumors, which are resistant to the traditional antitubulin drugs used in chemotherapy, such as Vinca alkaloids and taxanes. Furthermore, these compounds have shown significant antivascular and antiangiogenic activity, leading to the possibility of using them both as alternatives to or in combination with preexistent drugs, as already indicated in several published studies [213]. A lot of clinical trials were conducted to study microtubule-targeting agents. In particular, epothilones are in advanced phases of clinical development [214, 215]. In cancer therapy, microtubule-targeting agents can target angiogenesis, cell migration, and intracellular trafficking to prevent tumor growth and induce cancer cell apoptosis. These new agents, which impair or enhance tubulin polymerization, can be classified in two groups: natural and synthetic drugs. The natural compounds are derived from different species of uni- and multicellular organisms. To improve their pharmacodynamic and pharmacokinetic features some of these compounds are transformed in semisynthetic molecules. Other agents are produced by a totally synthetic procedure. The great diversity of natural and synthetic compounds capable of interacting with microtubules represents an important source for developing of novel potential anticancer agents. However, the effectiveness of these agents in cancer therapy has been impaired by various side effects and drug resistance. Phase I trials have allowed identifying more tolerable schedules with the most frequent toxicities represented by neutropenia and neurological, cardiovascular, and gastrointestinal effects. The main way of delivery is the i.v. infusion. Oral assumption was studied for the synthetic compounds D-24851 and ABT-751. The most evident efficacy was observed for rhizoxin, above all in NSCLC. For the other agents only minor or no responses were obtained. The identification of new schedules or the transformation in more potent analogues should allow overcoming these hurdles in their clinical advancement.
5. Conclusions
Data obtained up till now have allowed introducing some of these microtubule-targeting drugs into the clinical experimentation phase, whereas others, still in their preclinical phase, represent excellent candidates for a future use in cancer treatment, thus opening new roads towards the development of new, individual, and efficient therapeutic approaches.
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Wang L. G., Liu X. M., Kreis W., Budman D. R. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemotherapy and Pharmacology. 1999;44(5):355–361. doi: 10.1007/s002800050989. [DOI] [PubMed] [Google Scholar]
- 2.Vicente J. J., Wordeman L. Mitosis, microtubule dynamics and the evolution of kinesins. Experimental Cell Research. 2015;334(1):61–69. doi: 10.1016/j.yexcr.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desai A., Mitchison T. J. Microtubule polymerization dynamics. Annual Review of Cell and Developmental Biology. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 4.Bringmann H., Skiniotis G., Spilker A., Kandels-Lewis S., Vernos I., Surrey T. A kinesin-like motor inhibits microtubule dynamic instability. Science. 2004;303(5663):1519–1522. doi: 10.1126/science.1094838. [DOI] [PubMed] [Google Scholar]
- 5.Loong H. H., Yeo W. Microtubule-targeting agents in oncology and therapeutic potential in hepatocellular carcinoma. OncoTargets and Therapy. 2014;7:575–585. doi: 10.2147/OTT.S46019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klute K., Nackos E., Tasaki S., Nguyen D. P., Bander N. H., Tagawa S. T. Microtubule inhibitor-based antibody-drug conjugates for cancer therapy. OncoTargets and Therapy. 2014;7:2227–2236. doi: 10.2147/ott.s46887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kavallaris M., Verrills N. M., Hill B. T. Anticancer therapy with novel tubulin-interacting drugs. Drug Resistance Updates. 2001;4(6):392–401. doi: 10.1054/drup.2002.0230. [DOI] [PubMed] [Google Scholar]
- 8.Katayama K., Noguchi K., Sugimoto Y. Regulations of P-Glycoprotein/ABCB1/MDR1 in human cancer cells. New Journal of Science. 2014;2014:10. doi: 10.1155/2014/476974.476974 [DOI] [Google Scholar]
- 9.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nature Reviews Cancer. 2010;10(3):194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 10.Katsetos C. D., Herman M. M., Mörk S. J. Class III beta-tubulin in human development and cancer. Cell Motility and the Cytoskeleton. 2003;55(2):77–96. doi: 10.1002/cm.10116. [DOI] [PubMed] [Google Scholar]
- 11.Kavallaris M., Tait A. S., Walsh B. J., et al. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Research. 2001;61(15):5803–5809. [PubMed] [Google Scholar]
- 12.Field J. J., Kanakkanthara A., Miller J. H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorganic and Medicinal Chemistry. 2014;22(18):5050–5059. doi: 10.1016/j.bmc.2014.02.035. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y.-M., Chen H.-L., Lee H.-Y., Liou J.-P. Tubulin inhibitors: a patent review. Expert Opinion on Therapeutic Patents. 2014;24(1):69–88. doi: 10.1517/13543776.2014.859247. [DOI] [PubMed] [Google Scholar]
- 14.Moudi M., Go R., Yien C. Y. S., Nazre M. Vinca alkaloids. International Journal of Preventive Medicine. 2013;4(11):1231–1235. [PMC free article] [PubMed] [Google Scholar]
- 15.Gregory R. K., Smith I. E. Vinorelbine—a clinical review. British Journal of Cancer. 2000;82(12):1907–1913. doi: 10.1054/bjoc.2000.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vallo S., Michaelis M., Rothweiler F., et al. Drug-resistant urothelial cancer cell lines display diverse sensitivity profiles to potential second-line therapeutics. Translational Oncology. 2015;8(3):210–216. doi: 10.1016/j.tranon.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukhtar E., Adhami V. M., Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Molecular Cancer Therapeutics. 2014;13(2):275–284. doi: 10.1158/1535-7163.MCT-13-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subbaraju G. V., Golakoti T., Patterson G. M. L., Moore R. E. Three new cryptophycins from Nostoc sp. GSV 224. Journal of Natural Products. 1997;60(3):302–305. doi: 10.1021/np960700a. [DOI] [PubMed] [Google Scholar]
- 19.Weiss C., Sammet B., Sewald N. Recent approaches for the synthesis of modified cryptophycins. Natural Product Reports. 2013;30(7):924–940. doi: 10.1039/c3np70022d. [DOI] [PubMed] [Google Scholar]
- 20.Smith C. D., Zhang X. Mechanism of action of cryptophycin. Interaction with the Vinca alkaloid domain of tubulin. Journal of Biological Chemistry. 1996;271(11):6192–6198. doi: 10.1074/jbc.271.11.6192. [DOI] [PubMed] [Google Scholar]
- 21.Mooberry S. L., Busquets L., Tien G. Induction of apoptosis by cryptophycin 1, a new antimicrotubule agent. International Journal of Cancer. 1997;73(3):440–448. doi: 10.1002/(SICI)1097-0215(19971104)73:3<440::AID-IJC20>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 22.Panda D., Ananthnarayan V., Larson G., Shih C., Jordan M. A., Wilson L. Interaction of the antitumor compound cryptophycin-52 with tubulin. Biochemistry. 2000;39(46):14121–14127. doi: 10.1021/bi0010827. [DOI] [PubMed] [Google Scholar]
- 23.Shih C., Teicher B. A. Cryptophycins: a novel class of potent antimitotic antitumor depsipeptides. Current Pharmaceutical Design. 2001;7(13):1259–1276. doi: 10.2174/1381612013397474. [DOI] [PubMed] [Google Scholar]
- 24.Bolduc K. L., Larsen S. D., Sherman D. H. Efficient, divergent synthesis of cryptophycin unit A analogues. Chemical Communications. 2012;48(51):6414–6416. doi: 10.1039/c2cc32417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corbett T. H., Valeriote F. A., Demchik L., et al. Preclinical anticancer activity of cryptophycin-8. Journal of Experimental Therapeutics and Oncology. 1996;1(2):95–108. [PubMed] [Google Scholar]
- 26.Sessa C., Weigang-Köhler K., Pagani O., et al. Phase I and pharmacological studies of the cryptophycin analogue LY355703 administered on a single intermittent or weekly schedule. European Journal of Cancer. 2002;38(18):2388–2396. doi: 10.1016/s0959-8049(02)00489-6. [DOI] [PubMed] [Google Scholar]
- 27.Dumontet C., Jordan M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nature Reviews Drug Discovery. 2010;9(10):790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Agostino G., del Campo J., Mellado B., et al. A multicenter phase II study of the cryptophycin analog LY355703 in patients with platinum-resistant ovarian cancer. International Journal of Gynecological Cancer. 2006;16(1):71–76. doi: 10.1111/j.1525-1438.2006.00276.x. [DOI] [PubMed] [Google Scholar]
- 29.Al-Awar R. S., Corbett T. H., Ray J. E., et al. Biological evaluation of cryptophycin 52 fragment A analogues: effect of the multidrug resistance ATP binding cassette transporters on antitumor activity. Molecular Cancer Therapeutics. 2004;3(9):1061–1067. [PubMed] [Google Scholar]
- 30.Wagner M. M., Paul D. C., Shih C., Jordan M. A., Wilson L., Williams D. C. In vitro pharmacology of cryptophycin 52 (LY355703) in human tumor cell lines. Cancer Chemotherapy and Pharmacology. 1999;43(2):115–125. doi: 10.1007/s002800050871. [DOI] [PubMed] [Google Scholar]
- 31.Stevenson J. P., Sun W., Gallagher M., et al. Phase I trial of the cryptophycin analogue LY355703 administered as an intravenous infusion on a day 1 and 8 schedule every 21 days. Clinical Cancer Research. 2002;8(8):2524–2529. [PubMed] [Google Scholar]
- 32.Edelman M. J., Gandara D. R., Hausner P., et al. Phase 2 study of cryptophycin 52 (LY355703) in patients previously treated with platinum based chemotherapy for advanced non-small cell lung cancer. Lung Cancer. 2003;39(2):197–199. doi: 10.1016/s0169-5002(02)00511-1. [DOI] [PubMed] [Google Scholar]
- 33.Corbett T., Polin L., LoRusso P., et al. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Berlin, Germany: Springer; 2004. In Vivo methods for screening and preclinical testing; pp. 99–123. [DOI] [Google Scholar]
- 34.Liang J., Moore R. E., Moher E. D., et al. Cryptophycins-309, 249 and other cryptophycin analogs: preclinical efficacy studies with mouse and human tumors. Investigational New Drugs. 2005;23(3):213–224. doi: 10.1007/s10637-005-6729-9. [DOI] [PubMed] [Google Scholar]
- 35.Menon K., Alvarez E., Forler P., et al. Antitumor activity of cryptophycins: effect of infusion time and combination studies. Cancer Chemotherapy and Pharmacology. 2000;46(2):142–149. doi: 10.1007/s002800000135. [DOI] [PubMed] [Google Scholar]
- 36.Boinpally R. R., Polin L., Zhou S.-L., et al. Pharmacokinetics and tissue distribution of cryptophycin 52 (C-52) epoxide and cryptophycin 55 (C-55) chlorohydrin in mice with subcutaneous tumors. Cancer Chemotherapy and Pharmacology. 2003;52(1):25–33. doi: 10.1007/s00280-003-0621-0. [DOI] [PubMed] [Google Scholar]
- 37.Griggs J., Metcalfe J. C., Hesketh R. Targeting tumour vasculature: the development of combretastatin A4. The Lancet Oncology. 2001;2(2):82–87. doi: 10.1016/s1470-2045(00)00224-2. [DOI] [PubMed] [Google Scholar]
- 38.Dark G. G., Hill S. A., Prise V. E., Tozer G. M., Pettit G. R., Chaplin D. J. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Research. 1997;57(10):1829–1834. [PubMed] [Google Scholar]
- 39.Tozer G. M., Kanthou C., Parkins C. S., Hill S. A. The biology of the combretastatins as tumour vascular targeting agents. International Journal of Experimental Pathology. 2002;83(1):21–38. doi: 10.1046/j.1365-2613.2002.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nabha S. M., Mohammad R. M., Dandashi M. H., et al. Combretastatin-A4 prodrug induces mitotic catastrophe in chronic lymphocytic leukemia cell line independent of caspase activation and poly(ADP-ribose) polymerase cleavage. Clinical Cancer Research. 2002;8(8):2735–2741. [PubMed] [Google Scholar]
- 41.Vitale I., Antoccia A., Cenciarelli C., et al. Combretastatin CA-4 and combretastatin derivative induce mitotic catastrophe dependent on spindle checkpoint and caspase-3 activation in non-small cell lung cancer cells. Apoptosis. 2007;12(1):155–166. doi: 10.1007/s10495-006-0491-0. [DOI] [PubMed] [Google Scholar]
- 42.Friesen D. E., Barakat K. H., Semenchenko V., et al. Discovery of small molecule inhibitors that interact with γ-tubulin. Chemical Biology and Drug Design. 2012;79(5):639–652. doi: 10.1111/j.1747-0285.2012.01340.x. [DOI] [PubMed] [Google Scholar]
- 43.Katsetos C. D., Reddy G., Dráberová E., et al. Altered cellular distribution and subcellular sorting of γ-tubulin in diffuse astrocytic gliomas and human glioblastoma cell lines. Journal of Neuropathology and Experimental Neurology. 2006;65(5):465–477. doi: 10.1097/01.jnen.0000229235.20995.6e. [DOI] [PubMed] [Google Scholar]
- 44.Caracciolo V., D'Agostino L., Dráberová E., et al. Differential expression and cellular distribution of gamma-tubulin and betaIII-tubulin in medulloblastomas and human medulloblastoma cell lines. Journal of Cellular Physiology. 2010;223(2):519–529. doi: 10.1002/jcp.22077. [DOI] [PubMed] [Google Scholar]
- 45.Maounis N. F., Dráberová E., Mahera E., et al. Overexpression of γ-tubulin in non-small cell lung cancer. Histology and Histopathology. 2012;27(9):1183–1194. doi: 10.14670/HH-27.1183. [DOI] [PubMed] [Google Scholar]
- 46.Liu T., Niu Y., Yu Y., Liu Y., Zhang F. Increased γ-tubulin expression and P16INK4A promoter methylation occur together in preinvasive lesions and carcinomas of the breast. Annals of Oncology. 2009;20(3):441–448. doi: 10.1093/annonc/mdn651. [DOI] [PubMed] [Google Scholar]
- 47.Mita M. M., Sargsyan L., Mita A. C., Spear M. Vascular-disrupting agents in oncology. Expert Opinion on Investigational Drugs. 2013;22(3):317–328. doi: 10.1517/13543784.2013.759557. [DOI] [PubMed] [Google Scholar]
- 48.Tozer G. M., Prise V. E., Wilson J., et al. Combretastatin A-4 phosphate as a tumor vascular-targeting agent: early effects in tumors and normal tissues. Cancer Research. 1999;59(7):1626–1634. [PubMed] [Google Scholar]
- 49.Landuyt W., Verdoes O., Darius D. O., et al. Vascular targeting of solid tumoursa major ‘inverse’ volume-response relationship following combretastatin A-4 phosphate treatment of rat rhabdomyosarcomas. European Journal of Cancer. 2000;36(14):1833–1843. doi: 10.1016/S0959-8049(00)00173-8. [DOI] [PubMed] [Google Scholar]
- 50.Chaplin D. J., Hill S. A. The development of combretastatin A4 phosphate as a vascular targeting agent. International Journal of Radiation Oncology Biology Physics. 2002;54(5):1491–1496. doi: 10.1016/s0360-3016(02)03924-x. [DOI] [PubMed] [Google Scholar]
- 51.Beauregard D. A., Hill S. A., Chaplin D. J., Brindle K. M. The susceptibility of tumors to the antivascular drug combretastatin A4 phosphate correlates with vascular permeability. Cancer Research. 2001;61(18):6811–6815. [PubMed] [Google Scholar]
- 52.Kanthou C., Tozer G. M. The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood. 2002;99(6):2060–2069. doi: 10.1182/blood.V99.6.2060. [DOI] [PubMed] [Google Scholar]
- 53.Hussain A., Steimle M., Hoppeler H., Baum O., Egginton S. The vascular-disrupting agent combretastatin impairs splitting and sprouting forms of physiological angiogenesis. Microcirculation. 2012;19(4):296–305. doi: 10.1111/j.1549-8719.2012.00160.x. [DOI] [PubMed] [Google Scholar]
- 54.Vincent L., Kermani P., Young L. M., et al. Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling. Journal of Clinical Investigation. 2005;115(11):2992–3006. doi: 10.1172/JCI24586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks A. C., Kanthou C., Cook I. H., et al. The vascular targeting agent combretastatin A-4-phosphate induces neutrophil recruitment to endothelial cells in vitro. Anticancer Research. 2003;23(4):3199–3206. [PubMed] [Google Scholar]
- 56.Dachs G. U., Steele A. J., Coralli C., et al. Anti-vascular agent Combretastatin A-4-P modulates hypoxia inducible factor-1 and gene expression. BMC Cancer. 2006;6, article 280 doi: 10.1186/1471-2407-6-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li L., Rojiani A. M., Siemann D. W. Preclinical evaluations of therapies combining the vascular targeting agent combretastatin A-4 disodium phosphate and conventional anticancer therapies in the treatment of Kaposi's sarcoma. Acta Oncologica. 2002;41(1):91–97. doi: 10.1080/028418602317314127. [DOI] [PubMed] [Google Scholar]
- 58.Hill S. A., Chaplin D. J., Lewis G., Tozer G. M. Schedule dependence of combretastatin A4 phosphate in transplanted and spontaneous tumour models. International Journal of Cancer. 2002;102(1):70–74. doi: 10.1002/ijc.10655. [DOI] [PubMed] [Google Scholar]
- 59.Horsman M. R., Murata R. Combination of vascular targeting agents with thermal or radiation therapy. International Journal of Radiation Oncology Biology Physics. 2002;54(5):1518–1523. doi: 10.1016/S0360-3016(02)03926-3. [DOI] [PubMed] [Google Scholar]
- 60.Lankester K. J., Maxwell R. J., Pedley R. B., et al. Combretastatin A-4-phosphate effectively increases tumor retention of the therapeutic antibody, 131I-A5B7, even at doses that are sub-optimal for vascular shut-down. International Journal of Oncology. 2007;30(2):453–460. [PubMed] [Google Scholar]
- 61.Wildiers H., Ahmed B., Guetens G., et al. Combretastatin A-4 phosphate enhances CPT-11 activity independently of the administration sequence. European Journal of Cancer. 2004;40(2):284–290. doi: 10.1016/j.ejca.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 62.Young S. L., Chaplin D. J. Combretastatin A4 phosphate: Background and current clinical status. Expert Opinion on Investigational Drugs. 2004;13(9):1171–1182. doi: 10.1517/13543784.13.9.1171. [DOI] [PubMed] [Google Scholar]
- 63.Bilenker J. H., Flaherty K. T., Rosen M., et al. Phase I trial of combretastatin A-4 phosphate with carboplatin. Clinical Cancer Research. 2005;11(4):1527–1533. doi: 10.1158/1078-0432.CCR-04-1434. [DOI] [PubMed] [Google Scholar]
- 64.Cooney M. M., Radivoyevitch T., Dowlati A., et al. Cardiovascular safety profile of combretastatin a4 phosphate in a single-dose phase I study in patients with advanced cancer. Clinical Cancer Research. 2004;10(1, part 1):96–100. doi: 10.1158/1078-0432.ccr-0364-3. [DOI] [PubMed] [Google Scholar]
- 65.Siemann D. W., Chaplin D. J., Walicke P. A. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P) Expert Opinion on Investigational Drugs. 2009;18(2):189–197. doi: 10.1517/13543780802691068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Granata R., Locati L. D., Licitra L. Fosbretabulin for the treatment of anaplastic thyroid cancer. Future Oncology. 2014;10(13):2015–2021. doi: 10.2217/FON.14.154. [DOI] [PubMed] [Google Scholar]
- 67.Dowlati A., Robertson K., Cooney M., et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin A-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Research. 2002;62(12):3408–3416. [PubMed] [Google Scholar]
- 68.Anderson H. L., Yap J. T., Miller M. P., Robbins A., Jones T., Price P. M. Assessment of pharmacodynamic vascular response in a phase I trial of combretastatin A4 phosphate. Journal of Clinical Oncology. 2003;21(15):2823–2830. doi: 10.1200/jco.2003.05.186. [DOI] [PubMed] [Google Scholar]
- 69.Rustin G. J. S., Galbraith S. M., Anderson H., et al. Phase I clinical trial of weekly combretastatin A4 phosphate: clinical and pharmacokinetic results. Journal of Clinical Oncology. 2003;21(15):2815–2822. doi: 10.1200/jco.2003.05.185. [DOI] [PubMed] [Google Scholar]
- 70.Stevenson J. P., Rosen M., Sun W., et al. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. Journal of Clinical Oncology. 2003;21(23):4428–4438. doi: 10.1200/jco.2003.12.986. [DOI] [PubMed] [Google Scholar]
- 71.Nallamothu R., Wood G. C., Kiani M. F., Moore B. M., Horton F. P., Thoma L. A. A targeted liposome delivery system for combretastatin A4: formulation optimization through drug loading and in vitro release studies. PDA Journal of Pharmaceutical Science and Technology. 2006;60(3):144–155. [PubMed] [Google Scholar]
- 72.Sosa J. A., Elisei R., Jarzab B., et al. Randomized safety and efficacy study of fosbretabulin with paclitaxel/carboplatin against anaplastic thyroid carcinoma. Thyroid. 2014;24(2):232–240. doi: 10.1089/thy.2013.0078. [DOI] [PubMed] [Google Scholar]
- 73.Korpanty G., Smyth E., Carney D. N. Update on anti-angiogenic therapy in non-small cell lung cancer: are we making progress? Journal of Thoracic Disease. 2011;3(1):19–29. doi: 10.3978/j.issn.2072-1439.2010.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hua J., Sheng Y., Pinney K. G., et al. Oxi4503, a novel vascular targeting agent: effects on blood flow and antitumor activity in comparison to combretastatin A-4 phosphate. Anticancer Research. 2003;23(2):1433–1440. [PubMed] [Google Scholar]
- 75.Sheng Y., Hua J., Pinney K. G., et al. Combretastatin family member OXI4503 induces tumor vascular collapse through the induction of endothelial apoptosis. International Journal of Cancer. 2004;111(4):604–610. doi: 10.1002/ijc.20297. [DOI] [PubMed] [Google Scholar]
- 76.Staflin K., Järnum S., Hua J., Honeth G., Kannisto P., Lindvall M. Combretastatin A-1 phosphate potentiates the antitumor activity of carboplatin and paclitaxel in a severe combined immunodeficiency disease (SCID) mouse model of human ovarian carcinoma. International Journal of Gynecological Cancer. 2006;16(4):1557–1564. doi: 10.1111/j.1525-1438.2006.00627.x. [DOI] [PubMed] [Google Scholar]
- 77.Kirwan I. G., Loadman P. M., Swaine D. J., et al. Comparative preclinical pharmacokinetic and metabolic studies of the combretastatin prodrugs combretastatin A4 phosphate and A1 phosphate. Clinical Cancer Research. 2004;10(4):1446–1453. doi: 10.1158/1078-0432.ccr-0518-03. [DOI] [PubMed] [Google Scholar]
- 78.Patterson D. M., Zweifel M., Middleton M. R., et al. Phase I clinical and pharmacokinetic evaluation of the vascular-disrupting agent OXi4503 in patients with advanced solid tumors. Clinical Cancer Research. 2012;18(5):1415–1425. doi: 10.1158/1078-0432.CCR-11-2414. [DOI] [PubMed] [Google Scholar]
- 79.Cummings J., Zweifel M., Smith N., et al. Evaluation of cell death mechanisms induced by the vascular disrupting agent OXi4503 during a phase I clinical trial. British Journal of Cancer. 2012;106(11):1766–1771. doi: 10.1038/bjc.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar S., Mehndiratta S., Nepali K., et al. Novel indole-bearing combretastatin analogues as tubulin polymerization inhibitors. Organic and Medicinal Chemistry Letters. 2013;3(1, article 3) doi: 10.1186/2191-2858-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng S., Zhong Q., Mottamal M., et al. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-A4 as anticancer agents. Journal of Medicinal Chemistry. 2014;57(8):3369–3381. doi: 10.1021/jm500002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hori K., Saito S., Sato Y., Kubota K. Stoppage of blood flow in 3-methylcholanthrene-induced autochthonous primary tumor due to a novel combretastatin A-4 derivative, AC7700, and its antitumor effect. Medical Science Monitor. 2001;7(1):26–33. [PubMed] [Google Scholar]
- 83.Delmonte A., Sessa C. AVE8062: a new combretastatin derivative vascular disrupting agent. Expert Opinion on Investigational Drugs. 2009;18(10):1541–1548. doi: 10.1517/13543780903213697. [DOI] [PubMed] [Google Scholar]
- 84.Hori K., Saito S. Microvascular mechanisms by which the combretastatin A-4 derivative AC7700 (AVE8062) induces tumour blood flow stasis. British Journal of Cancer. 2003;89(7):1334–1344. doi: 10.1038/sj.bjc.6601261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morinaga Y., Suga Y., Ehara S., Harada K., Nihei Y., Suzuki M. Combination effect of AC-7700, a novel combretastatin A-4 derivative, and cisplatin against murine and human tumors in vivo. Cancer Science. 2003;94(2):200–204. doi: 10.1111/j.1349-7006.2003.tb01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blay J. Y., Pápai Z., Tolcher A. W., et al. Ombrabulin plus cisplatin versus placebo plus cisplatin in patients with advanced soft-tissue sarcomas after failure of anthracycline and ifosfamide chemotherapy: a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology. 2015;16(5):531–540. doi: 10.1016/s1470-2045(15)70102-6. [DOI] [PubMed] [Google Scholar]
- 87.Poncet J. The dolastatins, a family of promising antineoplastic agents. Current Pharmaceutical Design. 1999;5(3):139–162. [PubMed] [Google Scholar]
- 88.Erik F., Jayaram S. Anticancer Agents from Natural Products. 2nd. CRC Press; 2011. The dolastatins; pp. 263–290. [Google Scholar]
- 89.Krug L. M., Miller V. A., Kalemkerian G. P., et al. Phase II study of dolastatin-10 in patients with advanced non-small-cell lung cancer. Annals of Oncology. 2000;11(2):227–228. doi: 10.1023/A:1008349209956. [DOI] [PubMed] [Google Scholar]
- 90.Margolin K., Longmate J., Synold T. W., et al. Dolastatin-10 in metastatic melanoma: a phase II and pharmokinetic trial of the California Cancer Consortium. Investigational New Drugs. 2001;19(4):335–340. doi: 10.1023/a:1010626230081. [DOI] [PubMed] [Google Scholar]
- 91.Saad E. D., Kraut E. H., Hoff P. M., et al. Phase II study of dolastatin-10 as first-line treatment for advanced colorectal cancer. American Journal of Clinical Oncology: Cancer Clinical Trials. 2002;25(5):451–453. doi: 10.1097/00000421-200210000-00005. [DOI] [PubMed] [Google Scholar]
- 92.Hoffman M. A., Blessing J. A., Lentz S. S. A phase II trial of dolastatin-10 in recurrent platinum-sensitive ovarian carcinoma: a Gynecologic Oncology Group Study. Gynecologic Oncology. 2003;89(1):95–98. doi: 10.1016/s0090-8258(03)00007-6. [DOI] [PubMed] [Google Scholar]
- 93.Vaishampayan U., Glode M., Du W., et al. Phase II study of dolastatin-10 in patients with hormone-refractory metastatic prostate adenocarcinoma. Clinical Cancer Research. 2000;6(11):4205–4208. [PubMed] [Google Scholar]
- 94.Perez E. A., Hillman D. W., Fishkin P. A., et al. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Investigational New Drugs. 2005;23(3):257–261. doi: 10.1007/s10637-005-6735-y. [DOI] [PubMed] [Google Scholar]
- 95.Kindler H. L., Tothy P. K., Wolff R., et al. Phase II trials of dolastatin-10 in advanced pancreaticobiliary cancers. Investigational New Drugs. 2005;23(5):489–493. doi: 10.1007/s10637-005-2909-x. [DOI] [PubMed] [Google Scholar]
- 96.von Mehren M., Balcerzak S. P., Kraft A. S., et al. Phase II trial of dolastatin-10, a novel anti-tubulin agent, in metastatic soft tissue sarcomas. Sarcoma. 2004;8(4):107–111. doi: 10.1080/13577140400009163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Simmons T. L., Andrianasolo E., McPhail K., Flatt P., Gerwick W. H. Marine natural products as anticancer drugs. Molecular Cancer Therapeutics. 2005;4(2):333–342. [PubMed] [Google Scholar]
- 98.Watanabe J., Natsume T., Fujio N., Miyasaka K., Kobayashi M. Induction of apoptosis in human cancer cells by TZT-1027, an antimicrotubule agent. Apoptosis. 2000;5(4):345–353. doi: 10.1023/A:1009687609330. [DOI] [PubMed] [Google Scholar]
- 99.Schöffski P., Thate B., Beutel G., et al. Phase I and pharmacokinetic study of TZT-1027, a novel synthetic dolastatin 10 derivative, administered as a 1-hour intravenous infusion every 3 weeks in patients with advanced refractory cancer. Annals of Oncology. 2004;15(4):671–679. doi: 10.1093/annonc/mdh141. [DOI] [PubMed] [Google Scholar]
- 100.de Jonge M. J. A., van der Gaast A., Planting A. S. T., et al. Phase I and pharmacokinetic study of the dolastatin 10 analogue TZT-1027, given on days 1 and 8 of a 3-week cycle in patients with advanced solid tumors. Clinical Cancer Research. 2005;11(10):3806–3813. doi: 10.1158/1078-0432.ccr-04-1937. [DOI] [PubMed] [Google Scholar]
- 101.Tamura K., Nakagawa K., Kurata T., et al. Phase I study of TZT-1027, a novel synthetic dolastatin 10 derivative and inhibitor of tubulin polymerization, which was administered to patients with advanced solid tumors on days 1 and 8 in 3-week courses. Cancer Chemotherapy and Pharmacology. 2007;60(2):285–293. doi: 10.1007/s00280-006-0382-7. [DOI] [PubMed] [Google Scholar]
- 102.Greystoke A., Blagden S., Thomas A. L., et al. A phase I study of intravenous TZT-1027 administered on day 1 and day 8 of a three-weekly cycle in combination with carboplatin given on day 1 alone in patients with advanced solid tumours. Annals of Oncology. 2006;17(8):1313–1319. doi: 10.1093/annonc/mdl097. [DOI] [PubMed] [Google Scholar]
- 103.Horti J., Juhasz E., Monostori Z., Maeda K., Eckhardt S., Bodrogi I. Phase I study of TZT-1027, a novel synthetic dolastatin 10 derivative, for the treatment of patients with non-small cell lung cancer. Cancer Chemotherapy and Pharmacology. 2008;62(1):173–180. doi: 10.1007/s00280-007-0665-7. [DOI] [PubMed] [Google Scholar]
- 104.Patel S., Keohan M. L., Saif M. W., et al. Phase II study of intravenous TZT-1027 in patients with advanced or metastatic soft-tissue sarcomas with prior exposure to anthracycline-based chemotherapy. Cancer. 2006;107(12):2881–2887. doi: 10.1002/cncr.22334. [DOI] [PubMed] [Google Scholar]
- 105.Riely G. J., Gadgeel S., Rothman I., et al. A phase 2 study of TZT-1027, administered weekly to patients with advanced non-small cell lung cancer following treatment with platinum-based chemotherapy. Lung Cancer. 2007;55(2):181–185. doi: 10.1016/j.lungcan.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 106.Cruz-Monserrate Z., Mullaney J. T., Harran P. G., Pettit G. R., Hamel E. Dolastatin 15 binds in the vinca domain of tubulin as demonstrated by Hummel-Dreyer chromatography. European Journal of Biochemistry. 2003;270(18):3822–3828. doi: 10.1046/j.1432-1033.2003.03776.x. [DOI] [PubMed] [Google Scholar]
- 107.Steube K. G., Grunicke D., Pietsch T., Gignac S. M., Pettit G. R., Drexler H. G. Dolastatin 10 and dolastatin 15: effects of two natural peptides on growth and differentiation of leukemia cells. Leukemia. 1992;6(10):1048–1053. [PubMed] [Google Scholar]
- 108.Ali M. A., Rosati R., Pettit G. R., Kalemkerian G. P. Dolastatin 15 induces apoptosis and BCL-2 phosphorylation in small cell lung cancer cell lines. Anticancer Research. 1998;18(2):1021–1026. [PubMed] [Google Scholar]
- 109.Sato M., Sagawa M., Nakazato T., Ikeda Y., Kizaki M. A natural peptide, dolastatin 15, induces G2/M cell cycle arrest and apoptosis of human multiple myeloma cells. International Journal of Oncology. 2007;30(6):1453–1459. [PubMed] [Google Scholar]
- 110.Piekarz R. L., Frye R., Turner M., et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. Journal of Clinical Oncology. 2009;27(32):5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bates S. E., Eisch R., Ling A., et al. Romidepsin in peripheral and cutaneous T-cell lymphoma: mechanistic implications from clinical and correlative data. British Journal of Haematology. 2015;170(1):96–109. doi: 10.1111/bjh.13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.de Arruda M., Cocchiaro C. A., Nelson C. M., et al. LU103793 (NSC D-669356): a synthetic peptide that interacts with microtubules and inhibits mitosis. Cancer Research. 1995;55(14):3085–3092. [PubMed] [Google Scholar]
- 113.Jordan M. A., Walker D., de Arruda M., Barlozzari T., Panda D. Suppression of microtubule dynamics by binding of cemadotin to tubulin: possible mechanism for its antitumor action. Biochemistry. 1998;37(50):17571–17578. doi: 10.1021/bi9817414. [DOI] [PubMed] [Google Scholar]
- 114.Villalona-Calero M. A., Baker S. D., Hammond L., et al. Phase I and pharmacokinetic study of the water-soluble dolastatin 15 analog LU103793 in patients with advanced solid malignancies. Journal of Clinical Oncology. 1998;16(8):2770–2779. doi: 10.1200/JCO.1998.16.8.2770. [DOI] [PubMed] [Google Scholar]
- 115.Mross K., Berdel W. E., Fiebig H. H., Velagapudi R., von Broen I. M., Unger C. Clinical and pharmacologic phase I study of Cemadotin-HCl (LU103793), a novel antimitotic peptide, given as 24-hour infusion in patients with advanced cancer. A study of the Arbeitsgemeinschaft Internistische Onkologie (AIO) Phase I Group and Arbeitsgruppe Pharmakologie in der Onkologie und Haematologie (APOH) Group of the German Cancer Society. Annals of Oncology. 1998;9(12):1323–1330. doi: 10.1023/a:1008430515881. [DOI] [PubMed] [Google Scholar]
- 116.Supko J. G., Lynch T. J., Clark J. W., et al. A phase I clinical and pharmacokinetic study of the dolastatin analogue cemadotin administered as a 5-day continuous intravenous infusion. Cancer Chemotherapy and Pharmacology. 2000;46(4):319–328. doi: 10.1007/s002800000152. [DOI] [PubMed] [Google Scholar]
- 117.Smyth J., Boneterre M. E., Schellens J., et al. Activity of the dolastatin analogue, LU103793, in malignant melanoma. Annals of Oncology. 2001;12(4):509–511. doi: 10.1023/a:1011194910571. [DOI] [PubMed] [Google Scholar]
- 118.Kerbrat P., Dieras V., Pavlidis N., Ravaud A., Wanders J., Fumoleau P. Phase II study of LU 103793 (dolastatin analogue) in patients with metastatic breast cancer. European Journal of Cancer. 2003;39(3):317–320. doi: 10.1016/S0959-8049(02)00531-2. [DOI] [PubMed] [Google Scholar]
- 119.Marks R. S., Graham D. L., Sloan J. A., et al. A phase II study of the dolastatin 15 analogue LU 103793 in the treatment of advanced non-small-cell lung cancer. American Journal of Clinical Oncology. 2003;26(4):336–337. doi: 10.1097/00000421-200308000-00005. [DOI] [PubMed] [Google Scholar]
- 120.Rasila K. K., Verschraegen C. Tasidotin HCI genzyme. Current Opinion in Investigational Drugs. 2005;6(6):631–638. [PubMed] [Google Scholar]
- 121.Bai R., Edler M. C., Bonate P. L., et al. Intracellular activation and deactivation of tasidotin, an analog of dolastatin 15: correlation with cytotoxicity. Molecular Pharmacology. 2009;75(1):218–226. doi: 10.1124/mol.108.051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ray A., Okouneva T., Manna T., et al. Mechanism of action of the microtubule-targeted antimitotic depsipeptide tasidotin (formerly ILX651) and its major metabolite tasidotin C-carboxylate. Cancer Research. 2007;67(8):3767–3776. doi: 10.1158/0008-5472.can-06-3065. [DOI] [PubMed] [Google Scholar]
- 123.Garg V., Zhang W., Gidwani P., Kim M., Kolb E. A. Preclinical analysis of tasidotin HCl in Ewing's sarcoma, rhabdomyosarcoma, synovial sarcoma, and osteosarcoma. Clinical Cancer Research. 2007;13(18, part 1):5446–5454. doi: 10.1158/1078-0432.ccr-06-2661. [DOI] [PubMed] [Google Scholar]
- 124.Ebbinghaus S., Rubin E., Hersh E., et al. A phase I study of the dolastatin-15 analogue tasidotin (ILX651) administered intravenously daily for 5 consecutive days every 3 weeks in patients with advanced solid tumors. Clinical Cancer Research. 2005;11(21):7807–7816. doi: 10.1158/1078-0432.CCR-05-0909. [DOI] [PubMed] [Google Scholar]
- 125.Cunningham C., Appleman L. J., Kirvan-Visovatti M., et al. Phase I and pharmacokinetic study of the dolastatin-15 analogue tasidotin (ILX651) administered intravenously on days 1, 3, and 5 every 3 weeks in patients with advanced solid tumors. Clinical Cancer Research. 2005;11(21):7825–7833. doi: 10.1158/1078-0432.ccr-05-0058. [DOI] [PubMed] [Google Scholar]
- 126.Mita A. C., Hammond L. A., Bonate P. L., et al. Phase I and pharmacokinetic study of tasidotin hydrochloride (ILX651), a third-generation dolastatin-15 analogue, administered weekly for 3 weeks every 28 days in patients with advanced solid tumors. Clinical Cancer Research. 2006;12(17):5207–5215. doi: 10.1158/1078-0432.ccr-06-0179. [DOI] [PubMed] [Google Scholar]
- 127.Hendriks H. R., Plowman J., Berger D. P., et al. Preclinical antitumour activity and animal toxicology studies of rhizoxin, a novel tubulin-interacting agent. Annals of Oncology. 1992;3(9):755–763. doi: 10.1093/oxfordjournals.annonc.a058334. [DOI] [PubMed] [Google Scholar]
- 128.Prota A. E., Bargsten K., Diaz J. F., et al. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(38):13817–13821. doi: 10.1073/pnas.1408124111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tsuruo T., Oh-hara T., Iida H., et al. Rhizoxin, a macrocyclic lactone antibiotic, as a new antitumor agent against human and murine tumor cells and the vincristine-resistant sublines. Cancer Research. 1986;46(1):381–385. [PubMed] [Google Scholar]
- 130.Bissett D., Graham M. A., Setanoians A., et al. Phase I and pharmacokinetic study of rhizoxin. Cancer Research. 1992;52(10):2894–2898. [PubMed] [Google Scholar]
- 131.Kerr D. J., Rustin G. J., Kaye S. B., et al. Phase II trials of rhizoxin in advanced ovarian, colorectal and renal cancer. British Journal of Cancer. 1995;72(5):1267–1269. doi: 10.1038/bjc.1995.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hanauske A. R., Catimel G., Aamdal S., et al. Phase II clinical trials with rhizoxin in breast cancer and melanoma. The EORTC early clinical trials group. British Journal of Cancer. 1996;73(3):397–399. doi: 10.1038/bjc.1996.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Verweij J., Wanders J., Gil T., et al. Phase II study of rhizoxin in squamous cell head and neck cancer. British Journal of Cancer. 1996;73(3):400–402. doi: 10.1038/bjc.1996.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaplan S., Hanauske A. R., Pavlidis N., et al. Single agent activity of rhizoxin in non-small-cell lung cancer: a phase II trial of the EORTC early clinical trials group. British Journal of Cancer. 1996;73(3):403–405. doi: 10.1038/bjc.1996.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McLeod H. L., Murray L. S., Wanders J., et al. Multicentre phase II pharmacological evaluation of rhizoxin. Eortc early clinical studies (ECSG)/pharmacology and molecular mechanisms (PAMM) groups. British Journal of Cancer. 1996;74(12):1944–1948. doi: 10.1038/bjc.1996.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tolcher A. W., Aylesworth C., Rizzo J., et al. A phase I study of rhizoxin (NSC 332598) by 72-hour continuous intravenous infusion in patients with advanced solid tumors. Annals of Oncology. 2000;11(3):333–338. doi: 10.1023/A:1008398725442. [DOI] [PubMed] [Google Scholar]
- 137.Bacher G., Nickel B., Emig P., et al. D-24851, a novel synthetic microtubule inhibitor, exerts curative antitumoral activity in vivo, shows efficacy toward multidrug-resistant tumor cells, and lacks neurotoxicity. Cancer Research. 2001;61(1):392–399. [PubMed] [Google Scholar]
- 138.Wienecke A., Bacher G. Indibulin, a novel microtubule inhibitor, discriminates between mature neuronal and nonneuronal tubulin. Cancer Research. 2009;69(1):171–177. doi: 10.1158/0008-5472.can-08-1342. [DOI] [PubMed] [Google Scholar]
- 139.Ito H., Kanzawa T., Kondo S., Kondo Y. Microtubule inhibitor D-24851 induces p53-independent apoptotic cell death in malignant glioma cells through Bcl-2 phosphorylation and Bax translocation. International Journal of Oncology. 2005;26(3):589–596. [PubMed] [Google Scholar]
- 140.Stokvis E., Nan-Offeringa L. G. A. H., Ouwehand M., et al. Quantitative analysis of D-24851, a novel anticancer agent, in human plasma and urine by liquid chromatography coupled with tandem mass spectrometry. Rapid Communications in Mass Spectrometry. 2004;18(13):1465–1471. doi: 10.1002/rcm.1493. [DOI] [PubMed] [Google Scholar]
- 141.Kuppens I. E. L. M., Witteveen P. O., Schot M., et al. Phase I dose-finding and pharmacokinetic trial of orally administered indibulin (D-24851) to patients with solid tumors. Investigational New Drugs. 2007;25(3):227–235. doi: 10.1007/s10637-006-9027-2. [DOI] [PubMed] [Google Scholar]
- 142.Huang T.-H., Chiu S.-J., Chiang P.-H., et al. Antiproliferative effects of N-heterocyclic indolyl glyoxylamide derivatives on human lung cancer cells. Anticancer Research. 2011;31(10):3407–3415. [PubMed] [Google Scholar]
- 143.Wong V. K. W., Chiu P., Chung S. S. M., et al. Pseudolaric acid B, a novel microtubule-destabilizing agent that circumvents multidrug resistance phenotype and exhibits antitumor activity in vivo . Clinical Cancer Research. 2005;11(16):6002–6011. doi: 10.1158/1078-0432.ccr-05-0209. [DOI] [PubMed] [Google Scholar]
- 144.Gong X., Wang M., Tashiro S.-I., Onodera S., Ikejima T. Involvement of JNK-initiated p53 accumulation and phosphorylation of p53 in pseudolaric acid B induced cell death. Experimental and Molecular Medicine. 2006;38(4):428–434. doi: 10.1038/emm.2006.50. [DOI] [PubMed] [Google Scholar]
- 145.Tan W.-F., Zhang X.-W., Li M.-H., et al. Pseudolarix acid B inhibits angiogenesis by antagonizing the vascular endothelial growth factor-mediated anti-apoptotic effect. European Journal of Pharmacology. 2004;499(3):219–228. doi: 10.1016/j.ejphar.2004.07.063. [DOI] [PubMed] [Google Scholar]
- 146.Li M.-H., Miao Z.-H., Tan W.-F., et al. Pseudolaric acid B inhibits angiogenesis and reduces hypoxia-inducible factor 1alpha by promoting proteasome-mediated degradation. Clinical Cancer Research. 2004;10(24):8266–8274. doi: 10.1158/1078-0432.ccr-04-0951. [DOI] [PubMed] [Google Scholar]
- 147.Tong Y.-G., Zhang X.-W., Geng M.-Y., et al. Pseudolarix acid B, a new tubulin-binding agent, inhibits angiogenesis by interacting with a novel binding site on tubulin. Molecular Pharmacology. 2006;69(4):1226–1233. doi: 10.1124/mol.105.020537. [DOI] [PubMed] [Google Scholar]
- 148.Sun Q., Li Y. The inhibitory effect of pseudolaric acid B on gastric cancer and multidrug resistance via Cox-2/PKC-α/P-gp pathway. PLoS ONE. 2014;9(9) doi: 10.1371/journal.pone.0107830.e107830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yu F., Li K., Chen S., Liu Y., Li Y. Pseudolaric acid B circumvents multidrug resistance phenotype in human gastric cancer SGC7901/ADR cells by downregulating Cox-2 and P-gp expression. Cell Biochemistry and Biophysics. 2015;71(1):119–126. doi: 10.1007/s12013-014-0170-7. [DOI] [PubMed] [Google Scholar]
- 150.Jung H. J., Shim J. S., Lee H. B., et al. Embellistatin, a microtubule polymerization inhibitor, inhibits angiogenesis both in vitro and in vivo. Biochemical and Biophysical Research Communications. 2007;353(2):376–380. doi: 10.1016/j.bbrc.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 151.Kiselyov A., Balakin K. V., Tkachenko S. E., Savchuk N., Ivachtchenko A. V. Recent progress in discovery and development of antimitotic agents. Anti-Cancer Agents in Medicinal Chemistry. 2007;7(2):189–208. doi: 10.2174/187152007780058650. [DOI] [PubMed] [Google Scholar]
- 152.de Ines C., Leynadier D., Barasoain I., et al. Inhibition of microtubules and cell cycle arrest by a new 1-deaza-7,8- dihydropteridine antitumor drug, CI 980, and by its chiral isomer, NSC 613863. Cancer Research. 1994;54(1):75–84. [PubMed] [Google Scholar]
- 153.Hook K. E., Przybranowski S. A., Leopold W. R. Cellular transport of CI-980. Investigational New Drugs. 1996;14(4):341–347. doi: 10.1007/BF00180809. [DOI] [PubMed] [Google Scholar]
- 154.Sklarin N. T., Lathia C. D., Benson L., et al. A phase I trial and pharmacokinetic evaluation of CI-980 in patients with advanced solid tumors. Investigational New Drugs. 1997;15(3):235–246. doi: 10.1023/a:1005854510468. [DOI] [PubMed] [Google Scholar]
- 155.Meyers C. A., Kudelka A. P., Conrad C. A., Gelke C. K., Grove W., Pazdur R. Neurotoxicity of CI-980, a novel mitotic inhibitor. Clinical Cancer Research. 1997;3(3):419–422. [PubMed] [Google Scholar]
- 156.Bernstein M. L., Baruchel S., Devine S., et al. Phase I and pharmacokinetic study of CI-980 in recurrent pediatric solid tumor cases: a Pediatric Oncology Group study. Journal of Pediatric Hematology/Oncology. 1999;21(6):494–500. doi: 10.1097/00043426-199911000-00010. [DOI] [PubMed] [Google Scholar]
- 157.Rowinsky E. K., Long G. S., Noe D. A., et al. Phase I and pharmacological study of CI-980, a novel synthetic antimicrotubule agent. Clinical Cancer Research. 1997;3(3):401–407. [PubMed] [Google Scholar]
- 158.Pazdur R., Meyers C., Diaz-Canton E., et al. Phase II trial of intravenous CI-980 (NSC 370147) in patients with metastatic colorectal carcinoma. Model for prospective evaluation of neurotoxicity. American Journal of Clinical Oncology: Cancer Clinical Trials. 1997;20(6):573–576. doi: 10.1097/00000421-199712000-00008. [DOI] [PubMed] [Google Scholar]
- 159.Thomas J. P., Moore T., Kraut E. H., Balcerzak S. P., Galloway S., Vandre D. D. A phase II study of CI-980 in previously untreated extensive small cell lung cancer: an Ohio State University phase II research consortium study. Cancer Investigation. 2002;20(2):192–198. doi: 10.1081/cnv-120001146. [DOI] [PubMed] [Google Scholar]
- 160.Kudelka A. P., Hasenburg A., Verschraegen C. F., et al. Phase II study of i.v. CI-980 in patients with advanced platinum refractory epithelial ovarian carcinoma. Anti-Cancer Drugs. 1998;9(5):405–409. doi: 10.1097/00001813-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 161.Shan B., Medina J. C., Santha E., et al. Selective, covalent modification of β-tubulin residue Cys-239 by T138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(10):5686–5691. doi: 10.1073/pnas.96.10.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rubenstein S. M., Baichwal V., Beckmann H., et al. Hydrophilic, pro-drug analogues of T138067 are efficacious in controlling tumor growth in vivo and show a decreased ability to cross the blood brain barrier. Journal of Medicinal Chemistry. 2001;44(22):3599–3605. doi: 10.1021/jm000478d. [DOI] [PubMed] [Google Scholar]
- 163.Donehower R. C., Schwartz G., Wolf A. C., et al. Phase I and pharmacokinetic study of T138067 administered as a weekly 3-hour infusion. Clinical Cancer Research. 2000;6:4578s–4579s. [Google Scholar]
- 164.Schwartz G., Rowinsky E. K., O'Dwyer P., et al. Phase I and pharmacokinetic study of T138067, a synthetic microtubule depolymerizing agent, administered as a 3-hour infusion daily x 5 every 3 weeks. Clinical Cancer Research. 2000;6:4579s–4579s. [Google Scholar]
- 165.Kirby S., Gertler S. Z., Mason W., et al. Phase 2 study of T138067-sodium in patients with malignant glioma: trial of the National Cancer Institute of Canada Clinical Trials Group. Neuro-Oncology. 2005;7(2):183–188. doi: 10.1215/s1152851704000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Berlin J. D., Venook A., Bergsland E., Rothenberg M., Lockhart A. C., Rosen L. Phase II trial of T138067, a novel microtubule inhibitor, in patients with metastatic, refractory colorectal carcinoma. Clinical Colorectal Cancer. 2008;7(1):44–47. doi: 10.3816/ccc.2008.n.006. [DOI] [PubMed] [Google Scholar]
- 167.Gelmon K. A., Belanger K., Soulieres D., et al. A phase I study of T900607 given once every 3 weeks in patients with advanced refractory cancers; National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG) IND 130. Investigational New Drugs. 2005;23(5):445–453. doi: 10.1007/s10637-005-2904-2. [DOI] [PubMed] [Google Scholar]
- 168.Yoshimatsu K., Yamaguchi A., Yoshino H., Koyanagi N., Kitoh K. Mechanism of action of E7010, an orally active sulfonamide antitumor agent: inhibition of mitosis by binding to the colchicine site of tubulin. Cancer Research. 1997;57(15):3208–3213. [PubMed] [Google Scholar]
- 169.Funahashi Y., Koyanagi N., Kitoh K. Effect of E7010 on liver metastasis and life span of syngeneic C57BL/6 mice bearing orthotopically transplanted murine Colon 38 tumor. Cancer Chemotherapy and Pharmacology. 2001;47(2):179–184. doi: 10.1007/s002800000199. [DOI] [PubMed] [Google Scholar]
- 170.Iwamoto Y., Nishio K., Fukumoto H., Yoshimatsu K., Yamakido M., Saijo N. Preferential binding of E7010 to murine β3-tubulin and decreased β3-tubulin in E7010-resistant cell lines. Japanese Journal of Cancer Research. 1998;89(9):954–962. doi: 10.1111/j.1349-7006.1998.tb00654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hayakawa K., Tashiro T., Sato K., et al. Collaborative work to evaluate toxicity on male reproductive organs by repeated dose studies in rats: 17) testicular toxicity of e7010, a sulfonamide tubulin polymerization inhibitor. Journal of Toxicological Sciences. 2000;25:173–178. doi: 10.2131/jts.25.specialissue_173. [DOI] [PubMed] [Google Scholar]
- 172.Nihei Y., Suzuki M., Okano A., et al. Evaluation of antivascular and antimitotic effects of tubulin binding agents in solid tumor therapy. Japanese Journal of Cancer Research. 1999;90(12):1387–1395. doi: 10.1111/j.1349-7006.1999.tb00724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yamamoto K., Noda K., Yoshimura A., Fukuoka M., Furuse K., Niitani H. Phase I study of E7010. Cancer Chemotherapy and Pharmacology. 1998;42(2):127–134. doi: 10.1007/s002800050795. [DOI] [PubMed] [Google Scholar]
- 174.Yee K. W. L., Hagey A., Verstovsek S., et al. Phase 1 study of ABT-751, a novel microtubule inhibitor, in patients with refractory hematologic malignancies. Clinical Cancer Research. 2005;11(18):6615–6624. doi: 10.1158/1078-0432.ccr-05-0650. [DOI] [PubMed] [Google Scholar]
- 175.Fox E., Maris J. M., Widemann B. C., et al. A phase I study of ABT-751, an orally bioavailable tubulin inhibitor, administered daily for 21 days every 28 days in pediatric patients with solid tumors. Clinical Cancer Research. 2008;14(4):1111–1115. doi: 10.1158/1078-0432.ccr-07-4097. [DOI] [PubMed] [Google Scholar]
- 176.Mauer A. M., Cohen E. E. W., Ma P. C., et al. A phase II study of ABT-751 in patients with advanced non-small cell lung cancer. Journal of Thoracic Oncology. 2008;3(6):631–636. doi: 10.1097/JTO.0b013e318174e01f. [DOI] [PubMed] [Google Scholar]
- 177.Michels J., Ellard S. L., Le L., et al. A phase IB study of ABT-751 in combination with docetaxel in patients with advanced castration-resistant prostate cancer. Annals of Oncology. 2010;21(2):305–311. doi: 10.1093/annonc/mdp311. [DOI] [PubMed] [Google Scholar]
- 178.Rudin C. M., Mauer A., Smakal M., et al. Phase I/II study of pemetrexed with or without ABT-751 in advanced or metastatic non-small-cell lung cancer. Journal of Clinical Oncology. 2011;29(8):1075–1082. doi: 10.1200/jco.2010.32.5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ma T., Fuld A. D., Rigas J. R., et al. A phase i trial and in vitro studies combining ABT-751 with carboplatin in previously treated non-small cell lung cancer patients. Chemotherapy. 2012;58(4):321–329. doi: 10.1159/000343165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wani M. C., Taylor H. L., Wall M. E., Coggon P., McPhail A. T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. Journal of the American Chemical Society. 1971;93(9):2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 181.Kavallaris M., Kuo D. Y.-S., Burkhart C. A., et al. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. The Journal of Clinical Investigation. 1997;100(5):1282–1293. doi: 10.1172/jci119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.McCarroll J. A., Gan P. P., Liu M., Kavallaris M. BetaIII-tubulin is a multifunctional protein involved in drug sensitivity and tumorigenesis in non-small cell lung cancer. Cancer Research. 2010;70(12):4995–5003. doi: 10.1158/0008-5472.can-09-4487. [DOI] [PubMed] [Google Scholar]
- 183.Gan P. P., McCarroll J. A., Po'Uha S. T., Kamath K., Jordan M. A., Kavallaris M. Microtubule dynamics, mitotic arrest, and apoptosis: drug-induced differential effects of βIII-tubulin. Molecular Cancer Therapeutics. 2010;9(5):1339–1348. doi: 10.1158/1535-7163.mct-09-0679. [DOI] [PubMed] [Google Scholar]
- 184.Parker A. L., Kavallaris M., McCarroll J. A. Microtubules and their role in cellular stress in cancer. Frontiers in Oncology. 2014;4, article 153 doi: 10.3389/fonc.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Raspaglio G., Filippetti F., Prislei S., et al. Hypoxia induces class III beta-tubulin gene expression by HIF-1α binding to its 3' flanking region. Gene. 2008;409(1-2):100–108. doi: 10.1016/j.gene.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 186.De Donato M., Mariani M., Petrella L., et al. Class III beta-tubulin and the cytoskeletal gateway for drug resistance in ovarian cancer. Journal of Cellular Physiology. 2012;227(3):1034–1041. doi: 10.1002/jcp.22813. [DOI] [PubMed] [Google Scholar]
- 187.Ojima I., Chakravarty S., Inoue T., et al. A common pharmacophore for cytotoxic natural products that stabilize microtubules. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(8):4256–4261. doi: 10.1073/pnas.96.8.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.St. George M., Ayoub A. T., Banerjee A., et al. Designing and testing of novel taxanes to probe the highly complex mechanisms by which taxanes bind to microtubules and cause cytotoxicity to cancer cells. PLoS ONE. 2015;10(6) doi: 10.1371/journal.pone.0129168.e0129168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Höfle G., Bedorf N., Steinmetz H., Schomburg D., Gerth K., Reichenbach H. Epothilone A and B—novel 16-membered macrolides with cytotoxic activity: isolation, crystal structure, and conformation in solution. Angewandte Chemie. 1996;35(13-14):1567–1569. doi: 10.1002/anie.199615671. [DOI] [Google Scholar]
- 190.Wartmann M., Altmann K.-H. The biology and medicinal chemistry of epothilones. Current Medicinal Chemistry: Anti-Cancer Agents. 2002;2(1):123–148. doi: 10.2174/1568011023354489. [DOI] [PubMed] [Google Scholar]
- 191.Altmann K.-H., Bold G., Caravatti G., Flörsheimer A., Guagnano V., Wartmann M. Synthesis and biological evaluation of highly potent analogues of epothilones B and D. Bioorganic and Medicinal Chemistry Letters. 2000;10(24):2765–2768. doi: 10.1016/s0960-894x(00)00555-2. [DOI] [PubMed] [Google Scholar]
- 192.Bollag D. M., McQueney P. A., Zhu J., et al. Epothilones, a new class of microtubule-stabilizing agents with a taxol- like mechanism of action. Cancer Research. 1995;55(11):2325–2333. [PubMed] [Google Scholar]
- 193.Yang C.-P. H., Verdier-Pinard P., Wang F., et al. A highly epothilone B-resistant A549 cell line with mutations in tubulin that confer drug dependence. Molecular Cancer Therapeutics. 2005;4(6):987–995. doi: 10.1158/1535-7163.mct-05-0024. [DOI] [PubMed] [Google Scholar]
- 194.Peterson J. K., Tucker C., Favours E., et al. In vivo evaluation of ixabepilone (BMS247550), a novel epothilone B derivative, against pediatric cancer models. Clinical Cancer Research. 2005;11(19) part 1:6950–6958. doi: 10.1158/1078-0432.ccr-05-0740. [DOI] [PubMed] [Google Scholar]
- 195.Brogdon C. F., Lee F. Y., Canetta R. M. Development of other microtubule-stabilizer families: the epothilones and their derivatives. Anticancer Drugs. 2014;25(5):599–609. doi: 10.1097/cad.0000000000000071. [DOI] [PubMed] [Google Scholar]
- 196.Larkin J. M. G., Kaye S. B. Epothilones in the treatment of cancer. Expert Opinion on Investigational Drugs. 2006;15(6):691–702. doi: 10.1517/13543784.15.6.691. [DOI] [PubMed] [Google Scholar]
- 197.De Geest K., Blessing J. A., Morris R. T., et al. Phase II clinical trial of ixabepilone in patients with recurrent or persistent platinum- and taxane-resistant ovarian or primary peritoneal cancer: a gynecologic oncology group study. Journal of Clinical Oncology. 2010;28(1):149–153. doi: 10.1200/jco.2009.24.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mooberry S. L., Tien G., Hernandez A. H., Plubrukarn A., Davidson B. S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Research. 1999;59(3):653–660. [PubMed] [Google Scholar]
- 199.Pryor D. E., O'Brate A., Bilcer G., et al. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry. 2002;41(29):9109–9115. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 200.Churchill C. D., Klobukowski M., Tuszynski J. A. The unique binding mode of laulimalide to two tubulin protofilaments. Chemical Biology & Drug Design. 2015;86(2):190–199. doi: 10.1111/cbdd.12475. [DOI] [PubMed] [Google Scholar]
- 201.Lu H., Murtagh J., Schwartz E. L. The microtubule binding drug laulimalide inhibits vascular endothelial growth factor-induced human endothelial cell migration and is synergistic when combined with docetaxel (taxotere) Molecular Pharmacology. 2006;69(4):1207–1215. doi: 10.1124/mol.105.019075. [DOI] [PubMed] [Google Scholar]
- 202.Wender P. A., Hilinski M. K., Soldermann N., Mooberry S. L. Total synthesis and biological evaluation of 11-desmethyllaulimalide, a highly potent simplified laulimalide analogue. Organic Letters. 2006;8(7):1507–1510. doi: 10.1021/ol060233g. [DOI] [PubMed] [Google Scholar]
- 203.Prota A. E., Bargsten K., Northcote P. T., et al. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angewandte Chemie. 2014;53(6):1621–1625. doi: 10.1002/anie.201307749. [DOI] [PubMed] [Google Scholar]
- 204.Madiraju C., Edler M. C., Hamel E., et al. Tubulin assembly, taxoid site binding, and cellular effects of the microtubule-stabilizing agent dictyostatin. Biochemistry. 2005;44(45):15053–15063. doi: 10.1021/bi050685l. [DOI] [PubMed] [Google Scholar]
- 205.Vollmer L. L., Jiménez M., Camarco D. P., et al. A simplified synthesis of novel dictyostatin analogues with in vitro activity against epothilone B-resistant cells and antiangiogenic activity in zebrafish embryos. Molecular Cancer Therapeutics. 2011;10(6):994–1006. doi: 10.1158/1535-7163.mct-10-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Brunden K. R., Gardner N. M., James M. J., et al. MT-stabilizer, dictyostatin, exhibits prolonged brain retention and activity: potential therapeutic implications. ACS Medicinal Chemistry Letters. 2013;4(9):886–889. doi: 10.1021/ml400233e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lindel T., Jensen P. R., Fenical W., et al. ChemInform abstract: eleutherobin, a new cytotoxin that mimics paclitaxel (taxol) by stabilizing microtubules. ChemInform. 2010;28(51) doi: 10.1002/chin.199751201. [DOI] [Google Scholar]
- 208.Long B. H., Carboni J. M., Wasserman A. J., et al. Eleutherobin, a novel cytotoxic agent that induces tubulin polymerization, is similar to paclitaxel (Taxol) Cancer Research. 1998;58(6):1111–1115. [PubMed] [Google Scholar]
- 209.Hamel E., Day B. W., Miller J. H., et al. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Molecular Pharmacology. 2006;70(5):1555–1564. doi: 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]
- 210.Nakao Y., Yoshida S., Matsunaga S., Fusetani N. (Z)-sarcodictyin A, a new highly cytotoxic diterpenoid from the soft coral Bellonella albiflora . Journal of Natural Products. 2003;66(4):524–527. doi: 10.1021/np0205452. [DOI] [PubMed] [Google Scholar]
- 211.Risinger A. L., Giles F. J., Mooberry S. L. Microtubule dynamics as a target in oncology. Cancer Treatment Reviews. 2009;35(3):255–261. doi: 10.1016/j.ctrv.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kaur R., Kaur G., Gill R. K., Soni R., Bariwal J. Recent developments in tubulin polymerization inhibitors: an overview. European Journal of Medicinal Chemistry. 2014;87:89–124. doi: 10.1002/cmdc.201400036. [DOI] [PubMed] [Google Scholar]
- 213.Liu Z., Xu P., Wu T., Zeng W. Microtubule-targeting anticancer agents from marine natural substance. Anti-Cancer Agents in Medicinal Chemistry. 2014;14(3):409–417. doi: 10.2174/187152061403140207163402. [DOI] [PubMed] [Google Scholar]
- 214.Cortes J., Baselga J. Targeting the microtubules in breast cancer beyond taxanes: the epothilones. Oncologist. 2007;12(3):271–280. doi: 10.1634/theoncologist.12-3-271. [DOI] [PubMed] [Google Scholar]
- 215.Vahdat L. Ixabepilone: a novel antineoplastic agent with low susceptibility to multiple tumor resistance mechanisms. Oncologist. 2008;13(3):214–221. doi: 10.1634/theoncologist.2007-0167. [DOI] [PubMed] [Google Scholar]