

The herpesviruses are characterized by their ability to undergo a productive infection upon infection of the host organism and then spread to establish a latent infection where they persist in the host. Later, under conditions of stress or other environmental stimuli, they reactivate to undergo a productive infection and spread to new host organisms. Herpes simplex virus (HSV), in particular, undergoes a productive infection in the mucosal epithelium and spreads to sensory neurons where it undergoes a latent infection. Reactivation leads to virus shedding but can also lead to recurrent disease. Much of the herpetic disease occurs following reactivation from latent infection, and all of the available antivirals target lytic infection. Thus, an understanding of the mechanisms of establishment and reactivation of HSV from latent infection is important for devising approaches for controlling HSV establishment, maintenance, and reactivation from latent infection. In epithelial cells, the HSV genome is initially silenced by host epigenetic mechanisms, but the virion protein 16 (VP16) and immediate early (IE) infected cell protein 0 reverse the heterochromatin modifications and promote euchromatin modifications on viral chromatin to promote lytic gene transcription, allowing lytic infection to proceed (1). In sensory neurons, VP16 cannot localize to the nucleus and/or assemble into nuclear complexes to activate IE gene expression, so the lytic cascade of gene expression is not initiated. Instead, a neuron-specific promoter in the viral genome drives the expression of the latency-associated transcript (LAT), which is processed into a stable lariat intron and microRNAs (miRNAs) (2). LAT is reported to enhance neuronal survival in several ways: (i) down-regulating lytic gene expression (3) by LAT sequences acting as a long noncoding RNA and promoting epigenetic silencing of lytic genes (4, 5); (ii) serving as a precursor for viral miRNAs that inhibit lytic gene expression (6); (iii) inhibiting apoptosis (7); and (iv) promoting of survival of infected neurons by nonapoptotic mechanisms (8). Thus, understanding of the mechanisms regulating the expression of LAT is critical for antiviral strategies targeting latent infection. In PNAS, Shu et al. (9) identify Activating Transcription Factor 3 (ATF3) as a host factor that induces expression of LAT in infected cells in culture and plays a role in maintaining latent infection in murine trigeminal ganglion sensory neurons.
A novel approach used by Roizman and colleagues in this and previous studies (10, 11) to examine the function of host factors in viral pathogenesis is to express wild type (WT) and dominant negative mutant host proteins from the viral genome to determine the effect of the factor on viral infection in vivo. In this study, they inserted WT and mutant ATF3 genes into the viral genome and studied these viruses in a murine reactivation model. A virus expressing WT ATF3 showed lower levels of reactivating lytic gene expression, consistent with ATF3 maintaining latent infection. In contrast, a virus expressing a dominant negative mutant form of ATF3 showed enhanced lytic gene expression during latent infection, consistent with the mutant ATF3 blocking the normal function of ATF3. They propose that HSV infection of neurons, or the stress induced by viral infection, induces the expression of ATF3, which increases the level of LAT transcription, which then would promote latent infection, by any or all of the mechanisms listed above (Fig. 1). The identification of a host factor that is induced by HSV infection and that is involved in LAT expression and maintaining latent infection is an important advance in our knowledge of the mechanisms of HSV latent infection.
Fig. 1.
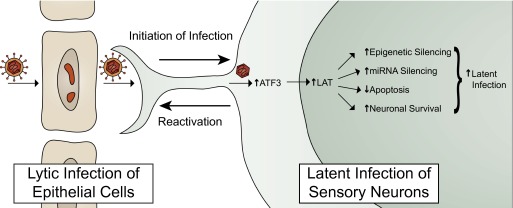
Model proposed by Shu et al. (9) for the role of ATF3 in HSV latent infection.
ATF3 is a transcription factor that is normally expressed at low levels in cells, but it is induced by a number of stimuli. It was originally considered to be a stress-induced transcription factor, but it is now considered to be a central part of a broader cellular adaptive response network to signals perturbing cellular homeostasis (12). It is a cAMP response element (CREB)-binding protein, and it can form homodimers and heterodimers with other CREB/ATF proteins. It can serve as a repressor in addition to its eponymous function on different promoters and in different cell types. There are several CREB sites on the HSV genome, including two in the LAT promoter (13, 14) named Cre1 and Cre2. Shu et al. (9) found that ATF3 can bind to both sites. Some mutational analyses of the viral genome have been conducted, but only the Cre1 site has been mutated. Therefore, the phenotypes might be expected to be only partial in nature. HSV-1 Cre1 mutant viruses show reduced reactivation in murine and rabbit ocular infection models but little change in LAT RNA levels (15–17).
It is important to consider the two different potential effects of ATF3 discussed in this paper. ATF3 could affect establishment and/or maintenance of latent infection, based on two observations in this paper. Shu et al. (9) show that HSV infection of cultured cells induces ATF3 RNA expression, which could then promote the induction of LAT. Expression of ATF3 from the virus does not affect lytic infection, presumably because the LAT promoter does not function in nonneuronal cells. However, Shu et al. also show that explant of latently infected sensory ganglia induces ATF3 RNA expression. Expression of ATF3 from the virus does reduce lytic gene expression due to reactivation, and a dominant negative ATF3 promotes lytic gene expression. Therefore, in neurons, ATF3 expression can inhibit lytic gene expression and promote maintenance of the latent infection. This, however, leaves us to explain the induction of ATF3 at the same time as reactivation is induced following explant. We do not know that ATF3 protein is induced, only that ATF3 transcripts are induced. Alternatively, the ATF3 that is induced during explant may be overridden by other signals inducing reactivation.
As with many publications that provide important new ideas, there are many new questions raised, and much remains to be done to substantiate this model. (i) Does ATF3 promote the expression of LAT in sensory neurons in vivo? Thus far, only lytic infection in cultured cells has been studied with the viruses expressing ATF3. (ii) Does ATF3 impact the level of establishment of latent infection? This study looks at the effect of mutant ATF3 molecules on reactivation but does not report on the level of latent infection. (iii) ATF3 is induced by explant of ganglia, but that does not normally prevent reactivation, if NGF is depleted. Therefore, the precise role of ATF3 in vivo remains to be determined. (iv) What HSV function(s) induce ATF3? ATF3 is induced by DNA damage repair and NF-κB, both of which are induced by HSV infection (2). Alternatively, HSV transactivators may induce expression of the cellular ATF3 gene; therefore, there are multiple possible ways by which HSV might induce ATF-3. (v) Is ATF3 induced during latent infection due to cellular mechanisms “sensing” the latent virus? The answers to these questions will greatly expand our understanding of the mechanisms of HSV latent infection.
All of the current herpes antiviral drugs target lytic infection functions; thus, the identification of a function that maintains latent infection provides an important target for possible intervention with a therapeutic. If the mechanism of induction of ATF3 were elucidated, this might provide a target for blocking induction of LAT and establishment of latent infection. For example, if it is due to NF-κB, numerous inhibitors of NF-κB exist (18). Furthermore, blocking induction of ATF3 during latent infection may result in reduced LAT expression and destabilization of latent infection. Alternatively, induction of ATF3 by NF-κB agonists could increase LAT during latent infection and promote maintenance of latent infection, thereby locking in latent infection, as others have proposed (19). Locking in latent infection may be safer for HSV than inducing reactivation, because reactivation from latent infection in the nervous system may lead to undesired disease outcomes. Indeed, this paper provides numerous possible future experiments on both the mechanisms of HSV latent infection and the approaches for treating latent infection.
Acknowledgments
Research in the authors’ laboratory on HSV latent infection is supported by National Institutes of Health Grant AI098681.
Footnotes
The authors declare no conflict of interest.
See companion article on page E5420.
References
- 1.Knipe DM, et al. Snapshots: Chromatin control of viral infection. Virology. 2013;435(1):141–156. doi: 10.1016/j.virol.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6th Ed. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 1823–1897. [Google Scholar]
- 3.Garber DA, Schaffer PA, Knipe DM. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol. 1997;71(8):5885–5893. doi: 10.1128/jvi.71.8.5885-5893.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q-Y, et al. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci USA. 2005;102(44):16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cliffe AR, Garber DA, Knipe DM. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009;83(16):8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Umbach JL, et al. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454(7205):780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perng GC, et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000;287(5457):1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 8.Thompson RL, Sawtell NM. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol. 2001;75(14):6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu M, Du T, Zhou G, Roizman B. Role of activating transcription factor 3 in the synthesis of latency-associated transcript and maintenance of herpes simplex virus 1 in latent state in ganglia. Proc Natl Acad Sci USA. 2015;112:E5420–E5426. doi: 10.1073/pnas.1515369112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu H, Roizman B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci USA. 2007;104(43):17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou G, Du T, Roizman B. HSV carrying WT REST establishes latency but reactivates only if the synthesis of REST is suppressed. Proc Natl Acad Sci USA. 2013;110(6):E498–E506. doi: 10.1073/pnas.1222497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hai T, Wolford CC, Chang YS. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. 2010;15(1):1–11. doi: 10.3727/105221610x12819686555015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leib DA, Nadeau KC, Rundle SA, Schaffer PA. The promoter of the latency-associated transcripts of herpes simplex virus type 1 contains a functional cAMP-response element: Role of the latency-associated transcripts and cAMP in reactivation of viral latency. Proc Natl Acad Sci USA. 1991;88(1):48–52. doi: 10.1073/pnas.88.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenny JJ, et al. Identification of a second ATF/CREB-like element in the herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) promoter. Virology. 1994;200(1):220–235. doi: 10.1006/viro.1994.1180. [DOI] [PubMed] [Google Scholar]
- 15.Rader KA, Ackland-Berglund CE, Miller JK, Pepose JS, Leib DA. In vivo characterization of site-directed mutations in the promoter of the herpes simplex virus type 1 latency-associated transcripts. J Gen Virol. 1993;74(Pt 9):1859–1869. doi: 10.1099/0022-1317-74-9-1859. [DOI] [PubMed] [Google Scholar]
- 16.Bloom DC, Stevens JG, Hill JM, Tran RK. Mutagenesis of a cAMP response element within the latency-associated transcript promoter of HSV-1 reduces adrenergic reactivation. Virology. 1997;236(1):202–207. doi: 10.1006/viro.1997.8723. [DOI] [PubMed] [Google Scholar]
- 17.Marquart ME, et al. A cAMP response element within the latency-associated transcript promoter of HSV-1 facilitates induced ocular reactivation in a mouse hyperthermia model. Virology. 2001;284(1):62–69. doi: 10.1006/viro.2001.0911. [DOI] [PubMed] [Google Scholar]
- 18.Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25(51):6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 19.Hill JM, et al. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci Transl Med. 2014;6(265):265ra169. doi: 10.1126/scitranslmed.3010643. [DOI] [PMC free article] [PubMed] [Google Scholar]