

Significance
Intensive rice breeding over the past 50 y has produced many high-performing cultivars, but our knowledge of the genomic changes associated with such improvement remains limited. By analyzing sequences of 1,479 rice accessions, this study identified genomic changes associated with breeding efforts, referred to as breeding signatures, involving 7.8% of the rice genome. Accumulation of selected regions is positively correlated with yield improvement. The number of selected regions in a line may be used for predicting agronomic potential, and the selected loci may provide useful targets for rice improvement.
Keywords: rice improvement, breeding signature, GWAS, resequencing
Abstract
Intensive rice breeding over the past 50 y has dramatically increased productivity especially in the indica subspecies, but our knowledge of the genomic changes associated with such improvement has been limited. In this study, we analyzed low-coverage sequencing data of 1,479 rice accessions from 73 countries, including landraces and modern cultivars. We identified two major subpopulations, indica I (IndI) and indica II (IndII), in the indica subspecies, which corresponded to the two putative heterotic groups resulting from independent breeding efforts. We detected 200 regions spanning 7.8% of the rice genome that had been differentially selected between IndI and IndII, and thus referred to as breeding signatures. These regions included large numbers of known functional genes and loci associated with important agronomic traits revealed by genome-wide association studies. Grain yield was positively correlated with the number of breeding signatures in a variety, suggesting that the number of breeding signatures in a line may be useful for predicting agronomic potential and the selected loci may provide targets for rice improvement.
Rice (Oryza sativa L.) is one of the most important cereal crops in the world. There have been landmark achievements in rice improvement over the past 50 y, especially in the indica subspecies. A major breakthrough resulted from the independent development of a series of semidwarf varieties in China and by the International Rice Research Institute (IRRI) in the 1950s and 1960s, leading to the “green revolution” in rice. Since then, semidwarfness has been a basic characteristic for almost all modern varieties. Based on semidwarf varieties, improvement for other traits, such as abiotic stress resistance, broad-spectrum resistances to biotic stresses, and better grain quality, has also been achieved. Another major breakthrough stemmed from the exploitation of hybrid vigor in China (1), resulting in the large-scale adoption of hybrid rice since the 1970s. Jointly, these breakthroughs have greatly increased rice productivity in the past several decades globally.
Genomic studies in recent years have identified a large number of loci that were under selection during rice domestication (2). However, there has been very limited study to identify loci or genomic regions that have been under selection due to breeding. Next-generation sequencing technologies have enabled sequencing of a large number of rice accessions at relatively low cost, providing opportunities to inspect the genomic regions selected in the history of crop improvement. Meanwhile, genome-wide association studies (GWAS) have provided an effective approach to analyze the genetic architecture of complex traits and allow identification of candidate genes for further improvement of agronomically important traits (3, 4).
In this study, we analyzed low-coverage sequencing data of 1,479 rice accessions, which revealed a large number of differentially selected regions associated with breeding efforts between two major subpopulations in indica. These selected regions are associated with agronomic performance of rice varieties and harbor many classes of known important genes. The results may have significant implications for rice improvement.
Results
Sequencing of Diverse Rice Varieties.
Data from two sets of Asian cultivated rice (O. sativa L.) germplasm consisting of 1,483 accessions, including both landraces and improved varieties from 73 countries, were analyzed in this study. The first set of 533 accessions was selected by us to represent both the genetic diversity in this species and their usefulness in rice improvement (Dataset S1). The sequence data were released in a previous study (5). The second set was the 950 accessions (Dataset S2) sequenced by Huang et al. (4) that were downloaded from the European Bioinformatics Institute (EBI) European Nucleotide Archive.
The details of SNP identification and imputation were described previously (5). Briefly, sequence reads were aligned to the rice reference genome [Nipponbare; Michigan State University (MSU), version 6.1]. After initial filtering, a total of 6,551,358 high-quality SNPs with the minor allele of each SNP shared by at least five accessions were identified. Three of the 533 accessions in the first set had excessive heterozygosity, and one had a low mapping rate; these accessions were excluded from further analysis. Sequences for the remaining 1,479 accessions were imputed using a linkage disequilibrium–k-nearest neighbor algorithm. After imputation, SNPs with missing data rates less than 20% of all of the accessions were selected, resulting in a total of 6,428,770 SNPs, with the overall missing data rate being about 0.38% (47.1% before imputation). We compared the imputed genotypes against relevant high-quality genome sequences in the database as well as our array-based genotypes, which showed that the accuracy of the imputed genotypes was >99% (SI Appendix, SI Result 1 and Table S1). SNPs and imputed genotypes can be queried on our website RiceVarMap (ricevarmap.ncpgr.cn) (6).
Genetic Structure and Diversity of the Rice Varieties.
The population structure of the 1,479 accessions was inferred using ADMIXTURE (7) (Methods). At K = 6, six distinct groups emerged (Fig. 1A): two indica groups referred to as indica I (IndI) and indica II (IndII), two japonica groups belonging to temperate japonica and tropical japonica, the Aus rice, and an intermediate group. The details of classification, values of subpopulation components, genome-wide distributions of sequence diversity, and patterns of linkage disequilibrium are given in SI Appendix, SI Result 2 and Datasets S1 and S2 and are displayed in SI Appendix, Figs. S1–S3.
Fig. 1.
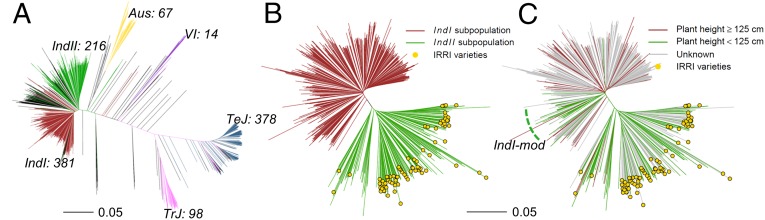
Genetic structure of the 1,479 rice accessions and substructure of indI and indII. (A) Neighbor-joining tree of 1,479 accessions constructed from the matching distance of 188,637 evenly distributed and randomly selected SNPs. The six different subpopulations, indica I (IndI), indica II (IndII), Aus, temperate japonica (TeJ), tropical japonica (TrJ), and an intermediate group (VI), are shown in different colors, and the numbers of accessions in each subpopulation are marked. [The tree reprinted from ref. 6, Copyright (2015), with permission from Oxford University Press.] (B and C) Neighbor-joining trees of accessions in IndI and IndII subpopulations. The gold circles indicate accessions developed by the IRRI. (B) IndI and IndII are colored red and green, respectively. (C) Distribution of the semidwarf trait in indica. A plant height of 125 cm in the 2011 rice-growing season in Wuhan, China was used as a threshold of semidwarfness, and accessions shorter than 125 cm were considered as modern varieties and are drawn in green. The dashed green line indicates a group of semidwarf varieties in IndI.
We subsequently focused our analysis on the 809 indica accessions, including 295 accessions sequenced by us and 514 from Huang et al. (4). It was found that 92.6% (353 of 381) of the accessions in the IndI group had germplasm of South China origin. Of the 386 Chinese indica landraces included in the study by Huang et al. (3), 71.0% (274 of 386) belonged to IndI and only nine belonged to IndII. Meanwhile, 95.1% (77 of 81) of the accessions from the IRRI were placed in the IndII group (Fig. 1B), and many accessions from Southeast Asia and some elite varieties bred in China were also placed in the IndII group. According to the pedigree information, almost all of the IndII accessions from Southeast Asia and China had parentage of IRRI varieties. In particular, many IndII accessions were restorer lines of widely used commercial three-line hybrids with wild-abortive cytoplasm (8). Another 212 indica accessions were classed as indica intermediate, 76.9% (163 of 212) of which were from China.
Because semidwarfness is the most obvious characteristic of modern cultivars, we used a cutoff plant height of 125 cm, measured in Wuhan, China in 2011, to classify the 294 indica accessions that were field-tested in this study (SI Appendix, Fig. S4). We observed that 95.2% (99 of 104) of the phenotyped IndII accessions were semidwarf, suggesting that this subpopulation mostly resulted from modern breeding programs. In contrast, only 36.7% (36 of 98) of the phenotyped IndI accessions were semidwarf. We divided IndI into two subgroups, IndI-land (landrace) and IndI-mod (cultivar), by placing the 36 semidwarf accessions in the IndI-mod group (Fig. 1C). The 36 IndI-mod accessions contained eight maintainer lines of commercial three-line hybrids, whereas the IndII group had many widely used restorer lines. These observations suggest that IndI-mod and IndII are consistent with the hypothetical two heterotic groups in the germplasms of indica rice: short-statured varieties of South China origin and medium-height lines of Southeast Asia origin, respectively (9).
We further explored the relevance between individual accessions and the allele frequency spectrum of IndI-mod and IndII groups, which may provide insights into the formations of IndI-mod and IndII groups. We inferred the “founder genotype (genome)” for each of the groups based on the assumption that the major allele of each SNP is more likely the allele of a hypothetical founder variety and the “founder genome” would be a combination of the major alleles of the SNPs across the genome. We then calculated the similarity between each indica accession and the inferred founder genomes of IndI-mod and IndII, respectively (SI Appendix, Figs. S5 and S6). In total, 3,217,614 SNPs with a minor allele frequency (MAF) of ≥0.05 in IndI, IndII, or all indica accessions were selected in this analysis. We found that the accession having the highest identity (93.9%; Z-score = 3.08, P = 0.002) with the inferred founder genome in the IndI-mod group was Aijiaonante, the first semidwarf variety in China released in 1956, which provided early semidwarf germplasm in China (10). The accession most similar to the inferred founder genome in the IndII group was IR 8 (89.4%; Z-score = 3.57, P = 0.0004), the first semidwarf variety released by the IRRI in 1966. Because the frequency of the major allele at each SNP was different, we also used a weighted scoring method in the analysis and obtained similar results (SI Appendix, SI Result 3). These results were consistent with the breeding history in rice that a lot of varieties were derived from a few widely cultivated elite varieties released in the early period of the green revolution (11), and the intensive use of limited breeding pools may have contributed to the recent emergence of IndI-mod and IndII.
We divided the rice genome into 10-kb segments and estimated the nucleotide diversity (π) in each segment based on the above population classification (SI Appendix, Fig. S2). The sequence diversities (π) of IndI and IndII were similar (0.0013 and 0.0016), indicating that modern breeding processes had not altered the overall genetic diversity significantly, which was similar to the situation in maize and wheat (12, 13). However, we observed that IndII had a far shorter linkage disequilibrium decay distance (78 kb) than IndI (142 kb) (SI Appendix, Fig. S3), indicating more recombination in IndII likely due to the efforts of intensive modern breeding. The details of nucleotide diversity and linkage disequilibrium in different subpopulations are described in SI Appendix, SI Result 2.
Differential Selection in the Two indica Groups.
To assess the extent of genetic differentiation between IndI and IndII, we used a cross-population likelihood method (XP-CLR) (14) to identify genomic regions differentially selected between the two groups (Fig. 2). Regions with the strongest 10th percentile of XP-CLR selection signals were considered. After filtering out regions around the centromeres or without SNPs with extreme differential allele frequency (Methods), 122 regions were identified as the most affected by selection in IndI in contrast to IndII (denoted as IndI-IndII) and 100 regions were identified from the reciprocal comparison (IndII-IndI). The selected regions of IndI-IndII and IndII-IndI contained 2,125 and 2,098 non–transposable element (TE)-related genes, respectively (Datasets S3 and S4), of which 70.9% also had strong positive selection evidence measured by the fixation index (FST), with highly differentiated alleles or long haplotype blocks (Methods). The selected regions of IndI-IndII had a mean size of 138 kb, covering ∼4.3% of the rice genome, whereas those selected regions of IndII-IndI covered 4.0% of the rice genome with a mean size of 157 kb. Moreover, 48.4% and 42.0% of the selected regions of IndI-IndII and IndII-IndI, respectively, contained no more than 10 genes; 14 selected regions each contained more than 50 genes; and the largest region encompassed 141 genes. Twenty selected regions of IndI-IndII overlapped with 16 selected regions of the IndII-IndI, containing 321 genes, which is greater than expected by chance [Fisher’s exact test (FET), odds ratio (OR) = 3.82; P = 8.1 × 10−74]. These results suggested that although different target genes were selected in different subpopulations, some of the targets were common to the different subpopulations. Overall selection in the two groups involved 200 regions that covered 7.8% of the rice genome. We compared the 200 selected regions with rice domestication-related regions and 58 quantitative trait loci (QTLs) for domestication traits identified in a previous study (2). The results showed that 21% of the selected regions overlapped with domestication-related regions and 11 QTLs for domestication traits matched the selected regions (Dataset S5), which included QTLs for seed- and panicle-related traits, indicating that a subset of domestication loci might have undergone additional selection for continued improvement of important agronomic traits.
Fig. 2.

Differential selection between subpopulations revealed by whole-genome screening of selection between IndI and IndII subpopulations using XP-CLR. IndII as the reference population and IndI as the object population (A) and IndI as the reference population and IndII as the object population (B) are shown. The XP-CLR scores from a genome-wide scan are plotted against the positions on the 12 chromosomes. The y-axis scores are limited to 200 to facilitate the display. Strong selection signals around SD1, Rf1, Xa4, and Xa26 are denoted.
Genes Under Selection.
Genes with a high XP-CLR score were regarded as candidate targets of selection, naturally and/or artificially, in the process of breeding and production (Datasets S3 and S4). We now present a few examples to show the actions and effects of selection. As expected, the locus of the well-known green revolution gene semi-dwarf1 (sd-1) was identified in the analysis. However, in contrast to the expectation that sd-1 was selected during modern breeding, we detected the sd-1 locus as being selected in IndI when using IndII (the semidwarf germplasms) as the reference population. We inspected haplotypes in the region and found that the frequency of a haplotype from a group of tall landraces was elevated in IndI with preferential geographic distribution, which was not observed in IndII (SI Appendix, Fig. S7E). However, another group of tall landraces in IndI showed similar haplotypes to IndII, agreeing with the report that the semidwarf genotype in indica mainly resulted from a deletion, which was not detected in the SNP analysis (15). The nucleotide diversities of both IndII and the selected haplotype of IndI around sd-1 were reduced dramatically (SI Appendix, Fig. S7 I–J). A previous study showed that the SD1 locus was also under selection during japonica rice domestication (16). These results suggested that this locus or the nearby region was independently under selection in different subpopulations.
Bacterial blight (BB) disease caused by Xanthomonus oryzea pv. oryzea (Xoo) is one of the most devastating diseases in rice production globally. Breeding for BB resistance has been one of the most important breeding objectives since the 1960s. A number of BB resistance genes (Xa3/Xa26, Xa4, Xa4b, Xa6, and Xa9) have been identified in the end of the long arm of chromosome 11, of which Xa4 is probably the most widely used BB resistance gene in rice breeding (17). A number of receptor-like kinase genes were found in this region that are arranged in tandem along the chromosome, and most of them showed strong selection signals in IndII (Fig. 3A). A receptor-like kinase near the cloned Xa3/Xa26 gene (18) had the highest XP-CLR score in the region. The not yet cloned Xa4 gene was also located within this region (18). In addition, the tandem receptor-like kinase genes may provide robust resistance to BB disease (19). We carried out GWAS for lesion length using a Xoo strain, PXO341 (Fig. 3B and SI Appendix, Fig. S8). The most significant lead SNP, sf1127718069 [linear mixed model (LMM): P = 1.9 × 10−16], was found within this region lying close to a receptor-like kinase gene (LOC_Os11g46980). This lead SNP had two alleles, G and T. The allele T was associated with higher resistance and existed almost exclusively in IndII (an allele frequency of 0.55 in IndII, whereas an allele frequency of only 0.01 in IndI), suggesting differential selection of this locus between the two indica groups.
Fig. 3.

Differential selection on chromosome 11 illustrated with the XP-CLR and GWAS results near the Xa4 and Xa26 regions (from 26.0 to 28.5 Mb on chromosome 11). (A) XP-CLR result by using IndI as the reference population and IndII as the object population. (B) Associations using the LMM on lesion length of a Xoo strain, PXO341. The blue point denotes the lead SNP sf1127718069 (P = 1.9 × 10−16). The colors of the other points represent the amounts of linkage disequilibrium for the lead SNP. Each arrow represents a receptor protein kinase gene. The vertical dashed line indicates the position of Xa26. Although Xa4 has not been cloned, a study has reported that it is tightly linked to Xa26 (18).
Cytoplasmic male sterility and nuclear fertility restorer (Rf) systems have facilitated the utilization of heterosis in rice. Consistent with the fact that most of restorer lines were in IndII, genomic regions near Rf1 (SI Appendix, Fig. S9) were strongly selected in IndII. This region contained a cluster of the pentatricopeptide repeat (PPR) gene family, including Rf1a/Rf5 and Rf1b, which were capable of restoring the pollen fertility disturbed by different male sterile cytoplasms (20). Because PPR genes perform important functions in posttranscriptional processes (21), this region may also provide other roles for plant performance. Interestingly, another fertility restoration-related gene, GRP162, was also strongly selected in IndII (20).
Increasing grain yield by optimizing yield component traits and plant architecture has been a major strategy for improving productivity in rice breeding. We observed that Gn1a (OsCKX2), encoding a cytokinin oxidase/dehydrogenase that regulates grain number by degrading bioactive cytokinin in inflorescence meristems (22), was strongly selected in IndI. LARGER PANICLE (LP), a gene that encodes a Kelch repeat-containing F-box protein (23), was also found to be significantly selected in IndI. In connection with the previous report that two allelic lp mutants showed an increased panicle size with more grains, it was suggested that LP might also be a very important target for improvement in rice breeding. One of the characteristics of modern cultivars is erect leaves. We found that OsBRI1, a gene-encoding a brassinosteroid receptor regulating plant height and leaf angle (24), was selected in both IndI and IndII. In addition, several genes encoding gibberellin 2-beta-dioxygenase (GA2ox3, GA2ox5, and GA2ox8); a GRAS family transcription factor gene, SLR1; a GA-stimulated transcript-related gene, OsGASR2; three glucosyltransferases genes; and several auxin-related genes (OsYUCCA1, COW1/YUCCA, and some carriers and responsive genes) were under strong selection. Referring to the similar results observed in maize population genomic studies (25), these results suggested very important roles of plant hormones during rice breeding.
Increased application of fertilizers, particularly nitrogen, enabled by the semidwarfness, is a key factor for the success of the green revolution. Both ammonium and nitrate are available forms of nitrogen for rice plants, and which one is the predominant form is dependent on soil conditions and fertilizer types (26). We observed that many genes involved in nitrogen assimilation were selected in either IndI or IndII (Fig. 4). OsAMT1;1, a gene encoding high-affinity ammonium transporter (27), was significantly selected in IndI, whereas three genes, OsNRT2.3, OsNAR2.2, and OsNiR1, either belonging to a high-affinity nitrate transporter family or encoding very important partner proteins or enzymes for nitrate uptake (26) were under strong selection in IndII. The two different selection patterns in nitrogen uptake by IndI and IndII may result from their different growth and cultivation conditions. In addition, we found that OsGS1;2 and OsGS1;3, which convert glutamate to glutamine, were significantly selected in IndII. We also observed many key genes involved in phosphate and potassium assimilation, such as OsSPX1, LOC_Os02g39750 (an inorganic phosphate transporter gene), OsPHO1;1, OsPHO1;2, OsK1.1, OsK2.1, OsK2.2, OsK4.1, OsK4.2, and OsHAK12, were under selection in IndI or IndII (Dataset S6).
Fig. 4.

Selected candidate genes involved in nitrogen assimilation. (A) Nitrogen assimilation pathway. The gene OsAMT1;1 strongly selected in IndI is indicated in red, and genes significantly selected in IndII (OsNRT2.3, OsNAR2.2, OsNiR1, OsGS1;2, and OsGS1;3) are indicated in blue. (B–G) XP-CLR values near the selected candidates. The candidates are indicated by the green arrows. Detailed information on selected candidates participating in nitrogen assimilation, phosphorus uptake and translocation, and potassium transporters can be found in Dataset S6.
In addition, we observed that genes reported to influence rice flowering time (heading date), such as OsLFL1, OsCOL4, OsMADS51, OsCRY2, PHYB, PHYA, and other five-CCT [CONSTANS (CO), CO-like, TOC1] domain family proteins, were in selected regions (Datasets S3 and S4), which is concordant with the fact that flowering time is an important trait in rice breeding. We also observed that small RNA loci and their targets might be selected during breeding (Datasets S3 and S4). We investigated further whether genes belonging to specific gene ontology categories were more likely to be selected, and the results showed that genes involved in hormone metabolic pathways were among the most significantly enriched listed in selected regions, suggesting their important roles during rice breeding (Dataset S7). More detailed information can be found in SI Appendix, SI Result 4 and SI Result 5.
Associations Between Grain Yield and Selected Haplotypes.
Yield improvement is the main objective in most crop breeding programs. Thus, a selected region resulting from the breeding process would be related to grain yield in one way or another. We obtained grain yield data of the 295 indica accessions from three field experiments in different years and locations (Methods and Dataset S8). We subtracted the mean value for each field experiment to normalize the data and then averaged the normalized data when multiple observations were available to obtain the normalized average grain yield. We carried out GWAS for the normalized average grain yield using both simple linear regression (LR) and the LMM (Fig. 5A and SI Appendix, Fig. S10). Seven loci were identified by the LMM using a significant threshold of 8.74 × 10−8 suggested by a previous study (5) on the same association panel. However, none of the seven loci was located within the selected regions, and all of them were isolated points, which were not in strong linkage disequilibrium with other SNPs in the local genomic regions (SI Appendix, Fig. S10). After checking the sequencing coverage of these loci, we found that six of them were located in copy number variation regions (SI Appendix, Fig. S11), suggesting possible false-positive results due to imputation errors. For LR, the most significant locus with a clear peak-like signal (sf0423098190) was located in a selected region (region ID 55 selected in IndII), and three of the 10 most significant loci or six of the 20 most significant loci were located in selected regions (SI Appendix, Figs. S12 and S13).
Fig. 5.

Correlation between yield and selected regions. (A) Genome-wide P values from association analysis of the normalized average yield of three field experiments using the simple LR model. SNPs located in the selected regions are shown in red. (B) Bean plot illustrating the distribution of BLUP breeding values of rice grain yield across accessions with different numbers of selected haplotypes (designated numDR, divided into groups by 20%, 40%, 60%, and 80% quantiles). Solid black bars denote the average for each group. (C) Bee swarm plot presenting the distribution of the difference of mean yield between Zhenshan 97 (ZS) and MH Minghui 63 (MH) genotypes marked as ΔYield (ZS-MH) from the RIL population across chromosome bins selected in ZS or MH. (Left) For the 96 bins only selected in MH, 27 bins show positive values of ΔYield (ZS-MH), whereas 69 bins show negative values. (Right) In the 75 bins only selected in ZS, 51 bins show positive values of ΔYield (ZS-MH), whereas only 24 bins show negative values. (D) Bee swarm plot presenting the distribution of ΔYield (ZS-MH) from the immortalized F2 population across chromosome bins selected in ZS or MH.
Grain yield is a complex trait composed of tillers per plant, grain number per panicle, and grain weight, each of which is controlled by many loci. Accordingly, there are numerous small-effect loci for grain yield, and most of them probably cannot exceed the significance threshold. We thus turned to analyze the genomic control factor λGC, which is defined as the ratio of the median of association test distribution to the expected value and is used to indicate the inflation of P values in GWAS due to population stratification. Higher values of λGC represent more significant (low P value) loci than expected (28). When using the LR method, we obtained a λGC of 3.30. However, when only the selected regions were considered, the λGC increased to 5.58. We randomly sampled 10,000 sets of genomic regions, each with the width and number the same as for the selected regions, and observed that none of the λGC values was greater than 5 (SI Appendix, Fig. S14). We also did the same analyses using the LMM method, which showed that the λGC values of the LMM of the whole genome and the selected regions were 0.93 and 1.15, respectively. Only 0.61% of 10,000 random samples gave a greater λGC value than the selected regions. These results indicated that the selected regions were enriched for loci for grain yield, although some loci did not reach the genome-wide significant threshold.
We further inspected the selected haplotypes in each accession. There were 2,161,733 SNPs polymorphic in indica (with a MAF greater than 0.05 in indica, IndI, or IndII) but nearly fixed in japonica (with a major allele frequency greater than 0.98 in japonica). We regarded the most frequent allele in japonica as the ancestral allele of each SNP and the other allele as the derived allele. Among them, ∼20.6% (446,593 SNPs, denoted as SNP set A) showed an allele frequency difference greater than 0.3 between IndI and IndII, of which 16.7% (74,724 SNPs, denoted as SNP set B) were located in the selected regions (7.8% of the rice genome) detected from the above analysis, suggesting that the selected regions were enriched for highly differentiated alleles between the two subpopulations (FET, OR = 3.63, P < 2.2 × 10−16; SI Appendix, Fig. S15). The derived allele of a set B SNP was regarded as selected. The number of derived alleles in set B SNPs per accession varied from 20,510 (27.4%) to 38,517 (51.5%). We grouped the derived alleles for the SNPs of each selected region to form a haplotype and calculated the number of selected regions with derived haplotypes in each accession [Methods, designated as the number of selected regions with derived haplotype in an accession (numDR)]. There are 65 (32.5%) to 134 (67%) of the 200 merged selected regions in a single accession with derived haplotypes (SI Appendix, Fig. S16).
We next calculated the Spearman’s rank correlation coefficients of yield with the number of SNPs with derived alleles within selected regions and numDR. For the 295 indica accessions we sequenced and phenotyped in field experiments, we found that the number of SNPs with derived alleles within the selected regions in an accession was significantly correlated with yield in all field experiments (Table 1), especially when calculated using the normalized average yield (ρ = 0.34, P = 1.6 × 10−9; SI Appendix, Fig. S17). In contrast, there was very little correlation between yield and the number of SNPs with derived alleles outside the selected regions (Table 1). We also observed a higher correlation (ρ = 0.38, P = 2.7 × 10−11) between numDR and the normalized average yield (SI Appendix, Fig. S17). More intriguingly, we found that the breeding values of accessions for yield obtained by ridge regression best linear unbiased prediction (BLUP) had a much higher correlation coefficient with numDR (ρ = 0.58, P = 6.8 × 10−28; Fig. 5B and SI Appendix, Fig. S17). Considering that the correlation of yield data was only 0.17 (P = 0.0032) between 2011 and 2012 in Wuhan and only 0.14 (P = 0.021) between 2012 in Lingshui and 2012 in Wuhan (Dataset S8), the high correlation between yield and the accumulation of derived haplotypes suggests that numDR could make an index to evaluate the potential of germplasms at the DNA sequence level.
Table 1.
Correlations between yield and selected regions
| Yield | N | RSNP-A | PSNP-A | RSNP-B | PSNP-B | RSNP-C | PSNP-C | RRegion | PRegion |
| Wuhan, 2011 | 289 | 0.085 | 0.15 | 0.26 | 5.5 × 10−6 | −0.020 | 0.74 | 0.26 | 9.1 × 10−6 |
| Wuhan, 2012 | 295 | 0.21 | 2.0 × 10−4 | 0.20 | 7.3 × 10−4 | 0.17 | 4.1 × 10−3 | 0.26 | 5.0 × 10−6 |
| Lingshui, 2012 | 283 | 0.19 | 1.6 × 10−3 | 0.24 | 6.0 × 10−5 | 0.13 | 3.1 × 10−2 | 0.24 | 4.2 × 10−5 |
| Normalized average | 295 | 0.23 | 5.1 × 10−5 | 0.34 | 1.3 × 10−9 | 0.12 | 3.4 × 10−2 | 0.38 | 2.7 × 10−11 |
| BLUP breeding value | 295 | 0.32 | 1.9 × 10−8 | 0.53 | 5.6 × 10−23 | 0.15 | 1.1 × 10−2 | 0.58 | 6.8 × 10−28 |
To obtain the normalized average for the yield data of three field experiments, we subtracted the mean value for each field experiment and averaged when multiple observations were available. BLUP breeding values for yield were obtained using ridge regression BLUP (49) from yield data of three field experiments. RSNP-A and PSNP-A were calculated based on 446,593 SNPs with an allele frequency difference (ΔDAF) greater than 0.3 between IndI and IndII; RSNP-B and PSNP-B were calculated based on 74,724 SNPs of SNP-A located in selected regions; RSNP-C and PSNP-C were calculated based on 312,497 SNPs of SNP-A departed from 100 kb of selected regions; and RRegion and PRegion were calculated using the number of selected regions in each accession containing derived haplotypes. N, number of phenotyped accessions; P, P value of Spearman's rank correlation test; R, Spearman’s rank correlation coefficient.
To obtain supporting evidence for the above-observed correlation between the selected haplotypes and grain yield, we examined such correlation in a recombinant inbred line (RIL) population constructed based on two indica varieties: Zhenshan 97, a member of IndI, and Minghui 63, belonging to IndII. The population had published phenotype data (29, 30) and was genotyped by resequencing (31, 32), which generated an ultra-high-density linkage map consisting of 1,619 chromosome bins. There were 84 and 102 regions with derived haplotypes, and thus considered to be selected in Zhenshan 97 (accession C145) and Minghui 63 (accession C147), respectively, based on XP-CLR analysis (Dataset S8). These selected regions were projected to 156 and 177 bins in the two varieties, respectively, of which 75 bins were only in Zhenshan 97 and 96 bins were only in Minghui 63. The number of bins with the selected genotypes was significantly correlated with grain yield in the RIL population (ρ = 0.30, P = 1.0 × 10−5, SI Appendix, Fig. S18 and Table S2); lines with more selected bins generally had a higher yield, and the selected genotype in a bin was more likely associated with a higher yield (Fig. 5C). We also observed similar results from an immortalized F2 population created by intercrossing the RILs (Fig. 5D and SI Appendix, Fig. S18 and Table S2).
Associations Between Other Traits and Selected Haplotypes.
We obtained additional data for morphological and metabolic traits from the 295 indica accessions we sequenced to evaluate a possible association with numDR.
A total of 840 metabolites were measured in our resequencing panel using the leaves of rice plants at the five-leaf stage (5). We calculated Spearman’s rank correlation coefficients between the metabolic traits and numDR. The 840 correlation coefficients showed an approximately normal distribution, with a mean value of 0.036 (SI Appendix, Fig. S19), and all were lower than the correlation coefficient between normalized average yield and numDR, suggesting that the metabolic traits as a class were not under selection during the rice breeding process. This distribution is in contrast to the high correlation between grain yield and numDR (ρ = 0.38, transformed to Z-score = 2.96, P = 3.1 × 10−3), further supporting that the selected regions contributed to yield improvement in breeding.
Using the recently developed high-throughput rice phenotyping facility for pot-grown plants (33), we obtained measurements for a range of morphological traits from the resequencing panel used in this study, including two newly defined traits, plant compactness and grain-projected area, that could not be scored by conventional means. A higher score of compactness indicates more compaction of the plant, and the grain-projected area is the pixel number of a 2D projected image of a grain. Although the normalized grain yield was not correlated with either of the two traits, plant compactness at the late booting stage was positively correlated with numDR (ρ = 0.27, P = 2.6 × 10−5), but grain-projected area was not (SI Appendix, Table S3). The results suggested that plant compactness is a useful trait for rice breeding and the association between numDR and a trait may reflect that the trait has some useful features in rice breeding that have been unrecognized.
Discussion
Resequencing of a large number of rice varieties provided opportunities to inspect the genetic and genomic changes reflecting the history of breeding, which we may consider as breeding signatures. In this study, we revealed various breeding signatures reflecting the complex genetic and genomic architecture from rice improvement, including the clear differentiation of two varietal groups in indica; identification of two most likely founder cultivars, Aijiaonante and IR8, in IndI-mod and IndII; and multiple targets of selection in IndI and IndII. Similar to a report on historical genomics in maize (12), our study suggests that the differentiation of IndI and IndII might be caused by geographic adaptation and accumulation of divergent selections in distinct breeding pools. However, the founder cultivars could be identified with high likelihood in our study, which is quite different from the report in maize (12). Furthermore, our study identified a number of candidate target selection regions in indica, which harbor thousands of uncharacterized genes with various putative biological functions, as well as those canonical genes associated with flowering time, plant architecture, disease resistance, nutrient assimilation, and yield components. The different target selection regions in IndI and IndII might reflect their adaption to local agricultural practices, such as breeders’ preference, local climates and ecological systems, and farming conditions. When comparing the selected regions with previously reported rice domestication regions and QTLs for domestication traits (2), we found that both domestication regions and our selected regions overlap with QTL intervals associated with yield-related traits, whereas QTLs for traits specific to domestication only match domestication regions. For example, the domestication regions encompass all five QTLs identified responsible for stigma exsertion and one QTL for seed shattering, but none of them resides in the selected regions identified here. These results suggest both similar and differential selection preference in rice domestication and subsequent breeding. The uncharacterized novel genes in the target selection regions would provide important entry points for future studies.
Our results may also have significant implications for predicting rice yield potential and hybrid performance to facilitate genomic selection. We found that higher yield and breeding values of yield are correlated with the accumulation of selected haplotypes in our resequencing panel, as well as in the RIL and immortalized F2 populations constructed based on Zhenshan 97 and Minghui 63, the parents of Shanyou 63. Shanyou 63 was the most widely cultivated hybrid in the late 1980s and 1990s in China. Although Zhenshan 97 and Minghui 63 had only moderate numbers of selected regions (84 and 102 regions, respectively; Dataset S8), the number of selected regions that the hybrid aggregates (139 regions) is greater than all of the RILs and varieties included in this study. Combining the genetic structure of indica accessions and XP-CLR results, we propose that the superior performance of the hybrid rice may have resulted from independent improvement of the two rice subgroups. Studies on the differentially selected target genes may shed light on the population genomic basis for hybrid vigor of rice and other species. Two parents with a higher number of different selected haplotypes may be more likely to have higher hybrid vigor. One may also expect that introgressing more selected haplotypes into an extant variety could improve its performance. Various genotyping platforms (34, 35) may be used to facilitate the application of these strategies.
Usually, one can identify beneficial haplotypes from GWAS based on genotype and phenotype data. However, for some complex traits showing strong interaction with environments, for example, adaptability, it is hard even to measure. There may be many such complex and important agronomic traits that have not yet been characterized. Our study demonstrates that identification of selected regions may provide an efficient way to find beneficial haplotypes without the need for extensive phenotyping. The number of selected haplotypes may serve as an indicator for evaluating the breeding potential of varieties to guide more efficient selection. A variety accumulating a whole complement of selected haplotypes might be an “ideotype” at the genomic level, which may be of both high-yielding and good adaptation to broad environmental conditions. However, considering the likely complexity of epistatic interactions between the selected loci and also the large numbers of genes in the selected regions (36, 37), the results obtained here should be regarded as the first step for revealing and possible utilization of selection signatures in breeding. The next step would be developing efficient statistical methods for genomic prediction (38).
Finally, it should be mentioned that the reference genome used here was from the japonica variety Nipponbare, which lacks all of the indica-specific genes and genome fragments. This factor may be limiting for the scope of the findings of this study.
Methods
Plant Materials and Sequencing Data.
The first set of 533 accessions was collected and sequenced by us as described previously (5). This set included 192 accessions from a core/minicore collection of O. sativa L. in China (39), 132 parental lines used in the International Rice Molecular Breeding Program (40), 148 accessions from a minicore subset of the US Department of Agriculture rice gene bank (41), 15 accessions used for SNP discovery in the OryzaSNP project (42), and 46 additional accessions from the Rice Germplasm Center at the IRRI. Information about the accessions, including names, countries of origin, geographical locations, and subpopulation classification, is listed in Dataset S1. The raw Illumina sequencing data could be downloaded from National Center for Biotechnology Information Sequence Read Archive under accession number PRJNA171289, which consisted of 6.7 billion 90-bp paired-end reads (more than 1 Gb for each accession).
Sequences of 950 accessions generated by Huang et al. (4) were downloaded from the EBI European Nucleotide Archive with accession numbers ERP000106 and ERP000729, which consisted of 4.6 billion 73-bp paired-end reads. The assembly release version 6.1 of genomic pseudomolecules of japonica cv. Nipponbare downloaded from the rice annotation database of MSU (rice.plantbiology.msu.edu/) was used as the reference genome.
Genetic Structure Analysis of the Population.
The model-based estimation of ancestry for the population was carried out using ADMIXTURE with default parameters (7) utilizing 188,637 evenly distributed SNPs. In SNP selection, we divided the genome into ∼3.8-kb regions; at most, two SNPs with a MAF ≥0.01 were randomly chosen for each region. The parameter of the number of ancient clusters K was set from two to seven to obtain different inferences. The inferred ancestry for each accession at K = 6 is given in Dataset S1. Each accession was classified based on its maximum subpopulation component. Accessions with the maximum subpopulation component value differing from the secondary value by less than 0.4 were classified as intermediate. The neighbor-joining tree was constructed using R package ape based on the simple matching distance of each pair of accessions calculated using the same random SNP set (43).
Screening of Differentially Selected Regions.
Whole-genome screening of selection was performed using XP-CLR, a method based on modeling the likelihood of multilocus allele frequency differentiation between two populations (14), following a previous study (25), with modifications. Genetic distances between SNPs were interpolated according to their physical distances in an ultra-high-density genetic map from a previous study (32). The program XP-CLR was run with parameters “-w1 0.0005 100 100 1 -p1 0.7” for each chromosome. After obtaining XP-CLR results, each chromosome was divided into 10-kb segments (approximates the average gene density of the rice genome) (44). An XP-CLR score was calculated for each 100 bp, and the average XP-CLR score was obtained for each 10-kb segment. Adjacent segments with an average of XP-CLR scores greater than the 80th percentile of the genome-wide average XP-CLR were then grouped as putatively selected regions. Putatively selected regions separated by no more than one low-score segment were merged. Each region was then given a score using the maximum of region-wise XP-CLR. Regions in the highest 10th percentile of these scores were considered as differentially selected regions. Regions less than 30 kb were filtered out because it is unlikely that such short regions could have been sorted out in such inbreeding populations, given the short history of modern breeding. Regions around the centromeres were also filtered out as suggested by a previous study (25). Eventually, regions with at least 10 SNPs with an allele frequency difference greater than 0.3 between IndI and IndII were considered as selected. For a segment with the annotated gene coordinates and an extension of flanking of 10 kb on each side, the selection signal for each gene was also quantified by four additional criteria: (i) the proportion of SNPs in the segment with Weir and Cockerham’s FST greater than the 90th percentile of all SNPs; (ii) the proportion of SNPs in the segment with an allele frequency difference between IndI and IndII greater than 0.3; (iii) the maximum absolute Rsb statistic value comparing IndI and IndII, which is an extended haplotype homozygosity-based test implemented in the R package rehh (45); and (iv) the permutation-based P value of XP-CLR. In general, genes with the top 20th percentile value were regarded as supported by these criteria.
Comparing Selected Regions and Domestication Features.
The rice domestication regions and QTLs for domestication traits were extracted from supplementary tables 9–12 of ref. 2. The genomic coordinates were transformed to MSU v6.1 by BLAST using border sequences with manual checks. To examine the overlap between QTLs for domestication traits and our selected regions, a 200-kb region flanking the peak position of a QTL (100-kb region on each side) was defined as the QTL region.
Inferring Derived Allele and Haplotype.
For SNPs with a MAF greater than 0.05 in indica, IndI, or IndII, and with a major allele frequency greater than 0.98 in japonica, we regarded the major allele in japonica as the ancestral allele and the other allele as the derived allele of each SNP. Among SNPs with determined derived alleles, only those SNPs showing an allele frequency difference greater than 0.3 between IndI and IndII were used to infer derived haplotypes. For each selected region, we identified the putatively advantageous haplotypes as those haplotypes carrying the most number of derived alleles and we regarded an accession as being one with a derived haplotype in this region only if this accession carried at least 10 SNPs with derived alleles and the number of SNPs with derived alleles was greater than half of the maximum number of SNPs with derived alleles in the 295 phenotyped indica accessions.
Phenotyping and Data Sources of Phenotype Data.
Field trials of yield-related traits were conducted in three environments. The rice seeds were sown in the Experimental Station of Huazhong Agricultural University, Wuhan, China in mid-May of 2011 and 2012 and, additionally, in the Experimental Station of Lingshui County of Hainan Island in mid-November of 2011. Seedlings about 25 d old were transplanted to the fields. The field planting followed a randomized complete block design with two replications. Each plot consisted of three rows with 10 plants each. The planting density was 16.5 cm between plants in a row, and the rows were 26 cm apart. Field management, including irrigation, fertilizer application, and pest control, followed essentially the normal agricultural practice. To prevent loss from overripening, each accession was harvested individually at its maturity. Five plants in the middle from the middle row of each accession were scored for the yield traits. Yield per plant was measured as the weight of all filled grains of the plant.
An additional field test was carried out in Wuhan in the summer of 2012 to evaluate BB resistance, in which the plants were inoculated with Philippine Xoo strain PXO341 (race 10) at the adult stage using the leaf-clipping method (18). Disease was scored by measuring the lesion length (centimeters) at 2 wk after inoculation.
To analyze associations between the grain yield and the selected haplotypes using an RIL population and an immortalized F2 population, the yield data were obtained from Xing et al. (29) and Hua et al. (30), and the genotype data were from Xie et al. (32).
To analyze associations between additional agronomic traits and metabolic traits and the selected haplotypes, metabolomics data were obtained from Chen et al. (5) and phenotype data of plant compactness and grain-projected area were obtained from Yang et al. (33).
Genome-Wide Association Analysis.
A total of 2,767,191 SNPs with a MAF ≥0.05 in all indica accessions evenly distributed in the genome were used to carry out GWAS. We performed GWAS using the LMM and the simple LR model provided by the FaST-LMM program (46). To control spurious associations, population structure was modeled as a random effect in the LMM using the K matrix. The SNP set used to analyze population structure was used to calculate K. The K coefficients were defined as the proportion of identical genotype for 188,165 SNPs (the same as those SNPs used to conduct genetic structure analysis) for each pair of individuals. The genome-wide significance threshold of the GWAS (8.74 × 10−8) was determined using Bonferroni correction based on the estimated effective number of independent SNPs in indica (47).
To examine the significance of overlap between the selected regions and the effective loci of GWAS for grain yield, a function called random.intervals in the R Bioconductor seqbias package (48) was used to generate random genomic intervals with the width and the number the same as in the selected regions.
Correlation Analysis.
Spearman’s rank correlation coefficient and the P value were calculated using the asymptotic t approximation by Fisher’s Z-transform implemented in the R function cor.test in the stats package.
Ridge Regression BLUP.
We obtained breeding values u of a specific trait with ridge regression BLUP using the function mixed.solve in the rrBLUP package (49) by fitting the following LMM:
where Y contains all observed yield data (length of 3n, where n is the sample size); X is a design matrix (3n × 3) for replicate effects; and Z is a design matrix (3n × n) for the breeding values u (length n). The covariance matrix (n × n) for u is the kinship matrix K used in GWAS.
Supplementary Material
Acknowledgments
We thank Prof. Shizhong Xu (University of California, Riverside) for help with statistical analysis. We also thank all the germplasm providers. This work was supported by the National High Technology Research and Development Program of China (Grant 2014AA10A602-3), the National Basic Research Program of China (2011CB100304), the transgenic project (2013ZX08009-001), the National Natural Science Foundation of China (Grant 31123009), and the Bill and Melinda Gates Foundation.
Footnotes
The authors declare no conflict of interest.
1W.X. and G.W. contributed equally to this work.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1515919112/-/DCSupplemental.
References
- 1.Yuan L, Yang Z, Yang J. 1994. Hybrid rice research in China. Hybrid Rice Technology: New Developments and Future Prospects (International Rice Research Institute, Philippines), pp 143–147.
- 2.Huang X, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490(7421):497–501. doi: 10.1038/nature11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang X, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 4.Huang X, et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet. 2012;44(1):32–39. doi: 10.1038/ng.1018. [DOI] [PubMed] [Google Scholar]
- 5.Chen W, et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet. 2014;46(7):714–721. doi: 10.1038/ng.3007. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, et al. RiceVarMap: A comprehensive database of rice genomic variations. Nucleic Acids Res. 2015;43(Database issue):D1018–D1022. doi: 10.1093/nar/gku894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Virmani SS. Heterosis and Hybrid Rice Breeding. Springer; Berlin: 1994. [Google Scholar]
- 9.Zhang Q, et al. A diallel analysis of heterosis in elite hybrid rice based on RFLPs and microsatellites. Theor Appl Genet. 1994;89(2-3):185–192. doi: 10.1007/BF00225139. [DOI] [PubMed] [Google Scholar]
- 10.Lin S, Min S. 1991. Rice Varieties and Their Genealogy in China (Shanghai Science and Technology Press, Shanghai, China). Chinese.
- 11.Shen J. Rice Improvement in China and Other Asian Countries. International Rice Research Institute; Philippines: 1980. Rice breeding in China; pp. 9–30. [Google Scholar]
- 12.van Heerwaarden J, Hufford MB, Ross-Ibarra J. Historical genomics of North American maize. Proc Natl Acad Sci USA. 2012;109(31):12420–12425. doi: 10.1073/pnas.1209275109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cavanagh CR, et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci USA. 2013;110(20):8057–8062. doi: 10.1073/pnas.1217133110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H, Patterson N, Reich D. Population differentiation as a test for selective sweeps. Genome Res. 2010;20(3):393–402. doi: 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spielmeyer W, Ellis MH, Chandler PM. Semidwarf (sd-1), “green revolution” rice, contains a defective gibberellin 20-oxidase gene. Proc Natl Acad Sci USA. 2002;99(13):9043–9048. doi: 10.1073/pnas.132266399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asano K, et al. Artificial selection for a green revolution gene during japonica rice domestication. Proc Natl Acad Sci USA. 2011;108(27):11034–11039. doi: 10.1073/pnas.1019490108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khush G, Mackill D, Sidhu G. 1989. Breeding rice for resistance to bacterial blight. Bacterial Blight of Rice (International Rice Research Institute, Philippines), pp 207–217.
- 18.Sun X, et al. Xa26, a gene conferring resistance to Xanthomonas oryzae pv. oryzae in rice, encodes an LRR receptor kinase-like protein. Plant J. 2004;37(4):517–527. doi: 10.1046/j.1365-313x.2003.01976.x. [DOI] [PubMed] [Google Scholar]
- 19.Li HJ, Li XH, Xiao JH, Wing RA, Wang SP. Ortholog alleles at Xa3/Xa26 locus confer conserved race-specific resistance against Xanthomonas oryzae in rice. Mol Plant. 2012;5(1):281–290. doi: 10.1093/mp/ssr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu J, et al. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell. 2012;24(1):109–122. doi: 10.1105/tpc.111.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitz-Linneweber C, Small I. Pentatricopeptide repeat proteins: A socket set for organelle gene expression. Trends Plant Sci. 2008;13(12):663–670. doi: 10.1016/j.tplants.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Ashikari M, et al. Cytokinin oxidase regulates rice grain production. Science. 2005;309(5735):741–745. doi: 10.1126/science.1113373. [DOI] [PubMed] [Google Scholar]
- 23.Li M, et al. Mutations in the F-box gene LARGER PANICLE improve the panicle architecture and enhance the grain yield in rice. Plant Biotechnol J. 2011;9(9):1002–1013. doi: 10.1111/j.1467-7652.2011.00610.x. [DOI] [PubMed] [Google Scholar]
- 24.Yamamuro C, et al. Loss of function of a rice brassinosteroid insensitive1 homolog prevents internode elongation and bending of the lamina joint. Plant Cell. 2000;12(9):1591–1606. doi: 10.1105/tpc.12.9.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hufford MB, et al. Comparative population genomics of maize domestication and improvement. Nat Genet. 2012;44(7):808–811. doi: 10.1038/ng.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu G, Fan X, Miller AJ. Plant nitrogen assimilation and use efficiency. Annu Rev Plant Biol. 2012;63(63):153–182. doi: 10.1146/annurev-arplant-042811-105532. [DOI] [PubMed] [Google Scholar]
- 27.Hoque MS, Masle J, Udvardi MK, Ryan PR, Upadhyaya NM. Over-expression of the rice OsAMT1-1 gene increases ammonium uptake and content, but impairs growth and development of plants under high ammonium nutrition. Funct Plant Biol. 2006;33(2):153–163. doi: 10.1071/FP05165. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, et al. GIANT Consortium Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19(7):807–812. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing Z, et al. Characterization of the main effects, epistatic effects and their environmental interactions of QTLs on the genetic basis of yield traits in rice. Theor Appl Genet. 2002;105(2-3):248–257. doi: 10.1007/s00122-002-0952-y. [DOI] [PubMed] [Google Scholar]
- 30.Hua J, et al. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA. 2003;100(5):2574–2579. doi: 10.1073/pnas.0437907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, et al. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One. 2011;6(3):e17595. doi: 10.1371/journal.pone.0017595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie W, et al. Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc Natl Acad Sci USA. 2010;107(23):10578–10583. doi: 10.1073/pnas.1005931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang W, et al. Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat Commun. 2014;5:5087. doi: 10.1038/ncomms6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, et al. A high-density SNP genotyping array for rice biology and molecular breeding. Mol Plant. 2014;7(3):541–553. doi: 10.1093/mp/sst135. [DOI] [PubMed] [Google Scholar]
- 35.Yu H, Xie W, Li J, Zhou F, Zhang Q. A whole-genome SNP array (RICE6K) for genomic breeding in rice. Plant Biotechnol J. 2014;12(1):28–37. doi: 10.1111/pbi.12113. [DOI] [PubMed] [Google Scholar]
- 36.Yu SB, et al. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA. 1997;94(17):9226–9231. doi: 10.1073/pnas.94.17.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou G, et al. Genetic composition of yield heterosis in an elite rice hybrid. Proc Natl Acad Sci USA. 2012;109(39):15847–15852. doi: 10.1073/pnas.1214141109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu S, Zhu D, Zhang Q. Predicting hybrid performance in rice using genomic best linear unbiased prediction. Proc Natl Acad Sci USA. 2014;111(34):12456–12461. doi: 10.1073/pnas.1413750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, et al. A core collection and mini core collection of Oryza sativa L. in China. Theor Appl Genet. 2011;122(1):49–61. doi: 10.1007/s00122-010-1421-7. [DOI] [PubMed] [Google Scholar]
- 40.Yu SB, et al. Molecular diversity and multilocus organization of the parental lines used in the International Rice Molecular Breeding Program. Theor Appl Genet. 2003;108(1):131–140. doi: 10.1007/s00122-003-1400-3. [DOI] [PubMed] [Google Scholar]
- 41.Agrama H, et al. Genetic assessment of a mini-core subset developed from the USDA rice genebank. Crop Sci. 2009;49(4):1336–1346. [Google Scholar]
- 42.McNally KL, et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc Natl Acad Sci USA. 2009;106(30):12273–12278. doi: 10.1073/pnas.0900992106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 44.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436(7052):793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 45.Tang K, Thornton KR, Stoneking M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007;5(7):e171. doi: 10.1371/journal.pbio.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lippert C, et al. FaST linear mixed models for genome-wide association studies. Nat Methods. 2011;8(10):833–835. doi: 10.1038/nmeth.1681. [DOI] [PubMed] [Google Scholar]
- 47.Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131(5):747–756. doi: 10.1007/s00439-011-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones DC, Ruzzo WL, Peng X, Katze MG. A new approach to bias correction in RNA-Seq. Bioinformatics. 2012;28(7):921–928. doi: 10.1093/bioinformatics/bts055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Endelman JB. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome. 2011;4(3):250–255. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.