

Significance
Nucleotides are biosynthesized from a ribose pyrophosphate species, with enzymes and magnesium ions as catalysts. Magnesium ions alone can perform this process with synthetic ribose pyrophosphates, by binding to the pyrophosphate groups and linking cyclic nitrogen bases to the ribose. In a kinase mimic ATP transfers the terminal phosphate to receptors catalyzed by magnesium ions, binding to pyrophosphate groups, but magnesium ion at low concentrations inhibits the process, and only at higher concentrations is it a catalyst. This affords great insight into the mechanism of ATP phosphorylation, a fundamental biological process. Our enzyme mimics could well operate on prebiotic earth before enzymes were present. This work strengthens the picture of how important components of life could have been formed prebiotically.
Keywords: ATP, ADP, phosphoribosyl pyrophosphate, nucleosides
Abstract
Derivatives of ribosyl pyrophosphate have been synthesized, and examined with magnesium salts in the coupling of the ribose unit to various nucleophiles, including pyrazole and 2-chloroimidazole. Only with the magnesium salt present did they generate the ribosyl cation by binding to the leaving group and then couple the ribose derivative with nucleophiles. The role of magnesium salts in phosphorylation of methanol by ATP was also examined. Here a remarkable effect was seen: phosphorylation by ATP was slowed with low concentrations of Mg2+ but accelerated by higher concentrations. Related effects were also seen in the effect of Mg2+ on phosphorylation by ADP. The likely mechanisms explain these effects.
Enzyme mimics, lacking protein catalysts, can afford insight into biochemical mechanisms because frequently the essential chemical paths are similar, with the enzyme protein furnishing greater velocity and selectivity. A good example from our own work includes the mechanisms used when thiamine pyrophosphate is the coenzyme; thiamine itself can perform the essential catalysis, but more slowly and less selectively than with the enzyme (1, 2). As another example, our mimics of ribonuclease use the imidazole groups that are biochemically part of the enzyme itself to catalyze cleavage of RNA (3, 4). We here describe mimics of the enzymes that attach base groups to a ribose unit. We also describe mimics of kinases that transfer a phosphate group from ATP.
The synthesis of nucleotides in modern biochemistry involves 5-phosphoribosyl pyrophosphate, formed by direct transfer of the pyrophosphoryl group from ATP. This attaches to a nitrogenous base to generate a new carbon nitrogen bond by the displacement of the pyrophosphate group with assistance by Mg2+ (Fig. 1). For purine nucleotides the nucleophile is an ammonia molecule that is generated by cleaving an amide. This generates ribosylamine-5-phosphate, and the rest of the base is constructed around this amino group. For the synthesis of pyrimidine nucleotides the nucleophile is orotic acid, which later undergoes decarboxylation and other steps to generate the modern nucleotides. It seemed likely that binding to magnesium ions activates the departure of the pyrophosphate leaving group. Simple binding of pyrophosphate ions to Mg2+ is well studied (5).
Fig. 1.
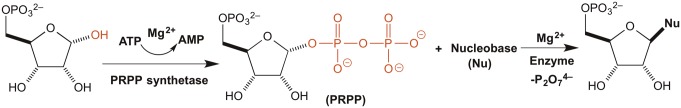
Simple pathway for nucleotide synthesis. In modern biochemistry the immediately bound nucleophile is further modified.
Similarly a pyrophosphate group (ADP) is the leaving group in ATP phosphorylations by ATP in kinase reactions (6). Some studies have been reported on mimics for this process, and it seemed likely that in this case also the departure of the ADP leaving group would be assisted by binding to magnesium ions (7). However, we now find that a more complex mechanism is involved in the mimic, and thus likely in the prebiotic world and in the modern enzymatic process itself. Of course the exact structures now used in biochemistry may almost certainly have evolved from much simpler structures on prebiotic earth, but the same overall general chemistry is likely.
The prebiotic synthesis of nucleotides can involve the direct reaction of nucleobases with ribose, but in quite low yield (8). Another proposed route involves an arabinose amino-oxazoline intermediate that later inverts to ribonucleotide (9). Because the phosphate esters and anhydrides dominate the living world (10), and currently nucleotides are biosynthetically synthesized from phosphoribosyl pyrophosphate, we believe that nucleotides could have formed prebiotically by the reaction of ribose 1-pyrophosphate (RPP) or possibly the 5-phosphate derivative with the corresponding nucleobases in the presence of Mg2+ ions. d-Ribose can be available in the prebiotic world, and there are many credible ways in which phosphorylations could occur, with three phosphorylations leading to 5-phosphoribosyl pyrophosphate. We have examined such a nucleotide synthesis catalyzed by Mg2+. This is similar to the current biosynthetic pathways, but without the enzymes.
We synthesized ribose with protecting groups on the 2-, 3-, and 5-hydroxyl groups, so only the hydroxyl group on position 1 was available, and then synthesized protected ribose 1-phosphate (RP) and protected RPP as model compounds. To our knowledge, there were no previous reports of the synthesis of RPP derivatives nonenzymatically. For simple analogs we synthesized 2,3,5-tribenzoylribose 1-pyrophosphate and 2,3,5-Tris-phenylpropylribose 1-pyrophosphate.
Synthesis of 2,3,5-Tribenzoylribose 1-Phosphate 5
1-O-Acetyl-2,3,5-Tris-O-benzoyl-β-d-ribofuranose 1, on treatment with HBr-AcOH (11), afforded the corresponding ribosylbromide 2, which on reaction with dibenzyl phosphate 3 formed 2,3,5-trisbenzoylribose dibenzylphosphate 4 (12). Compound 4 on reduction with Pd-C afforded the title compound 5 (Fig. 2).
Fig. 2.

Synthesis of 2,3,5-tribenzoylribose 1-phosphate 5.
Dibenzylphosphonate 6, on reaction with N-chlorosuccinimide, afforded dibenzylphosphoryl chloride 7 (13), which on reaction with previously synthesized ribosyl phosphate 5 afforded the corresponding ribosyl dibenzylpyrophosphate derivative 8. Compound 8 on reduction with Pd-C and triethylamine afforded the title compound 9 (Fig. 3).
Fig. 3.

Synthesis of 2,3,5-tribenzoylribose 1-pyrophosphate 9.
We examined the reaction of RP 5 and RPP 9 with and without magnesium ions in methanol (used as solvent, Fig. 4). We found that 1-methylriboside 10 was formed from ribosyl pyrophosphate with MgCl2 in almost 50% yield in 48 h at room temperature with no significant increase after longer times. No methylriboside was obtained in the absence of magnesium ions (Table 1, entry 3 and 4). Under our conditions no methylriboside was obtained with RP both with and without magnesium ions (Table 1, entry 1 and 2).
Fig. 4.
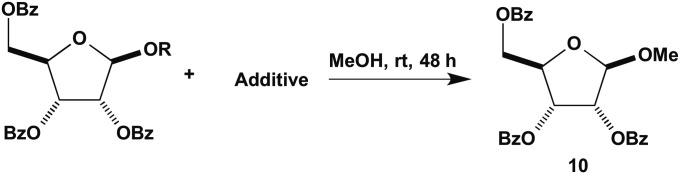
Reaction of RP and RPP with methanol.
Table 1.
Reaction of RP and RPP in methanol with and without MgCl2
| Entry | Starting material | Additive | Yield, % |
| 1 | (OBz)3 RP (5) | No additive | No reaction |
| 2 | (OBz)3 RP (5) | MgCl2 | Trace |
| 3 | (OBz)3 RPP (9) | No additive | No reaction |
| 4 | (OBz)3 RPP (9) | MgCl2 | 50 |
Compound 5 or 9 (1 equiv, 0.05 M) and MgCl2 (2 equiv, 0.1 M) for entry 2 and 4.
We also examined the formation of a C–N bond of a cyclic nitrogen compound to the ribosyl pyrophosphate, but because the benzoyl protecting groups reacted in the model study we synthesized 2,3,5-Tris-phenylpropyl substituted RPP. Accordingly d-ribose 11, on treatment with methanol and catalytic conc. sulfuric acid furnished the methyl ribofuranoside 12 (14), which on treatment with sodium hydride and phenylpropyl bromide 13 furnished 2,3,5-trisprotected methylribofuranoside, 14 (15). Compound 14 on treatment with HBr-AcOH (16) gave the corresponding ribosylbromide 15, which on reaction with dibenzyl phosphate 3 gave ribose-dibenzylphosphate 16. Compound 16 on reduction with Pd–C gave protected RP 17, which on reaction with dibenzylphosphoryl chloride 7 yielded the corresponding ribosyl dibenzylpyrophosphate derivative 18. Compound 18 on reduction with Pd–C produced the title compound 19 (Fig. 5).
Fig. 5.

Synthesis of phenylpropyl substituted RPP 19 and its reaction with methanol using Mg2+ catalyst.
We found that compound 19 (1 equiv, 0.05 M), MgI2 (2 equiv, 0.1 M) in methanol at room temperature for 48 h afforded 78% yield of methyl ribofuranoside 14 in the presence of MgI2 as additive, whereas there was no reaction in the absence of MgI2 (used also with other nucleophiles as more soluble than MgCl2).
We examined the reaction with two amine nucleophiles, pyrazole and 2-chloroimidazole, in dimethylformamide, the best solvent for all of the reactants (Fig. 6). The reaction of pyrazole (2 equiv, 0.2 M) and magnesium iodide (4 equiv, 0.4 M) with ribosyl pyrophosphate 19 (1 equiv, 0.1 M) at 60 °C for 40 h gave a 30% yield of ribose-pyrazole derivative 20. The reaction gave no desired product in the absence of magnesium iodide. Similarly, the reaction of 2-chloroimidazole (8 equiv, 0.8 M) and magnesium iodide (4 equiv, 0.4 M) with ribosyl pyrophosphate 19 (1 equiv, 0.1 M) at 60 °C for 40 h afforded 48% yield of the ribose-imidazole derivative 21. Only a trace of compound 21 was obtained in the absence of magnesium iodide (Table 2). We found that the relatively weak base pyrazole and 2-chloroimidazole added to the ribosyl unit but the more basic imidazole was not incorporated, nor was simple ammonia. Apparently the basic imidazole or ammonia binds to the magnesium ions, blocking catalysis, and with an excess of magnesium the imidazole or ammonia itself is bound and thus not a nucleophile.
Fig. 6.
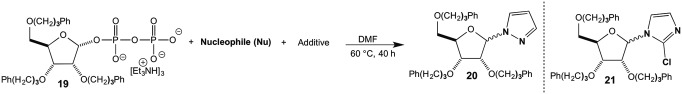
Reaction of RPP 19 with pyrazole and with 2-chloroimidazole.
Table 2.
Reaction of RPP 19 with pyrazole and with 2-chloroimidazole nucleophiles in the presence and absence of magnesium iodide
| Entry | Nucleophiles | Additive | Product | Yield, % |
| 1* | Pyrazole | No additive | 20 | — |
| 2* | Pyrazole | MgI2 | 20 | 30 |
| 3† | 2-Chloroimidazole | No additive | 21 | Trace |
| 4† | 2-Chloroimidazole | MgI2 | 21 | 48 |
Compound 19 (0.1 M), MgI2 (0.4 M), pyrazole (0.2 M), DMF (solvent).
Compound 19 (0.1 M), MgI2 (0.4 M), 2-chloro imidazole (0.8 M), DMF (solvent).
We also examined the possible role of Mg2+ in phosphorylations by ATP (adenosine triphosphate) 21 (Fig. 7). We performed the reaction in 50% vol/vol of water/methanol (where MgCl2 is quite soluble) and compared the yields of methyl phosphate with various concentrations of added MgCl2 under the same conditions. The results are listed in Table 3; the yield as a function of time is shown in Table 4 and plotted in Fig. 8. The findings were at first sight remarkable. Under our standard conditions the presence of 10 equivalents of MgCl2 (0.25 M) led to a decrease in the product formed, so Mg2+ was an inhibitor. With higher concentrations of Mg2+ there was an increase in product formed, so here the Mg2+ furnished positive catalysis. These results are not unexpected, considering the likely roles the magnesium ions play.
Fig. 7.

Structure of ATP with the phosphate residues numbered.
Table 3.
Phosphorylation of methanol with ATP
| Entry | Phosphorylating agent | Concentration of Mg2+, M | Yield, %* |
| 1 | ATP.2Na | No MgCl2 | 36 ± 1 |
| 2 | ATP.2Na | 0.25 | 23 ± 1 |
| 3 | ATP.2Na | 1.0 | 32 ± 1 |
| 4 | ATP.2Na | 2.0 | 50 ± 0.5 |
ATP (0.025 M), MgCl2, H2O:MeOH (1:1), 60 °C, 48 h.
NMR yield.
Table 4.
Rate of methanol phosphorylation with ATP
| Entry | Time, h | Yield, %* |
| 1 | 12 | 8 |
| 2 | 24 | 15.6 |
| 3 | 36 | 23.7 |
| 4 | 48 | 30 |
| 5 | 60 | 36.2 |
ATP (0.025 M), MgCl2 (1 M), H2O:MeOH (1:1), 60 °C.
NMR yield.
Fig. 8.

Rate of methanol phosphorylation at 1.0 M MgCl2 concentration with ATP (0.025 M) at 60 °C.
In ATP 22 phosphate #3, the terminal phosphate, is transferred as phosphate #1 and #2 are pyrophosphate leaving groups, and this should be accelerated by Mg2+ binding of pyrophosphates. However, the first Mg2+ would surely preferentially bind to the pyrophosphate unit of phosphates #2 and #3. Phosphate #3 will bind more strongly because it is a dianion, the others are monoanions, and the result is that transfer of phosphate #3 is inhibited because it will require breaking up the magnesium-linked #2–#3 pyrophosphate group. Thus, the phosphate transfer is slowed. However, with increasing Mg2+ concentrations a second magnesium can then bind in the #1–#2 position, furnishing the driving force anticipated when a magnesium-bound pyrophosphate is the leaving group. At some point this can apparently be enough to overcome the inhibition effect from the binding at the first #2–#3 pyrophosphate site.
To add to this picture, we examined phosphorylation of methanol by ADP, under the same conditions. As Table 5 shows, ADP is a phosphorylating agent even without added Mg2+, and has this role to some extent in modern enzyme-catalyzed biology, but it is poorer than ATP (Table 3). Addition of Mg2+ has a significantly smaller effect, and here there is also a decrease in yield at low concentration when the chelate is present and then an increase at higher concentrations. Its affinity for the first magnesium ion should be less than for ATP, and at high concentrations a second Mg2+ is bound. It does not have a pyrophosphate leaving group, but breaking up a chelated magnesium pyrophosphate with added magnesium ion could activate phosphorylation simply by partially neutralizing the negative charges that make the phosphorus less reactive.
Table 5.
Phosphorylation of methanol with ADP
| Entry | Phosphorylating agent | Concentration of Mg2+, M | Yield, %* |
| 1 | ADP.2Na | No MgCl2 | 10.5 ± 0.5 |
| 2 | ADP.2Na | 0.25 | 8.5 ± 0.5 |
| 3 | ADP.2Na | 1.0 | 12.3 ± 0.2 |
| 4 | ADP.2Na | 2.0 | 16.8 ± 0.2 |
ADP (0.025 M), MgCl2, H2O:MeOH (1:1), 60 °C, 48 h.
NMR yield.
Conclusion
Here we see the other aspect of magnesium cation binding, activating a phosphate group by simple charge neutralization. This effect could also play a role with ATP, but we think binding the 1–2 pyrophosphate leaving group, as we invoked for ATP, is the more likely explanation there.
How could enzymes play roles in these processes to accelerate them? In both the systems binding of the substrates would of course help, but with ATP we propose an extra effect. The reason that the first magnesium ion is an inhibitor is that it should spontaneously go to the 2–3 pyrophosphate group, but an enzyme could bind it selectively to favor the magnesium 1–2 pyrophosphate structure. There the binding of the pyrophosphate leaving group would be fully effective, as in our ribosyl pyrophosphate systems. Thus, we propose that our results could be clues to prebiotic chemistry where enzymes did not yet exist, and they give insights into the modern enzyme systems for which they are simple mimics.
Acknowledgments
This work was supported by a grant from NASA.
Footnotes
The authors declare no conflict of interest.
References
- 1.Breslow R. On the mechanism of thiamine action. IV. Evidence from studies on model systems. J Am Chem Soc. 1958;80(14):3719–3726. [Google Scholar]
- 2.Breslow R, McNelis E. Studies on model systems for thiamine action. Synthesis of reactive intermediates, and evidence on the function of the pyrimidine ring. J Am Chem Soc. 1959;81(12):3080–3082. [Google Scholar]
- 3.Cheng L, Abhilash KG, Breslow R. Binding and biomimetic cleavage of the RNA poly(U) by synthetic polyimidazoles. Proc Natl Acad Sci USA. 2012;109(32):12884–12887. doi: 10.1073/pnas.1210846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng L, Mahendran A, Gonzalez RL, Jr, Breslow R. Deoxypolypeptides bind and cleave RNA. Proc Natl Acad Sci USA. 2014;111(22):7920–7924. doi: 10.1073/pnas.1407295111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert SM, Watters JI. The complexes of magnesium ion with pyrophosphate and triphosphate ions. J Am Chem Soc. 1957;79(21):5606–5608. [Google Scholar]
- 6.Lillo AM, Tetzlaff CN, Sangari FJ, Cane DE. Functional expression and characterization of EryA, the erythritol kinase of Brucella abortus, and enzymatic synthesis of L-erythritol-4-phosphate. Bioorg Med Chem Lett. 2003;13(4):737–739. doi: 10.1016/s0960-894x(02)01032-6. [DOI] [PubMed] [Google Scholar]
- 7.Ramirez F, Marecek JF. Coordination of magnesium with adenosine 5′-diphosphate and triphosphate. Biochim Biophys Acta. 1980;589(1):21–29. doi: 10.1016/0005-2728(80)90129-2. [DOI] [PubMed] [Google Scholar]
- 8.Fuller WD, Sanchez RA, Orgel LE. Studies in prebiotic synthesis. VI. Synthesis of purine nucleosides. J Mol Biol. 1972;67(1):25–33. doi: 10.1016/0022-2836(72)90383-x. [DOI] [PubMed] [Google Scholar]
- 9.Powner MW, Gerland B, Sutherland JD. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature. 2009;459(7244):239–242. doi: 10.1038/nature08013. [DOI] [PubMed] [Google Scholar]
- 10.Zubay G, Mui T. Prebiotic synthesis of nucleotides. Orig Life Evol Biosph. 2001;31(1-2):87–102. doi: 10.1023/a:1006722423070. [DOI] [PubMed] [Google Scholar]
- 11.Gao M, Chen Y, Tan S, Reibenspies JH, Zingaro RA. Syntheses of 1-thio-D-xylose and D-ribose esters of diorganoarsinous acids and their anticancer activity. Heteroatom Chem. 2008;19(2):199–206. [Google Scholar]
- 12.Kraft MB, Martinez Farias MA, Kiessling LL. Synthesis of lipid-linked arabinofuranose donors for glycosyltransferases. J Org Chem. 2013;78(5):2128–2133. doi: 10.1021/jo302507p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doussin S, Birlirakis N, Georgin D, Taran F, Berthault P. Novel zwitterionic reverse micelles for encapsulation of proteins in low-viscosity media. Chemistry. 2006;12(15):4170–4175. doi: 10.1002/chem.200501422. [DOI] [PubMed] [Google Scholar]
- 14.Wang WB, et al. A practical synthesis of sugar-derived cyclic nitrones: Powerful synthons for the synthesis of iminosugars. Synlett. 2010;3:488–492. [Google Scholar]
- 15.Martinez GV, et al. Demonstration of a sucrose-derived contrast agent for magnetic resonance imaging of the GI tract. Bioorg Med Chem Lett. 2013;23(7):2061–2064. doi: 10.1016/j.bmcl.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Addepalli H, et al. Modulation of thermal stability can enhance the potency of siRNA. Nucleic Acids Res. 2010;38(20):7320–7331. doi: 10.1093/nar/gkq568. [DOI] [PMC free article] [PubMed] [Google Scholar]