

Significance
Centromeres are the sites where chromosomes attach to microtubules during mitosis, and they are necessary for chromosome segregation. We discovered an unusual type of centromere in the yeast Candida lusitaniae, which is an occasional human pathogen. These centromeres are similar to many eukaryotic centromeres in that they are not specified by a particular sequence. However, unlike other centromeres of this type, C. lusitaniae centromeres are not flanked by a compact chromatin structure, known as pericentromeric heterochromatin. This finding reveals that, although pericentromeric heterochromatin is generally important for promoting proper centromere function, it is not universally necessary. This unusual centromere structure could contribute to stress-induced errors in chromosome segregation that are observed in Candida species.
Keywords: centromere, heterochromatin, Candida, CSE4, Sir2
Abstract
Point centromeres are specified by a short consensus sequence that seeds kinetochore formation, whereas regional centromeres lack a conserved sequence and instead are epigenetically inherited. Regional centromeres are generally flanked by heterochromatin that ensures high levels of cohesin and promotes faithful chromosome segregation. However, it is not known whether regional centromeres require pericentromeric heterochromatin. In the yeast Candida lusitaniae, we identified a distinct type of regional centromere that lacks pericentromeric heterochromatin. Centromere locations were determined by ChIP-sequencing of two key centromere proteins, Cse4 and Mif2, and are consistent with bioinformatic predictions. The centromeric DNA sequence was unique for each chromosome and spanned 4–4.5 kbp, consistent with regional epigenetically inherited centromeres. However, unlike other regional centromeres, there was no evidence of pericentromeric heterochromatin in C. lusitaniae. In particular, flanking genes were expressed at a similar level to the rest of the genome, and a URA3 reporter inserted adjacent to a centromere was not repressed. In addition, regions flanking the centromeric core were not associated with hypoacetylated histones or a sirtuin deacetylase that generates heterochromatin in other yeast. Interestingly, the centromeric chromatin had a distinct pattern of histone modifications, being enriched for methylated H3K79 and H3R2 but lacking methylation of H3K4, which is found at other regional centromeres. Thus, not all regional centromeres require flanking heterochromatin.
Centromeres are the chromosomal sites of spindle microtubule attachment and are necessary for chromosome segregation during mitosis. In budding yeast, point centromeres are defined by a short (120 bp) consensus sequence that recruits kinetochore proteins. In contrast, in most eukaryotes, regional centromeres span thousands to millions of base pairs and lack a conserved sequence that seeds kinetochore formation (1, 2). Instead, regional centromeres are epigenetically inherited, such that the prior presence of centromeric proteins mediates new centromere formation upon DNA replication (2, 3). Regional centromeres are flanked by pericentromeric heterochromatin, which ensures high levels of cohesin to hold sister chromatids together in the face of tension from the spindle microtubules (4–6). Pericentromeric heterochromatin is important for promoting faithful chromosome segregation (7, 8) and the assembly of centromeric chromatin (9, 10). In contrast to regional centromeres, point centromeres in budding yeast lack pericentromeric heterochromatin and have alternate mechanisms for recruiting cohesin (11, 12). However, it is not known whether the larger size and/or epigenetic inheritance of regional centromeres necessitates the presence of pericentromeric heterochromatin.
Centromeric DNA is packaged in a distinct chromatin structure containing a histone H3 variant, known as Cse4 in budding yeast and CENP-A in mammals (13, 14). This centromere-specific H3 is interspersed with canonical histone H3 that is methylated on lysine 4 (15, 16). For regional centromeres, the core centromeric chromatin is flanked by heterochromatin with a distinct molecular structure (17, 18). This heterochromatin is characterized by histones that are hypoacetylated and methylated on H3K9. In addition, the chromodomain protein HP1 binds methylated H3K9. This type of heterochromatin is best characterized in Schizosaccharomyces pombe, where heterochromatin is established in part through siRNA-presenting Argonaute proteins (19) and is maintained by histone deacetylases, such as Sir2 and Clr3 (20, 21).
An intermediate type of centromere occurs in the Candida clade of yeasts. In Candida albicans and Candida dubliniensis, Cse4/CENP-A is associated with 3–5 kbp of DNA (22–24). This length is much greater than the point centromeres of S. cerevisiae but not as long as the 10- to 15-kbp centromeric core of S. pombe. C. albicans centromeric cores have no common sequence and are epigenetically inherited (23, 25). However, it is not clear whether these centromeres are flanked by heterochromatin. In fact, Candida species and other hemiascomycete budding yeasts lack conventional mechanisms for generating heterochromatin (26). In particular, these species lack orthologs of HP1 and the methyltransferase specific for H3K9, although deacetylases are retained. Thus, Candida species could have a distinct type of centromere, which could impact the fidelity of chromosome segregation and contribute to the high rates of chromosome loss and aneuploidy observed in response to stress (27–31).
Candida lusitaniae (teleomorph Clavispora lusitaniae) is a particularly good candidate to have regional centromeres lacking heterochromatin. It is predicted to have centromeres spanning several kilobase pairs (32). However, not only does C. lusitaniae lack orthologs of heterochromatin-forming proteins such as HP1, a methyltransferase specific for H3K9, and Argonaute, but it also lacks heterochromatin-forming capacities that are present in the related yeast S. cerevisiae. In particular, an ortholog of the deacetylase Sir2, which generates heterochromatin in S. cerevisiae, does not form subtelomeric heterochromatin or repress transcription in C. lusitaniae (33). Therefore, understanding the centromere structure in C. lusitaniae could provide insights into genome stability in this occasional pathogen.
We identified the centromeres of C. lusitaniae by localizing two conserved centromeric proteins through ChIP followed by high-throughput sequencing (ChIP-Seq). These centromeres coincide with GC-poor troughs predicted to be centromeres (32, 34) and are comprised of 4–4.5 kbp of unique sequences. Despite having a size and sequence features consistent with epigenetically inherited regional centromeres, no indication of flanking heterochromatin was observed. Transcription was not repressed, the histones lacked modification patterns consistent with heterochromatin, and the deacetylase Sir2, which is associated with heterochromatin in other yeast, was not present at centromeres. Thus, a regional centromere does not require flanking heterochromatin.
Results
Identification of Centromeres in C. lusitaniae.
To determine the locations of centromeres in C. lusitaniae, we used two centromere-associated proteins as markers. Cse4 (orthologous to mammalian CENP-A) is a centromere-specific variant of histone H3, and Mif2 (orthologous to mammalian CENP-C) is an inner kinetochore component. Both proteins are associated with centromeres in other yeast species (13, 23, 35). We generated strains expressing myc-tagged alleles of Cse4 and Mif2 from the endogenous chromosomal loci, and these strains were used for ChIP-Seq. As a control, a mock immunoprecipitation (IP) was conducted using an untagged strain and the same antibody. Sequence reads were aligned to the reference genome (36), and the fold difference in reads mapped between the IP and mock IP samples was calculated at each genomic position.
Analysis of the ChIP-Seq signal revealed one locus on each of the eight chromosomes that had high enrichment of both Cse4 and Mif2 (Fig. 1, triangles). To confirm this enrichment, we analyzed separate ChIP samples by quantitative PCR (ChIP-qPCR) using primers distributed across the presumed centromeres 3 and 7. Cse4 and Mif2 were highly enriched at both loci relative to a control locus and had similar enrichment patterns to those observed by ChIP-Seq (Fig. 2 and Fig. S1). The eight Cse4- and Mif2-enriched loci detected by ChIP-Seq coincide with computationally predicted centromeres (32, 34).
Fig. 1.

Centromeres in C. lusitaniae coincide with GC-poor troughs. Heat maps of the eight chromosomes depict the relative enrichment of Cse4 (Top row) and Mif2 (Second row) based on ChIP-Seq analysis, with high enrichment in red. The average GC content (Third row) was calculated for sliding windows of 5 kbp. The average GC3 content (Bottom row) was calculated for sliding windows of 15 genes (32). Dashed boxes indicate regions for which GC3 content was not calculated because there are fewer than 15 genes between that point and the end of the chromosome. Triangles indicate centromeres.
Fig. 2.

Histone H3 is depleted at C. lusitaniae CEN3. (A) The ChIP-Seq signal for Cse4 and Mif2 across CEN3. (B) The relative enrichment of Cse4 or Mif2 at CEN3 compared with a control locus (PRI2) was determined by quantitative PCR. Chromatin IP was conducted using two independently constructed strains of myc-CSE4 (LRY2995, LRY2996) and MIF2-myc (LRY2997, LRY2998) and an untagged strain (LRY2826). (C) The ratio of H3 to H4, as measured by chromatin IP, was normalized to the ratio at a control locus (PRI2) expected to have a 1:1 ratio of H3:H4. Chromatin IP was conducted using WT yeast (LRY2826).
Fig. S1.

Cse4 and Mif2 are enriched at C. lusitaniae centromere 7. (A) The ChIP-Seq signal for Cse4 and Mif2 across centromere 7. (B) The relative enrichment of Cse4 or Mif2 at centromere 7 compared with a control locus (PRI2) was determined by quantitative PCR analysis. Chromatin IP was conducted using two independently constructed strains of myc-CSE4 (LRY2995, LRY2996) and MIF2-myc (LRY2997, LRY2998) and an untagged strain (LRY2826).
To estimate the size of the C. lusitaniae centromeres, we determined the lengths of the Cse4-enriched regions. These regions ranged from 4,018 to 4,619 bp, with an average of 4,298 bp (Table S1). This size is in keeping with the sizes of centromeres in C. albicans and is intermediate between S. cerevisiae and S. pombe centromeres.
Table S1.
Coordinates of centromeres in C. lusitaniae
| Centromere | Start (bp) | End (bp) | Length (bp) |
| CEN1 | 1,056,291 | 1,060,764 | 4,473 |
| CEN2 | 1,805,590 | 1,809,986 | 4,396 |
| CEN3 | 1,158,517 | 1,162,639 | 4,122 |
| CEN4 | 140,396 | 145,015 | 4,619 |
| CEN5 | 286,818 | 290,900 | 4,082 |
| CEN6 | 278,633 | 282,799 | 4,166 |
| CEN7 | 374,923 | 378,941 | 4,018 |
| CEN8 | 158,537 | 163,041 | 4,504 |
The boundaries of each centromere were defined as the points where the fold-enrichment of Cse4 is equal to 3. The coordinates of each boundary are provided, based on the reference genome from the Broad Institute, now available at wiki.biomisc.org/Supplementary_for_Candida_lusitaniae.
Cse4 Replaces H3 at Centromeres.
Because Cse4 is a variant of histone H3, it is expected to replace H3 in some of the centromeric nucleosomes. To test this prediction, the enrichment of H3 relative to H4 was determined by ChIP-qPCR across CEN3. This ratio was then compared with a control locus, ClPRI2, where H3 and H4 should be present in a 1:1 ratio. We took this approach because we had observed a reduced enrichment of H4 at centromeres relative to other genomic locations, indicating either that nucleosomes are less abundant within centromeres or that a technical issue, such as the presence of a kinetochore, reduces the efficiency of immunoprecipitation. Nevertheless, by examining the ratio of H3 to H4, it is possible to determine whether centromeric nucleosomes are lacking H3. Indeed, deviations from the ratio observed at the control locus indicate that H3 is depleted relative to H4 at CEN3 (Fig. 2C). The locations with the greatest depletion of H3 coincide with the regions most enriched for Cse4 and Mif2. These results are consistent with Cse4 replacing H3 in the centromere nucleosomes in C. lusitaniae.
Centromere Sequences Are Unique.
To determine whether a consensus sequence defines the centromeres in C. lusitaniae, we compared each centromere sequence to the entire genome by BLASTN (Fig. S2A). The positions and lengths of significant BLASTN hits were projected onto each sequence. If a consensus sequence was present, each centromere would have seven significant hits of similar lengths at the position of the consensus sequence. However, no such pattern was observed. Four of the eight centromeres shared no sequence with other centromeres. Centromeres 4 and 8 had related 300-bp sequences within the Cse4-enriched region, and centromeres 2 and 5 had related sequences in the pericentromeric regions (blue boxes in Fig. S2A). These observations indicate that no consensus sequence specifies the centromeres in C. lusitaniae. Therefore, these centromeres are likely to be inherited epigenetically.
Fig. S2.
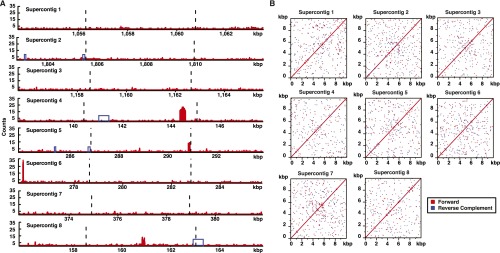
Centromeres in C. lusitaniae have unique sequences and are not flanked by repetitive elements. (A) The positions and lengths of significant BLASTN hits to C. lusitaniae genomic sequence were projected to the corresponding segment on the centromere sequence and piled up. Numbers of pileups show how many times the same segment can be aligned in the genome. Each centromere is represented by 10 kbp of the sequence centered on the middle of the Cse4-enriched region. Dashed lines indicate the Cse4-enriched regions. Sequences that can be reciprocally aligned with other centromeres are marked in blue boxes. (B) The same centromere sequences analyzed in A were compared with themselves in dot-matrix plots. Dots indicate a match of 10-bp words through forward (red) or reverse complement (blue) alignment. The red diagonal line in each plot indicates self-identity.
Regional centromeres, such as those in S. pombe, are flanked by repetitive sequences that assemble into heterochromatin through an RNA-mediated pathway. In particular, short RNAs complementary to the repeats are associated with Argonaute, which targets heterochromatin proteins to the repeated loci (19). In C. albicans, the nature of the pericentromeric chromatin has not been reported. However, the centromeres either lack repeats or are flanked by inverted repeats unique to a single centromere (22, 23). To determine whether centromeres in C. lusitaniae are flanked by repetitive sequences, we compared the sequence of each centromere region against itself using a dot-matrix plot (Fig. S2B). All identical 10-bp words, by either forward or reverse complement alignment, were plotted as dots using the EMBOSS dottup program (37). If repeats were present, they would appear as parallel diagonal lines in the dot plot, with direct repeats in red and inverted repeats in blue. However, the dot plot revealed no repetitive sequences within or flanking the centromeres. This observation is consistent with the absence of Argonaute proteins in C. lusitaniae and suggests that the centromeres may not be flanked by heterochromatin.
Centromeres Coincide with GC-Poor Troughs.
The centromeres in C. lusitaniae were previously predicted to coincide with GC-poor troughs (32). In their original analysis, Lynch and colleagues calculated for each gene the percentage of G or C bases at the third positions of all codons except the stop and CTG codons. This property was termed GC3 content. When the average GC3 content for sliding windows of 15 consecutive genes was plotted, one major GC3-poor trough was observed per chromosome (32), as indicated by the blue color in the GC3 row (Fig. 1). In this analysis, the GC content of the actual centromeres was not evaluated because only coding regions were considered. Therefore, we calculated the percentage of G or C bases across each chromosome in sliding windows of 5 kbp, with a moving step of 1 kbp. A trough in GC content coincided precisely with the region of Cse4 and Mif2 enrichment on each chromosome (Fig. 1). In fact, the centromeres represent the point of lowest GC content on each chromosome (Fig. S3). Thus, the centromeres in C. lusitaniae are particularly poor in GC content. These GC-poor troughs are surrounded by regions with GC contents somewhat higher than the remainder of the chromosome.
Fig. S3.
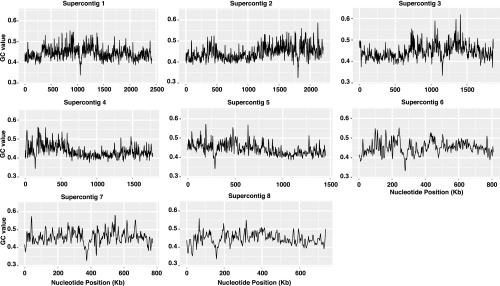
Centromeres occur at the point of lowest GC content on each chromosome. The GC content is plotted as a function of chromosomal position for each of the C. lusitaniae chromosomes.
Centromeres Display a Skew in Base Composition.
In C. albicans, the centromeres are the earliest replicating regions on each chromosome (34). Consequently, sequences flanking the centromeric replication origin are consistently replicated in the same way with respect to the leading and lagging strands. This replication pattern leads to a skew in base composition between the two strands (34), proposed to result from different rates of nucleotide substitutions during leading and lagging strand synthesis (38). Moreover, the skew in base composition switches polarity at the origin, where the strand that had been the leading strand becomes the lagging strand. A similar skew in base composition has been described for C. lusitaniae (34). To align the skew with the Cse4-enriched regions, we calculated GC skew as (G − C)/(G + C) for each 100-bp window in the 20-kbp region surrounding each centromere. We found a skew in G vs. C that switched polarity near the center of the region of Cse4 enrichment on each chromosome, and a similar skew in A vs. T (Fig. S4). These results suggest that C. lusitaniae resembles C. albicans in having an early replication origin associated with its centromeres.
Fig. S4.
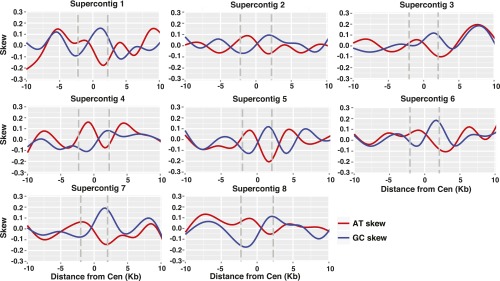
A skew in nucleotide composition is observed at C. lusitaniae centromeres. For each centromere, GC skew [(G − C)/(G + C) (red line)] and AT skew [(A − T)/(A + T) (blue line)] were calculated in sliding windows of 100 bp with a 1-bp step for the 20 kbp surrounding the region of Cse4 enrichment. The gray lines indicate the boundaries of the centromere.
Genes Near Centromeres Are Not Transcriptionally Repressed.
Heterochromatin represses the transcription of resident genes. Therefore, if the pericentromeric regions of C. lusitaniae were associated with heterochromatin, the genes in these regions would be poorly expressed. We analyzed RNA-sequencing data generated from a WT strain grown in rich medium (33) and calculated the normalized read count in FPKM (fragments per kilobase of exons per million mapped reads) for each annotated gene. We then calculated the average FPKM for genes in cumulative bins of 1 kbp from the centromeres and compared these values to the genome-wide average (Fig. 3A). Genes adjacent to centromeres did not display significantly different expression levels compared with the genome-wide average. This absence of gene repression in pericentromeric regions is consistent with a lack of pericentromeric heterochromatin.
Fig. 3.

Genes flanking centromeres do not display reduced expression. (A) Normalized read counts (FPKM) of all genes in C. lusitaniae were calculated from RNA-Seq data for WT cells (LRY2826). The average log2 (FPKM + 1) was determined for cumulative bins of 1 kbp for all genes within 10 kbp of a centromere and for the entire genome. The middle line represents the median value, and the lower and upper lines represent the first and third quartiles. Expression of genes within 1.5 times the interquartile range are shown by the whiskers. Dots represent outliers. Overlapping notches indicate medians are not significantly different. The hinges in the second boxplot are due to the small number of samples in this group. (B) A URA3 reporter was inserted between the Cse4-enriched region of CEN3 and the flanking gene. Expression was assessed by growth in the absence of uracil or in the presence of 5-FOA for three independent integration events (LRY3073–3075). Strains lacking URA3 (LRY2826) or with URA3 at the endogenous locus (CL143) were included as controls.
A Reporter Gene Placed at the Centromere Is Not Repressed.
The presence of heterochromatin can be detected by the repression of a reporter gene inserted into a block of heterochromatin. Such genes often display variegated expression, in which they are repressed in some cells but expressed in others. To determine whether transcription is repressed or variegated immediately adjacent to a centromere, we inserted the C. lusitaniae URA3 gene between the Cse4-enriched region and the first gene flanking CEN3. Three independent insertions were tested, and in all cases, URA3 was expressed, as determined by the ability of cells to grow in the absence of uracil (Fig. 3B). Moreover, no variegation of expression was detected, as the cells did not grow on 5-fluoroorotic acid (5-FOA), a drug that is specifically toxic to cells expressing URA3. Thus, there was no evidence that transcription is repressed adjacent to CEN3, as would be expected if heterochromatin occurs in this location.
The Deacetylase Hst1 Was Not Associated with Centromeres.
In S. cerevisiae, the deacetylase Sir2 is essential for the formation of heterochromatin at subtelomeres and cryptic mating-type loci (39). Similarly, in S. pombe Sir2 plays a significant role in the assembly and maintenance of heterochromatin at centromeres and other loci (20, 21). Thus, if the pericentromeric regions of C. lusitaniae were heterochromatic, a Sir2 ortholog might be enriched in these regions. The only member of the Sir2/Hst1 subfamily in C. lusitaniae is CLUG_01277, annotated as HST1 (33). To determine whether Hst1 associates with centromeres, we conducted ChIP-qPCR. We found no enrichment of Hst1-myc at CEN3 relative to a control locus (Fig. 4A). Importantly, Hst1-myc was enriched at the rDNA locus (Fig. 4B) as previously observed (33), indicating that the ChIP was successful. We also analyzed the genome-wide distribution of Hst1 by ChIP-Seq and found no significant enrichment at centromeres.
Fig. 4.

The sirtuin deacetylase Hst1 is not enriched at C. lusitaniae centromeres. (A) The relative enrichment of Hst1-myc at CEN3 compared with a control locus (PRI2) was determined by quantitative PCR analysis of chromatin IP samples from HST1-myc (LRY2858) and untagged (LRY2826) cells. (B) The relative enrichment of Hst1 at the rDNA locus was determined for the same chromatin IP samples.
Chromatin Flanking Centromeres Is Not Hypoacetylated.
Heterochromatin is typically associated with hypoacetylated histones. Thus, if the pericentromeric regions of C. lusitaniae are heterochromatic, the histones should be hypoacetylated in the regions flanking the Cse4-enriched centromeric core. To examine the distribution of acetylation across the centromeres, we conducted ChIP-qPCR of several acetylated histone lysines. We normalized the enrichment to the recovery of total histone H3 or H4. H4K8ac and H4K16ac were more abundant in the flanking regions than within the Cse4-enriched core of CEN3 (Fig. 5A). In contrast, acetylation of lysines 5 and 12 on H4 and lysines 9, 14, 18, 23, and 56 on H3 were relatively unchanged across the region surveyed (Fig. 5B). Thus, the flanking regions were not hypoacetylated compared with the Cse4-enriched core, suggesting a lack of heterochromatin.
Fig. 5.

The regions flanking Cse4-enriched centromere cores are not hypoacetylated. (A) The relative enrichment of acetylated H4K8 and H4K16 compared with total histone H4 was determined by chromatin IP. The abundance of each PCR amplicon was first normalized to primer set 6 (Table S5), and then the ratio of modified histone to total H4 was calculated. Chromatin IP was conducted using WT strain (LRY2826) and antibodies described in Table S4. (B) The relative enrichment of acetylated H4K5, H4K12, H3K9, H3K14, H3K18, H3K23, and H3K56 compared with total histone (H4 or H3) was determined as described for A.
Centromeric Chromatin Lacks H3K4 Methylation and Is Enriched for H3K79 and H3R2 Methylation.
In addition to the presence of Cse4/CENP-A, centromere cores have a distinct chromatin composition. In particular, methylation of H3K4 occurs on those nucleosomes harboring canonical histone H3 (15, 16). To determine whether a similar chromatin structure exists in C. lusitaniae centromeres, we examined the enrichment of methylated H3K4. Surprisingly, we found that neither H3K4me2 nor H3K4me3 was enriched in the Cse4-associated region compared with flanking regions of CEN3 (Fig. S5A).
Fig. S5.

Methylated H3K79 and H3R2 are enriched at centromeres. (A) The relative enrichment of di- and trimethylated H3K4 compared with total histone H3 was determined as described for Fig. S5A. (B) The relative enrichment of di- and trimethylated H3K79 and symmetric and asymmetric dimethylated H3R2 compared with total histone H3 was determined. (C) The relative enrichment of H2A.Z compared with total histone H4 was determined as described for Fig. S5A. Anti-myc antibody was used to immunoprecipitate proteins from HTZ1-myc (LRY3062) yeast.
To identify histone modifications that are associated with centromeres, we conducted ChIP using a panel of antibodies. We observed enrichment of H3K79me2, H3K79me3, and H3R2me2 in the same region enriched for Cse4 and Mif2 (Fig. S5B). For H3R2me2, we used antibodies specific for both symmetric and asymmetric methylation and found that both methylation patterns were enriched. These results are consistent with C. lusitaniae centromeres having a distinct chromatin composition.
Discussion
C. lusitaniae Centromeres Are Consistent with Regional Centromeres.
The centromeres of C. lusitaniae span 4–4.5 kbp, and each have a unique sequence (Fig. S2). This size and the lack of a conserved sequence are also observed for C. albicans and C. dubliniensis and distinguish the centromeres in the Candida clade from the point centromeres of other hemiascomycete yeasts, including Kuraishia capsulata (40) and Yarrowia lipolytica (41) that lie outside both the Saccharomyces and Candida clades. Based on their size and lack of a consensus sequence, C. lusitaniae centromeres are likely to be epigenetically inherited, as are those in C. albicans (23, 25). Consistent with this idea, we found that a plasmid containing C. lusitaniae CEN7 transformed C. lusitaniae poorly compared with an integrating cassette.
The centromeres in C. lusitaniae display several interesting sequence features. First, the centromeres coincide with regions of particularly low GC content that are surrounded by areas of high GC content (Fig. 1). Low GC content is also reported for the centromeres of other yeast, such as Scheffersomyces stipites, Y. lipolytica, and K. capsulata (32, 34, 40). Another interesting feature of the centromere sequences in C. lusitaniae is a strand-specific skew in G vs. C and A vs. T composition (Fig. S4). This property suggests an early replication origin associated with the centromeres. Such early replication origins could contribute to epigenetic mechanisms for inheritance of centromeres. These sequence features of low GC content and skew in base composition might be used to develop algorithms for predicting centromeres in other yeast species.
C. lusitaniae Centromeres Lack Pericentromeric Heterochromatin.
This study reveals an example of a regional centromere that is not flanked by heterochromatin. In particular, H3 and H4 were not hypoacetylated (Fig. 5) nor was the sirtuin deacetylase Hst1 present in regions flanking the Cse4-enriched core (Fig. 4). Additionally, there was no transcriptional repression of neighboring genes or a reporter gene inserted adjacent to the centromere (Fig. 3). Moreover, repetitive sequences flanking the centromere were absent (Fig. S2). These findings indicate that C. lusitaniae has a distinct centromere organization compared with other species with regional centromeres and that the pericentromeric chromatin more closely resembles that of S. cerevisiae, which has point centromeres.
In many species, pericentromeric heterochromatin is important for ensuring high levels of cohesin and promoting faithful segregation (4–8), and it is not known what alternative mechanisms exist for recruiting cohesin in species such as C. lusitaniae that lack heterochromatin. However, such mechanisms must exist, as the rates of chromosome loss in S. cerevisiae and C. albicans are lower than in S. pombe (8, 42–44). The pericentromeric regions in Candida species may function similarly to those in S. cerevisiae, which are not heterochromatic but are associated with high levels of cohesin (11, 12).
Interestingly, some neocentromeres in human and chicken cells also lack flanking heterochromatin (45, 46). Unlike most metazoan centromeres, these neocentromeres are not embedded in repetitive DNA sequences, suggesting that heterochromatin is correlated with the presence of repetitive sequences rather than the centromere per se (46). Thus, it may be that in the pericentromeric regions the presence of topology adjusters, such as cohesin and condensin, is more critical than a particular pattern of histone modifications and chromatin proteins (47).
C. lusitaniae Centromeres Have a Distinct Chromatin Structure.
We found that, as in other eukaryotic species, the conserved centromeric protein Cse4/CENP-A replaced canonical histone H3 at the centromere core in C. lusitaniae (Fig. 2). However, the centromere core appeared to have a different chromatin structure compared with other described species, as it lacked methylation of H3K4 and instead was associated with methylated H3K79 and H3R2 (Fig. S5). The absence of methylated H3K4 has also been reported for some neocentromeres (46), plant centromeres (48), and the filamentous fungus Neurospora crassa (49). Thus, there may be significant flexibility in the pattern of histone modifications that can occur within a centromeric core.
There is little information on whether methylation of H3K79 or H3R2 occurs at the centromeres of other species. H3K79 methylation is associated with euchromatin and transcriptional activity (50–52), but paradoxically loss of H3K79 methylation perturbs heterochromatin formation in budding yeast and mouse cells (51, 53). Symmetric methylation of H3R2 is also associated with euchromatin (54). In contrast, asymmetric methylation of H3R2 is associated with heterochromatin and repressed euchromatic genes (55). The unexpected presence of both symmetric and asymmetric H3R2 methylation, as well as H3K79 methylation, suggests a distinctive chromatin structure at centromeres in C. lusitaniae.
Materials and Methods
Yeast Strains and Growth.
The C. lusitaniae strains used in this study (Table S2) are based on a derivative of CL143 (31) in which the URA3 and HIS1 genes were deleted to generate LRY2826 (33). Details of strain construction are provided in SI Materials and Methods and Table S3. Yeast strains were grown at 30 °C in YPD [1% yeast extract, 2% (wt/vol) peptone, and 2% (wt/vol) glucose]. For the URA3 reporter assay, strains were grown overnight, serially diluted 10-fold, and spotted on plates with a starting concentration of 10 OD/mL. Transformation by electroporation was conducted as previously described (33).
Table S2.
Strains used in this study
| Strain | Genotype | Source |
| CL143 | MATa chxr (congenic to ATCC 42720) | Reedy et al. 2009 |
| LRY2826 | CL143 ura3∆ his1∆::FRT | Froyd et al. 2013 |
| LRY2858 | LRY2826 HST1::myc-HIS1 | Froyd et al. 2013 |
| LRY2995 | LRY2826 myc-CSE4::HIS1 | |
| LRY2996 | LRY2826 myc-CSE4::HIS1 | |
| LRY2997 | LRY2826 MIF2::myc-HIS1 | |
| LRY2998 | LRY2826 MIF2::myc-HIS1 | |
| LRY3062 | LRY2826 HTZ1::myc-HIS1 | |
| LRY3073 | LRY2826 CEN3::URA3 | |
| LRY3074 | LRY2826 CEN3::URA3 myc-CSE4::HIS1 | |
| LRY3075 | LRY2826 CEN3::URA3 HTZ1::myc-HIS1 |
Table S3.
Plasmids used in this study
| Construct | Oligonucleotides used | Restriction sites used |
| myc-CSE4::HIS1 (pLR 1040) | ||
| CSE4 from LRY2826 in pRS414 (pLR989) | SacI, SacII | |
| ClHIS1 from pLR950 in pLR989 (pLR1034) | ccgagtcacgtatccctcaaCTATTCCCAACCAATGCCG | |
| gcttcgcggacatctctgaCGGGAATCTCGGTCGTAATG | ||
| myc tag from pLR950 in pLR1034 (pLR1040) | gaaaaaaccttcatttactactcccatgGGGGAACAGAAGCTTATATCCG | |
| gttgttggtggaaagacgggcCCCTAGGTCTTCCTCTGATATG | ||
| MIF2::myc-HIS1 (pLR1038) | ||
| MIF2 from LRY2826 in pRS414 (pLR1031) | SpeI, PstI | |
| myc-ClHIS1 from pLR950 in pLR1031 (pLR1038) | cagcttggcacagcagagaaaGGGGAACAGAAGCTTATATCCG | |
| cccagctcgtatggtgaatagCTATTCCCAACCAATGCCG | ||
| HTZ1::myc-HIS1 (pLR1039) | ||
| HTZ1 from LRY2826 in pRS414 (pLR1032) | SacI, EagI | |
| myc-ClHIS1 from pLR950 in pLR1032 (pLR1039) | gagagaaagaagggccagaagGGGGAACAGAAGCTTATATCCG | |
| gtacatagggcccagcgCTATTCCCAACCAATGCCG | ||
| CEN3::URA3 (pLR1111) | ||
| CEN3 from LRY2826 in pRS414 (pLR1110) | ClaI, SacI | |
| GGGGAGCTCGTCTTCATTTATGGTAATGATGGCC | ||
| ClURA3 from pLR787 in pLR1110 (pLR1111) | ggagtgaggaaagtgcatccCTTCCTCCTTCTTCCGC | |
| gttcctcttactcctataagttttgatacGGCATTGGTGGTCTAAGTAGG |
These plasmids were intermediate steps in the construction of yeast strains, as described in the SI Materials and Methods. For PCR stitching, the oligonucleotides listed were used to amplify the region to be inserted into the indicated plasmid, with the uppercase portion of oligonucleotide annealing to the template. Then, the resulting PCR product was stitched into the indicated plasmid, guided by the lowercase portion of the oligonucleotide.
ChIP.
Approximately 100 OD of logarithmically growing cells were harvested at OD600 =3–4. Cells were cross-linked with 1% formaldehyde for 45 min. Preparation of soluble chromatin and immunoprecipitation was conducted as previously described (33) with 5 µL antibody against the myc tag (Millipore 06–549) or modifications of H3 or H4 (Table S4). ChIP samples were analyzed by real-time PCR using oligonucleotides listed in Table S5. For myc-tagged proteins, the amount of immunoprecipitated DNA at experimental loci and the control locus, ClPRI2, was calculated relative to a standard curve prepared from input DNA. For histone modifications, the abundance of each PCR amplicon was normalized to primer set 6 (Table S5), and then the ratio of modified histone to total histone was calculated. The SE of the ratio was obtained using BootstRatio (56).
Table S4.
Antibodies used for chromatin-IP
| Histone | Modification | Catalog no. |
| H4 | α-Histone H4 | Millipore 04–858 |
| Acetyl-lysine 5 | Millipore 07–327 | |
| Acetyl-lysine 8 | Millipore 07–328 | |
| Acetyl-lysine 12 | Millipore 06–761 | |
| Acetyl-lysine 16 | Millipore 07–329 | |
| H3 | α-Histone H3 | Millipore 05–928 |
| Dimethyl lysine 4 | Millipore 07–030 | |
| Trimethyl lysine 4 | Millipore 04–745 | |
| Dimethyl lysine 79 | Millipore 07–366 | |
| Trimethyl lysine 79 | Millipore 07–952-S | |
| Dimethyl (Arg2) asymmetric | Millipore ABE377 | |
| Dimethyl (Arg2) symmetric | Millipore ABE460 | |
| Acetyl lysine 9 | Millipore 06–942 | |
| Acetyl lysine 14 | Upstate 07–353 | |
| Acetyl lysine 18 | Upstate 07–354 | |
| Acetyl lysine 23 | Upstate 07–355 | |
| Acetyl lysine 56 | Upstate 07–677 |
Table S5.
Oligonucleotides used for qPCR analysis of ChIP samples
| C. lusitaniae target | Oligonucleotides |
| ClPRI2 (CLUG_00368) | CGGAGCAAATCGCAAGTACC |
| GTGTTGTTGAAGACGCGTAGTG | |
| rDNA (NTS2) | CTCGGATCAAGCCCTAATTAGG |
| GGCACACCACAAAATTACGAAAACG | |
| Centromere 3 | |
| 1 | CTGAAATGGCTCAGCGTTGG |
| GCCATCCGTCTACTACAAACG | |
| 2 | CCTCTGTCTCAGCTCAGG |
| CCAAGGTTCAGGTCTGCG | |
| 3 (promoter CLUG_02872) | GAGTTCTTGAGGTGCATTTCGC |
| CACTTCTTTGAACCGCCCTC | |
| 4 | GTTTGGACGCGCCCAC |
| GTGTTATGCTTGGCCACTCG | |
| 5 | GTCAACTGGCGGATCGTAG |
| GCGAATTGGTTTCCTGTGAAGC | |
| 6 | CAAAGAATATGGGACGCTTATCAG |
| GTCTTCATTTATGGTAATGATGGCC | |
| 7 | CAATGTTCTGTGGCTGATGTTTCTTG |
| CAGTTCCAAAGCCCTAATTTAGATC | |
| 8 | GGTGGAAGTCATTGGGTTAGG |
| GTTCATCCAGCAATTGACAAGAGATAG | |
| 9 | GGATTGACGCCAAAGCAC |
| GGGCTCTGCCAATGCG | |
| 10 | CTCGGCGTCACCAAACCTG |
| CCAAAGCAAACACGAACCAGAAG | |
| 11 (promoter CLUG_02873) | CCTGTACTTCGATGAACACTC |
| CGTGCAGATAACGTTGAGC | |
| 12 | CTATTTCCTCGGCTTCCTCG |
| CCATTCTGGTCCCAAGTTG | |
| Centromere 7 | |
| 1 | GGCAGGATCAGACTCATTGGAGC |
| CCTAACAAAGGGGGCAAGTTG | |
| 2 | CTTCTCCAGTCCCGTCC |
| GCTGATACTGTTAGTCGCC | |
| 3 (promoter CLUG_05420) | GGACGGGACTGGAGAAG |
| CTAGATTGTAGTGTGGACATGG | |
| 4 | GCACGGGGTTAGGCACG |
| GACTTGGACAGTTGCGTCC | |
| 5 | CGGGTTAACATCCATGACTTGG |
| GACTAAATGGTGCCATGGTCC | |
| 6 | GAGCCACTCTTTATTGTCTGTTAACG |
| CAGGAGAGAATTCGCTTTTGGTATTG | |
| 7 | CTGAACATAAGGACAAAGTACGAAGG |
| GTGTACCCTTATAAATCCCATCCC | |
| 8 | GAGTCTCATCTCTCATTCACTCTAC |
| GAAACAACCGTATACTTCTATAAGCC | |
| 9 | GGGAAGGGAGCAAGGTACT |
| CGTTTATGATCCCGACAAGC | |
| 10 | GGCCCCAGCAAAATGGTG |
| CGGGCCGCAATGGAATC | |
| 11 (promoter CLUG_05421) | CCAGTGAACCAAAGCACAGTC |
| CATAAGAAGGCTAATTGATGTCCG | |
| 12 | GTCAAGCTCTTCTAATCTTCTCC |
| CAGTATCCACAACGGAAACGC |
ChIP Sequencing.
For ChIP sequencing, immunoprecipitated DNA was prepared as described above with two modifications. The immunoprecipitation was conducted with 10 μL Protein A agarose beads in the absence of the usual blocking agents BSA and salmon sperm DNA, and 20 µg/mL RNaseA was added to the last wash. The immunoprecipitated DNA was processed for deep sequencing at the Next-Generation Sequencing and Expression Analysis facility at University at Buffalo. Data are available at the Gene Expression Omnibus (GEO) repository (GSE71667).
For analysis, the genome sequence of C. lusitaniae was downloaded from the Broad Institute, now available at wiki.biomisc.org/Supplementary_for_Candida_lusitaniae. ChIP-Seq reads were mapped to the reference genome using BWA v0.7.7-r441 (57). For calling enriched regions, MACS2 v2.1.0 (58) was used with mock-IP samples as control. The parameters used in MACS2 were “–nomodel–extsize 200 -q 0.01 -g 12114892–keep-dup 10”. The maximum number of duplicates to keep was selected according to assumed Poisson distribution of read coverage. MACS2 “bdgcmp” function was used to generate genome-wide fold-enrichment profile with parameters “-m FE”. To define the boundaries of Cse4-enriched peaks, we analyzed the average length of predicted centromeres with different cutoff values of fold-enrichment. The cutoff of 3 corresponded to stable length prediction (Fig. S6) and was selected for downstream analysis.
Fig. S6.

Calculation of centromere lengths. The average lengths of the eight centromeres were calculated as a function of the fold-enrichment used as a cutoff.
Gene Expression Analysis Within Region of Centromere.
RNA sequencing of C. lusitaniae WT cells was previously reported (33), and the data are available at the GEO repository (GSE71667). The raw reads obtained from paired-end RNA-Seq were mapped to the C. lusitaniae reference genome, allowing no more than two mismatches using tophat v2.0.10 (59). Gene expression levels were calculated as FPKM by cufflinks v2.2.1 (60) based on gene annotation obtained from the Broad Institute, now available at wiki.biomisc.org/Supplementary_for_Candida_lusitaniae.
SI Materials and Methods
Yeast Strain Construction.
The strains used in this study (Table S2) are based on a derivative of CL143 (31) in which the URA3 and HIS1 genes were deleted to generate LRY2826 (33). For tagging, each gene plus 800–1,000 bp upstream and downstream of the ORF was amplified by PCR from genomic DNA. The genomic sequences of the genes CSE4 (CLUG_01355), MIF2 (CLUG_04953), and HTZ1 (CLUG_00615) were obtained from the Broad Institute database, now available at wiki.biomisc.org/Supplementary_for_Candida_lusitaniae. The PCR products were ligated into an S. cerevisiae shuttle vector pRS414. These plasmids were subsequently modified (Table S3) by inserting a myc tag and HIS1 marker from pLR950 (33) just after the last nonstop codon of the ORF. For Cse4, placing the myc tag at the C terminus was unsuccessful and may have inactivated the protein. Therefore, the epitope tag was placed at the N terminus, and the HIS1 marker was inserted downstream of the gene. The correct integration of the tags was checked by sequencing. Finally, the tagging cassettes were integrated into the C. lusitaniae genome by homologous recombination, colonies were screened for correct integrants by PCR, and expression of tagged protein was confirmed by immunoblotting.
To insert the URA3 reporter gene between the Cse4-enriched region of centromere 3 and the flanking gene, a portion of CEN3 was first subcloned into pRS414. Then, URA3 along with its promoter was amplified from pLR787 (33) and incorporated by PCR stitching into the CEN3 plasmid (Table S3). Finally, this cassette was integrated into the yeast chromosome by homologous recombination.
Analysis of Centromere Sequences.
To search for repetitive sequences associated with centromeres, 10 kbp of the sequence centered at the middle of the Cse4-enriched region of each centromere was compared with the whole genome using NCBI BLASTN (61). The maximum target sequences to be reported was set to 100, the Expect threshold was set to 10, and the length of the seed that initiates the alignment was set as 11. The reward and penalty for matching and mismatching bases were 2 and −3, and the cost for gap opening was 5 and for gap extension was 2. The low complexity regions were filtered. The positions and lengths of significant BLASTN hits to the C. lusitaniae genomic sequence were projected to the corresponding segment on the centromere sequence and were then piled up and plotted using R Sushi package (62).
Dot-matrix was used to determine whether centromeres in C. lusitaniae are flanked by repetitive sequences. Two runs of the dottup program from EMBOSS compared each 10-kbp centromere to itself and its reverse complementary sequence separately using a 10-bp sliding window, and then the two dot-matrix plots were merged using red dots to indicate forward and blue to indicate reverse complementary alignments.
GC3 content values were obtained from ref. 32. GC content values were calculated using a sliding window method, with a window size of 5 kbp and a moving step of 1 kbp. If a window has unspecified (N) bases, the average GC content was calculated from the remaining bases. For windows at the end of each chromosome, the GC content was calculated for the actual window size, even though the size was less than 5 kbp.
GC skew was calculated as (G − C)/(G + C) and AT skew as (A − T)/(A + T). The GC and AT skews were calculated in a sliding window of 100 bp with a moving step of 1 bp, and then each was smoothed using the “geom_smooth” function in R ggplot2 (63) using the “gam” method.
Acknowledgments
We thank Dr. Michael Buck and the University at Buffalo Next Generation Sequencing and Expression Analysis Facility for assistance with ChIP sequencing; members of the L.N.R. laboratory for discussion; and Drs. Robin Allshire, Kerry Bloom, Alessia Buscaino, Shiv Grewal, and Beth Sullivan for helpful comments on the manuscript. This work was supported by National Science Foundation Grant MCB-1121569.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE71667).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508749112/-/DCSupplemental.
References
- 1.Fukagawa T, Earnshaw WC. The centromere: Chromatin foundation for the kinetochore machinery. Dev Cell. 2014;30(5):496–508. doi: 10.1016/j.devcel.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falk SJ, Black BE. Centromeric chromatin and the pathway that drives its propagation. Biochim Biophys Acta. 2013;1819(3-4):313–321. [PubMed] [Google Scholar]
- 3.Catania S, Allshire RC. Anarchic centromeres: Deciphering order from apparent chaos. Curr Opin Cell Biol. 2014;26:41–50. doi: 10.1016/j.ceb.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernard P, et al. Requirement of heterochromatin for cohesion at centromeres. Science. 2001;294(5551):2539–2542. doi: 10.1126/science.1064027. [DOI] [PubMed] [Google Scholar]
- 5.Nonaka N, et al. Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast. Nat Cell Biol. 2002;4(1):89–93. doi: 10.1038/ncb739. [DOI] [PubMed] [Google Scholar]
- 6.Peters AH, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107(3):323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 7.Kellum R, Alberts BM. Heterochromatin protein 1 is required for correct chromosome segregation in Drosophila embryos. J Cell Sci. 1995;108(Pt 4):1419–1431. doi: 10.1242/jcs.108.4.1419. [DOI] [PubMed] [Google Scholar]
- 8.Allshire RC, Nimmo ER, Ekwall K, Javerzat JP, Cranston G. Mutations derepressing silent centromeric domains in fission yeast disrupt chromosome segregation. Genes Dev. 1995;9(2):218–233. doi: 10.1101/gad.9.2.218. [DOI] [PubMed] [Google Scholar]
- 9.Folco HD, Pidoux AL, Urano T, Allshire RC. Heterochromatin and RNAi are required to establish CENP-A chromatin at centromeres. Science. 2008;319(5859):94–97. doi: 10.1126/science.1150944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakashima H, et al. Assembly of additional heterochromatin distinct from centromere-kinetochore chromatin is required for de novo formation of human artificial chromosome. J Cell Sci. 2005;118(Pt 24):5885–5898. doi: 10.1242/jcs.02702. [DOI] [PubMed] [Google Scholar]
- 11.Glynn EF, et al. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol. 2004;2(9):E259. doi: 10.1371/journal.pbio.0020259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber SA, et al. The kinetochore is an enhancer of pericentric cohesin binding. PLoS Biol. 2004;2(9):E260. doi: 10.1371/journal.pbio.0020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meluh PB, Yang P, Glowczewski L, Koshland D, Smith MM. Cse4p is a component of the core centromere of Saccharomyces cerevisiae. Cell. 1998;94(5):607–613. doi: 10.1016/s0092-8674(00)81602-5. [DOI] [PubMed] [Google Scholar]
- 14.Warburton PE, et al. Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Curr Biol. 1997;7(11):901–904. doi: 10.1016/s0960-9822(06)00382-4. [DOI] [PubMed] [Google Scholar]
- 15.Sullivan BA, Karpen GH. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol. 2004;11(11):1076–1083. doi: 10.1038/nsmb845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cam HP, et al. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet. 2005;37(8):809–819. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
- 17.Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8(1):35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 18.Ebert A, Lein S, Schotta G, Reuter G. Histone modification and the control of heterochromatic gene silencing in Drosophila. Chromosome Res. 2006;14(4):377–392. doi: 10.1007/s10577-006-1066-1. [DOI] [PubMed] [Google Scholar]
- 19.Verdel A, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303(5658):672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buscaino A, et al. Distinct roles for Sir2 and RNAi in centromeric heterochromatin nucleation, spreading and maintenance. EMBO J. 2013;32(9):1250–1264. doi: 10.1038/emboj.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alper BJ, et al. Sir2 is required for Clr4 to initiate centromeric heterochromatin assembly in fission yeast. EMBO J. 2013;32(17):2321–2335. doi: 10.1038/emboj.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishra PK, Baum M, Carbon J. Centromere size and position in Candida albicans are evolutionarily conserved independent of DNA sequence heterogeneity. Mol Genet Genomics. 2007;278(4):455–465. doi: 10.1007/s00438-007-0263-8. [DOI] [PubMed] [Google Scholar]
- 23.Sanyal K, Baum M, Carbon J. Centromeric DNA sequences in the pathogenic yeast Candida albicans are all different and unique. Proc Natl Acad Sci USA. 2004;101(31):11374–11379. doi: 10.1073/pnas.0404318101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Padmanabhan S, Thakur J, Siddharthan R, Sanyal K. Rapid evolution of Cse4p-rich centromeric DNA sequences in closely related pathogenic yeasts, Candida albicans and Candida dubliniensis. Proc Natl Acad Sci USA. 2008;105(50):19797–19802. doi: 10.1073/pnas.0809770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baum M, Sanyal K, Mishra PK, Thaler N, Carbon J. Formation of functional centromeric chromatin is specified epigenetically in Candida albicans. Proc Natl Acad Sci USA. 2006;103(40):14877–14882. doi: 10.1073/pnas.0606958103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hickman MA, Froyd CA, Rusche LN. Reinventing heterochromatin in budding yeasts: Sir2 and the origin recognition complex take center stage. Euk Cell. 2011;10(9):1183–1192. doi: 10.1128/EC.05123-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perepnikhatka V, et al. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J Bacteriol. 1999;181(13):4041–4049. doi: 10.1128/jb.181.13.4041-4049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janbon G, Sherman F, Rustchenko E. Monosomy of a specific chromosome determines L-sorbose utilization: A novel regulatory mechanism in Candida albicans. Proc Natl Acad Sci USA. 1998;95(9):5150–5155. doi: 10.1073/pnas.95.9.5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barton RC, Gull K. Isolation, characterization, and genetic analysis of monosomic, aneuploid mutants of Candida albicans. Mol Microbiol. 1992;6(2):171–177. doi: 10.1111/j.1365-2958.1992.tb01998.x. [DOI] [PubMed] [Google Scholar]
- 30.Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313(5785):367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reedy JL, Floyd AM, Heitman J. Mechanistic plasticity of sexual reproduction and meiosis in the Candida pathogenic species complex. Curr Biol. 2009;19(11):891–899. doi: 10.1016/j.cub.2009.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lynch DB, Logue ME, Butler G, Wolfe KH. Chromosomal G + C content evolution in yeasts: Systematic interspecies differences, and GC-poor troughs at centromeres. Genome Biol Evol. 2010;2:572–583. doi: 10.1093/gbe/evq042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Froyd CA, Kapoor S, Dietrich F, Rusche LN. The deacetylase Sir2 from the yeast Clavispora lusitaniae lacks the evolutionarily conserved capacity to generate subtelomeric heterochromatin. PLoS Genet. 2013;9(10):e1003935. doi: 10.1371/journal.pgen.1003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koren A, et al. Epigenetically-inherited centromere and neocentromere DNA replicates earliest in S-phase. PLoS Genet. 2010;6(8):e1001068. doi: 10.1371/journal.pgen.1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi K, Chen ES, Yanagida M. Requirement of Mis6 centromere connector for localizing a CENP-A-like protein in fission yeast. Science. 2000;288(5474):2215–2219. doi: 10.1126/science.288.5474.2215. [DOI] [PubMed] [Google Scholar]
- 36.Butler G, et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459(7247):657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 38.Sernova NV, Gelfand MS. Identification of replication origins in prokaryotic genomes. Brief Bioinform. 2008;9(5):376–391. doi: 10.1093/bib/bbn031. [DOI] [PubMed] [Google Scholar]
- 39.Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 40.Morales L, et al. Complete DNA sequence of Kuraishia capsulata illustrates novel genomic features among budding yeasts (Saccharomycotina) Genome Biol Evol. 2013;5(12):2524–2539. doi: 10.1093/gbe/evt201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamane T, Ogawa T, Matsuoka M. Derivation of consensus sequence for protein binding site in Yarrowia lipolytica centromere. J Biosci Bioeng. 2008;105(6):671–674. doi: 10.1263/jbb.105.671. [DOI] [PubMed] [Google Scholar]
- 42.Forche A, et al. Stress alters rates and types of loss of heterozygosity in Candida albicans. MBio. 2011;2(4):e00129-11. doi: 10.1128/mBio.00129-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall IM, Noma K, Grewal SI. RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc Natl Acad Sci USA. 2003;100(1):193–198. doi: 10.1073/pnas.232688099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumaran R, Yang SY, Leu JY. Characterization of chromosome stability in diploid, polyploid and hybrid yeast cells. PLoS One. 2013;8(7):e68094. doi: 10.1371/journal.pone.0068094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alonso A, Hasson D, Cheung F, Warburton PE. A paucity of heterochromatin at functional human neocentromeres. Epigenetics Chromatin. 2010;3(1):6. doi: 10.1186/1756-8935-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shang WH, et al. Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev Cell. 2013;24(6):635–648. doi: 10.1016/j.devcel.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bloom KS. Centromeric heterochromatin: The primordial segregation machine. Annu Rev Genet. 2014;48:457–484. doi: 10.1146/annurev-genet-120213-092033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin W, et al. Histone modifications associated with both A and B chromosomes of maize. Chromosome Res. 2008;16(8):1203–1214. doi: 10.1007/s10577-008-1269-8. [DOI] [PubMed] [Google Scholar]
- 49.Smith KM, Phatale PA, Sullivan CM, Pomraning KR, Freitag M. Heterochromatin is required for normal distribution of Neurospora crassa CenH3. Mol Cell Biol. 2011;31(12):2528–2542. doi: 10.1128/MCB.01285-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: A potential mechanism for position-effect variegation. Proc Natl Acad Sci USA. 2003;100(4):1820–1825. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109(6):745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 52.Steger DJ, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28(8):2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones B, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4(9):e1000190. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuan CC, et al. Histone H3R2 symmetric dimethylation and histone H3K4 trimethylation are tightly correlated in eukaryotic genomes. Cell Reports. 2012;1(2):83–90. doi: 10.1016/j.celrep.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirmizis A, et al. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature. 2007;449(7164):928–932. doi: 10.1038/nature06160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clèries R, et al. BootstRatio: A web-based statistical analysis of fold-change in qPCR and RT-qPCR data using resampling methods. Comput Biol Med. 2012;42(4):438–445. doi: 10.1016/j.compbiomed.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 57.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim D, et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Altschul SF, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phanstiel DH, Boyle AP, Araya CL, Snyder MP. Sushi.R: Flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics. 2014;30(19):2808–2810. doi: 10.1093/bioinformatics/btu379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer; New York: 2009. [Google Scholar]