

Abstract
It is well known that hydroxyurea can cause pancytopaenia secondary to bone marrow suppression, which is reversible with short-term discontinuation of the therapy. However, it is important to note that bone marrow suppressive effects caused by hydroxyurea could be easily potentiated in patients with sickle cell anaemia complicated by chronic kidney disease (CKD). We present a case of a Jehovah's Witness with sickle cell anaemia, who developed severe bone marrow suppression due to the combined effects of hydroxyurea and CKD, resulting in a prolonged recovery period after discontinuation of hydroxyurea.
Background
Hydroxyurea and long-term transfusion therapy are the currently accepted agents as disease-modifying therapies in sickle cell disease (SCD).1 Hydroxyurea, a ribonucleotide reductase inhibitor, is well-tolerated, safe and efficacious in the management of SCD.2 It is used to prevent acute and chronic complications of SCD1 and to improve survival.3 Since hydroxyurea is myelosuppressive, the occurrence of cytopenias requiring temporary discontinuation of therapy is expected in most patients with SCD on hydroxyurea.4 In addition, severe myelotoxicity related to hydroxyurea is seen more commonly in patients with sickle cell anaemia, complicated by nephropathy, unless the dose is adjusted, because hydroxyurea is partly eliminated in an unchanged form by renal excretion.5
Case presentation
A 48-year-old African-American man with sickle cell anaemia (Haemoglobin SS) presented to the hospital with a 1-day history of generalised body pain, which was typical of his usual painful vaso-occlusive crisis. He had three similar episodes of painful vaso-occlusive crisis over the past 1 year. He was a Jehovah’s Witness and so refused to receive blood transfusion, as it is forbidden in that religion. His medical history was significant for stage 4 chronic kidney disease (CKD) secondary to sickle cell nephropathy, a recent episode of unprovoked pulmonary embolism and cholelithiasis. Physical examination on presentation revealed marked conjunctival pallor and a functional murmur at the pulmonic area. Admission laboratory tests were noted as haemoglobin of 7.7 g/dL (baseline haemoglobin of 7–8 g/dL), white cell count (WCC) 4.7×103 cells/µL, platelet count 130×103/µL and serum creatinine 3.2 mg/dL (282.9 µmol/L on International Standard of Units conversion; estimated glomerular filtration rate (eGFR) of 25 mL/min/1.73 m2). The patient was managed with hydration, adequate pain control with hydromorphone and incentive spirometry. Hydroxyurea 2000 mg/day (30 mg/kg/day), warfarin and subcutaneous injection of erythropoietin were continued.
During the hospital stay, the patient's haemoglobin gradually trended down to 2.7 g/dL. Owing to his persistent refusal to accept a blood transfusion and severe anaemia, paediatric tubes were used for blood collection to minimise sampling-associated blood loss. Hydroxyurea was discontinued on day 21 when the haemoglobin went down from 4.3 to 4 g/dL. Owing to severely low haemoglobin, warfarin was also stopped, and an inferior vena cava filter was placed on day 21. There was no active or occult bleeding throughout the hospital stay.
Investigations
Peripheral blood smear examination demonstrated macrocytosis and occasional sickled red blood cells (RBCs) with unremarkable findings on WCCs and platelet counts. Anaemia work up revealed findings consistent with anaemia of kidney disease in combination with chronic inflammation. Trends of haemoglobin, WCC and reticulocyte counts are illustrated in figure 1. Haemoglobin electrophoresis revealed Haemoglobin S 76.3%, Haemoglobin A2 2.2% and Haemoglobin F 21.5%. Throughout the hospital stay, the patient had platelet counts ranging from 130 to 160×103/µL, lactate dehydrogenase 280–500 U/L (normal range 125–220 U/L), haptoglobin <8 mg/dL (normal range 14–250 mg/dL) and mild elevation indirect bilirubin to 2 mg/dL (remaining tests for hepatic function were within the reference ranges). Mean corpuscular volume (MCV) was 118 fL, but measurement for serum levels of vitamin B12 was 693 pg/mL (reference range 213–816 pg/mL) and folic acid was 18.3 ng/mL (normal value >5.4 ng/mL). The patient's eGFR was stable, ranging from 23 to 27 mL/min/1.73 m2 during this hospitalisation.
Figure 1.
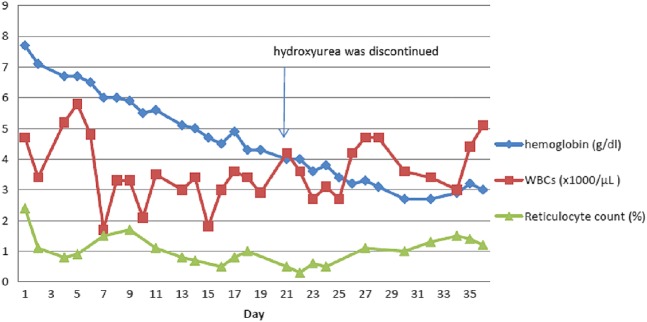
Timeline of haemoglobin, white cell count and reticulocyte count over the period of hospitalisation.
Bone marrow biopsy (figure 2) was performed on day 22 due to unusually low haemoglobin. The biopsy findings were consistent with ineffective erythropoiesis due to maturation defect likely secondary to hydroxyurea. Parvovirus IgG was positive but IgM was negative.
Figure 2.
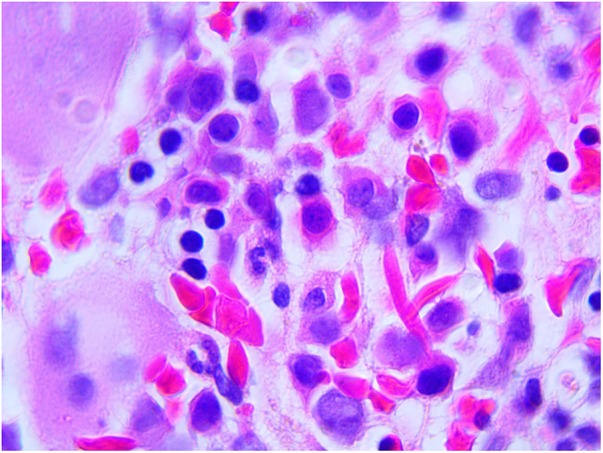
Bone marrow biopsy reveals maturation arrest (Wright-Giemsa, original magnification ×100).
Differential diagnosis
Megaloblastic macrocytic anaemia, which results from inhibition of DNA synthesis required for production of RBCs, can be seen in vitamin B12 deficiency, folic acid deficiency and pernicious anaemia, and can also be caused by chemotherapeutic agents that interfere with DNA metabolism, such as hydroxyurea, methotrexate, cyclophosphamide, etc; in addition, it can result from inherited disorders of DNA metabolism including Lesch-Nyhan syndrome and thiamine-responsive megaloblastic anaemia.6 In our patient, the cause of severe megaloblastic macrocytic anaemia was most likely secondary to the use of hydroxyurea.
Outcome and follow-up
Although the hydroxyurea was discontinued for 2 weeks before the patient signed out against medical advice, there was only minimal improvement in his haemoglobin (3 g/dL). He was off hydroxyurea for approximately 3 weeks after he left the hospital (off for a total duration of 5 weeks). He was then restarted with a lower dose of hydroxyurea and was noted to have a follow-up haemoglobin of 7.2 g/dL on hydroxyurea 1500 mg/day (22 mg/kg/day).
Discussion
SCD is a form of hereditary haemolytic anaemia caused by homozygosity in the sickle cell mutation (Haemoglobin S) or by a compound heterozygote for sickle haemoglobin and β-thalassaemia, haemoglobin C and other less common β-globin mutations.7 There are about 90 000 to 100 000 cases in the USA, with incidence of 1 in 500 newborns of African heritage.8 Approximately 12% of patients with SCD have chronic renal failure, which is associated with significant morbidity and mortality.9 Use of hydroxyurea may be beneficial to prevent the progression of sickle cell nephropathy10 by decreasing the degree of proteinuria,11 which is perhaps correlated with increased Haemoglobin F.12 Hydroxyurea reduces the production of RBCs with predominant Haemoglobin S by inhibiting ribonucleotide reductase, with subsequent compensatory increase in RBC with Haemoglobin F formation by activating the γ- gene.4 Moreover, release of nitric oxide induced by hydroxyurea increases fetal haemoglobin production and causes local vasodilation.13 Increasing the concentration of fetal haemoglobin is the primary effect of hydroxyurea, with resultant decrease in frequency of vaso-occlusive crisis.4 There is a possible but unproven risk of carcinogenicity, such as leucaemia,4 with long-term use of hydroxyurea. However, it is important to note that there are studies suggesting uncertain risk of carcinogenicity attributable to hydroxyurea.14 15
Although there is some degree of hydroxyurea-induced myelosuppression, the haemoglobin level increases slightly on hydroxyurea treatment by decreasing the degree of haemolysis. Even though hydroxyurea is safe for most patients with SCD, caution should be taken in managing patients with nephropathy because of increased myelotoxicity. As was seen in our patient, myelosuppressive effects may continue (haemoglobin dropped further) even after stopping hydroxyurea. The expert panel1 on SCD recommends starting hydroxyurea at 5–10 mg/kg of body weight per day for patients with CKD (usual starting dose is 15 mg/kg/day). Hydroxyurea should be stopped temporarily if absolute neutrophil count is <2000/μL, platelet count <80 000/μL, haemoglobin level <4.5 g/dL or the absolute reticulocyte count <80 000/μL.4 Despite the recommendation to discontinue hydroxyurea temporarily when haemoglobin is <4.5 g/dL, it is important to consider early discontinuation of hydroxyurea for a Jehovah's Witness patient who also has CKD.
When the counts fall to very low range (myelosuppression), stop hydroxyurea temporarily and monitor cell counts weekly. After recovery of cell counts, reintroduce hydroxyurea at dose reduction by 5 mg/kg/day from the prior dosage, and increase by 5 mg/kg/day every 8 weeks until maximum tolerated dose (MTD) is achieved.1 Blood counts can be monitored every 2–3 months after achieving a stable MTD; the usual maximum dose of hydroxyurea is 35 mg/kg/day. MCV and fetal haemoglobin levels can be used to evaluate laboratory response to hydroxyurea; however, failure to increase in MCV, fetal haemoglobin, or both, is not an indication to stop therapy.1 Long-term hydroxyurea therapy is indicated for patients who have a clinical response.1
Learning points.
The use of hydroxyurea may prevent the progression of sickle cell nephropathy that complicates sickle cell disease (SCD).
The myelotoxic effect of hydroxyurea is increased in patients with impaired renal function.
Frequent, closer monitoring of cell counts and timely adjustment of hydroxyurea dosage are crucial in patient with SCD who have renal insufficiency, to avoid severe bone marrow suppression.
Higher peripheral-blood thresholds for discontinuation of hydroxyurea should be considered when managing Jehovah's Witness SCD patients with CKD.
Footnotes
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Yawn BP, Buchanan GR, Afenyi-Annan AN et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. 10.1001/jama.2014.10517 [DOI] [PubMed] [Google Scholar]
- 2.Ware RE, Aygun B. Advances in the use of hydroxyurea. Hematology Am Soc Hematol Educ Program 2009:62–9. 10.1001/asheducation-2009.1.62 [DOI] [PubMed] [Google Scholar]
- 3.Steinberg MH, Barton F, Castro O et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment . JAMA 2003;289:1645–51. 10.1001/jama.289.13.1645 [DOI] [PubMed] [Google Scholar]
- 4.Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med 2008;358:1362–9. 10.1056/NEJMct0708272 [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez GI, Kuhn JG, Weiss GR et al. A bioavailability and pharmacokinetic study of oral and intravenous hydroxyurea. Blood 1998;911533.–. [PubMed] [Google Scholar]
- 6.Lee G, Wintrobe M. Clinical hematology. 9th edn Philadelphia: Lea & Febiger, 1993. [Google Scholar]
- 7.Litchtman M, Kipps T, Williams WJ et al. Williams hematology. 7th edn McGRAW-HILL, 2006. [Google Scholar]
- 8.US Centers for Disease Control and Prevention. Sickle cell disease: data and statistics. http://www.cdc.gov/ncbddd/sicklecell/data.html (accessed 8 Mar 2014).
- 9.Powars DR, Chan LS, Hiti A et al. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84:363–76. 10.1097/01.md.0000189089.45003.52 [DOI] [PubMed] [Google Scholar]
- 10.Alvarez O, Miller ST, Wang WC et al. Effect of hydroxyurea treatment on renal function parameters: results from the multi-center placebo-controlled BABY HUG clinical trial for infants with sickle cell anemia. Pediatr Blood Cancer 2012;59:668–74. 10.1002/pbc.24100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silva Junior GB, Vieira AP, Couto Bem AX et al. Proteinuria in adults with sickle-cell disease: the role of hydroxycarbamide (hydroxyurea) as a protective agent. Int J Clin Pharm 2014;36:766–70. 10.1007/s11096-014-9955-4 [DOI] [PubMed] [Google Scholar]
- 12.Emokpae MA, Onaiwu MA. Association of proteinuria with fetal hemoglobin in adult sickle cell anemia. Niger J Exp Clin Biosci 2014;2:120–4. 10.4103/2348-0149.144853 [DOI] [Google Scholar]
- 13.King SB. Nitric oxide production from hydroxyurea. Free Radic Biol Med 2004;37:737–44. 10.1016/j.freeradbiomed.2004.02.073 [DOI] [PubMed] [Google Scholar]
- 14.McGann PT, Howard TA, Flanagan JM et al. Chromosome damage and repair in children with sickle cell anaemia and long-term hydroxycarbamide exposure. Br J Haematol 2011;154:134–40. 10.1111/j.1365-2141.2011.08698.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finazzi G, Caruso V, Marchioli R et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70. 10.1182/blood-2004-09-3426 [DOI] [PubMed] [Google Scholar]