

Abstract
Krabbe disease is a rare autosomal recessive leucodystrophy, with <5% of the cases having an adolescent-onset form. A 30-year-old woman with a history of a subacute episode of gait impairment at 14 years of age, and mild spastic paraparesis since then, was followed with an initial diagnosis of multiple sclerosis. After 10 years of slow disease progression without response to treatment, the initial diagnosis was reviewed, and an extensive metabolic work up revealed decreased activity of galactocerebrosidase. Genetic testing of the GALC gene proved the diagnosis of Krabbe disease and found a novel mutation. This case highlights the value of a critical eye in the initial differential diagnosis, mainly in the presence of atypical findings.
Background
Krabbe disease, also known as globoid cell leucodystrophy (OMIM #245200) is a rare (1/100 000) autosomal recessive lysosomal storage disease caused by a deficiency of galactocerebrosidase, which causes progressive demyelination of the central and peripheral nervous system.1 The adolescent-onset form is very rare, accounting for <5% of the cases, and includes patients with their first neurological symptoms occurring between 10 and 20 years of age.2 It has been mainly described in a few small case reports, with heterogeneous phenotypes and, usually, a slow progression of symptoms.3 4 The most common clinical features are dysarthria, pyramidal tract dysfunction and peripheral neuropathy, which lead to include other demyelinating disorders in the differential diagnosis. We present a case of adolescent-onset Krabbe disease with an initial diagnosis of multiple sclerosis (MS). After more than 10 years, the initial diagnosis was reviewed and an alternative cause was considered.
Case presentation
A 14-year-old Portuguese girl had a subacute episode of asymmetric weakness in the inferior limbs, with subsequent spasticity and gait impairment, without any other symptoms. Her medical and family history was unremarkable and her parents were non-consanguineous. At 30 years of age, she was referred to our neurology department for the first time, and neurological examination revealed mild asymmetric spastic paraparesis, patellar and ankle clonus, and bilateral extensor plantar responses. Also evident was a bilateral optic pallor, a spastic gait, no superficial or deep sensory loss and a negative Romberg's sign. Brain MRI revealed extensive areas of T2 and fluid-attenuated inversion recovery hyperintensity in the semiovale and posterior periventricular white matter, consistent with demyelination, and no infratentorial or medullary lesions. An IgG oligoclonal band pattern was restricted to the cerebrospinal fluid. Arylsulfatase enzymatic activity testing for metachromatic leucodystrophy returned normal results. Somatosensory-evoked potentials (EPs) of the lower limbs showed symmetrical bilateral delayed latencies, with normal upper limbs and visual EPs, and normal electromyography and nerve conduction studies. Given the clinical symptoms (retrospective reference of a subacute attack lasting more than 24 h), the paraclinical evidence of the lesions, obtained by EPs, and the presence of cerebrospinal fluid oligoclonal bands, the diagnosis of laboratory-supported definite MS was made based on the Poser criteria.5 Despite several therapeutic attempts with steroids and immunosuppressors, follow-up revealed no benefit. Paraparesis evolved a few years later to tetraparesis with mild involvement of the upper limbs and appearance of spastic dysarthria. The patient did not develop cerebellar ataxia or peripheral abnormalities (clinical or electromyographic). Subsequent serial MRIs showed no evolution of the existing lesions and no appearance of new lesions. In 2002, when the patient was 40 years of age, and after the publication of the 2001 McDonald criteria,6 a diagnosis re-evaluation was carried out. All the previous examinations were repeated, with similar results. After the review of the new MRI, which showed a relatively symmetric extensive bilateral demyelination (figure 1) with pyramidal tract involvement (figure 2), a metabolic panel and enzymatic studies for metabolic diseases were performed, revealing decreased activity of galactocerebrosidase (0.52 nmoL/h/mg in leucocytes and 0.72 nmoL/h/mg in fibroblasts). Finally, genetic testing of the GALC gene found two heterozygous mutations, del30kb and p.L293F (the latter being a novel mutation), confirming the diagnosis of Krabbe disease.
Figure 1.
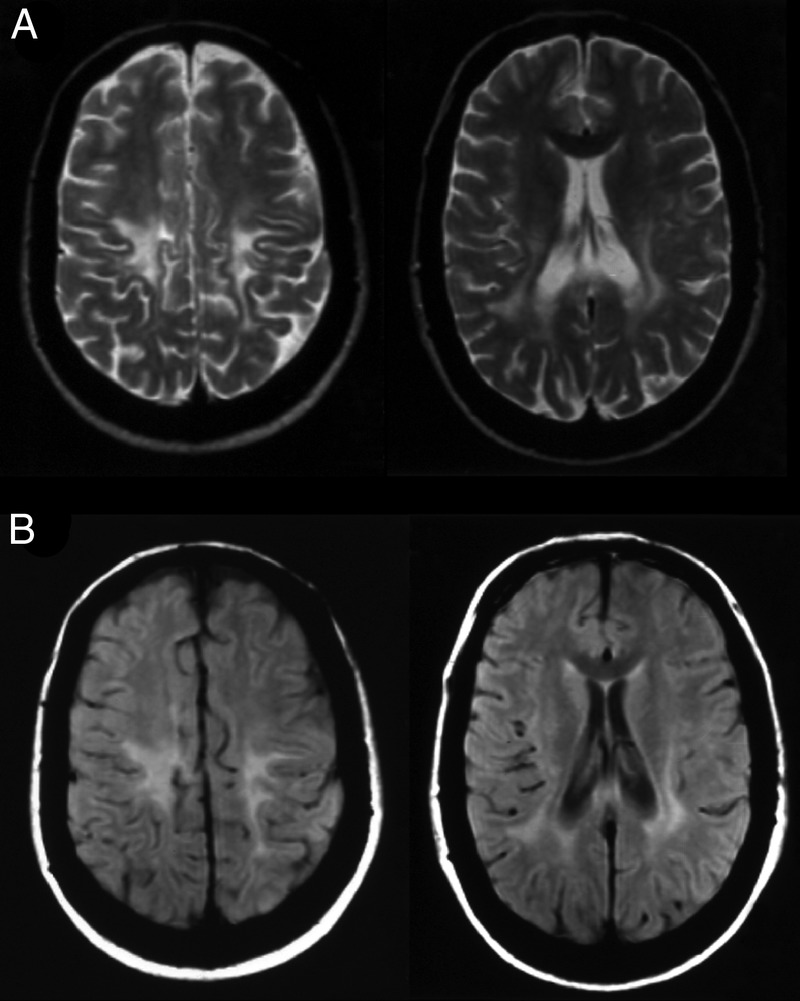
Axial T2-weighted (A) and axial fluid-attenuated inversion recovery-weighted (B) images showing posterior periventricular and deep white matter hyperintensity, consistent with demyelination.
Figure 2.
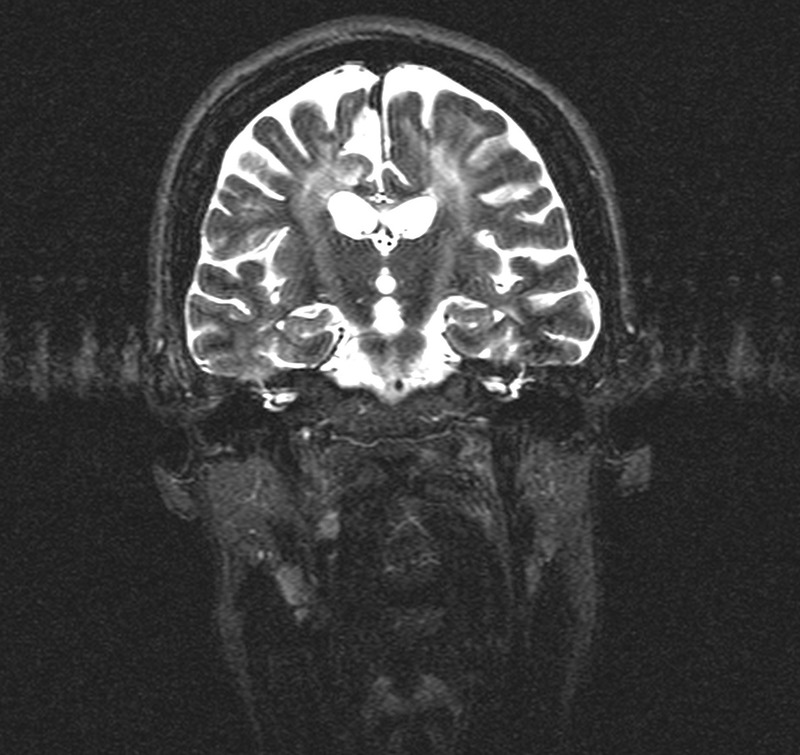
Coronal T2-weighted image showing bilateral symmetric areas of T2 hyperintensity in the corticospinal tracts.
Outcome and follow-up
After the diagnosis, the patient maintained a slow progression of her symptoms, and the therapeutic approach was based on physiotherapy and symptomatic medication. The possibility of haematopoietic cell transplantation was discussed but the patient refused. At the time of writing, the patient has tetraparesis with severe lower limb spasticity, generalised hyper-reflexia, dysarthria and dysphonia. The symptoms seem to have stabilised, and the patient maintains a socially active life despite her clinical limitations and limited degree of dependence.
Discussion
During the first evaluation of the patient, a misdiagnosis of MS was made based on the Poser criteria. Although the patient met the necessary criteria, a broader differential diagnosis should have been made, including autoimmune or genetically determined conditions. The initial presentation provided some clues to consider an alternative to MS, namely the age at presentation, the atypical demyelination pattern in the MRI (symmetric posterior white matter hyperintensity along the corticospinal tracts), and the symmetric and pure pyramidal symptoms and signs on neurological examination.
The incidence of adolescent-onset Krabbe disease appears to be low, with only a few cases reported.2
Krabbe disease has a heterogeneous phenotype, and the patient reported here had the common presenting symptoms in most reports: limb weakness and gait disturbance.4 7 8 However, there was no peripheral neuropathy, a common but inconsistent finding in adolescent and adult Krabbe patients.4 9 The semiovale and posterior periventricular locations of the lesions in the MRI were compatible with adolescent-onset and adult-onset Krabbe disease,10 representing preferential pyramidal tract demyelination.
Genetic studies have shown that the most common mutation in adolescent-onset and adult-onset Krabbe disease is the del30kb associated with a 502C>T polymorphism.4 The patient had two heterozygous mutations, del30kb and p.L293F, and, at the time of writing this report, the latter mutation has not been described in adolescent-onset Krabbe disease.
In conclusion, this case depicts the importance of a critical eye even when a patient meets the diagnostic criteria for a disease, and that other causes have to be reconsidered in the presence of some atypical clues, because a correct diagnosis can avoid an inaccurate management approach or even iatrogenesis.
Learning points.
Krabbe disease and other genetically determined leucodystrophies should be considered in the differential diagnosis of progressive paraparesis, particularly in young patients with typical brain MRI findings.
A metabolic panel can be useful in those cases.
The adolescent-onset form of Krabbe disease is very rare and has been described mainly in isolated case reports.
Footnotes
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat 1997;10: 268–79. [DOI] [PubMed] [Google Scholar]
- 2.Abdelhalim AN, Alberico RA, Barczykowski AL et al. Patterns of magnetic resonance imaging abnormalities in symptomatic patients with Krabbe disease correspond to phenotype. Pediatr Neurol 2014;50:127–34. 10.1016/j.pediatrneurol.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 3.Duffner PK, Barczykowski A, Kay DM et al. Later onset phenotypes of Krabbe disease: results of the world-wide registry. Pediatr Neurol 2012;46: 298–306. 10.1016/j.pediatrneurol.2012.02.023 [DOI] [PubMed] [Google Scholar]
- 4.Debs R, Froissart R, Aubourg P et al. Krabbe disease in adults: phenotypic and genotypic update from a series of 11 cases and a review. J Inherit Metab Dis 2013;36:859–68. 10.1007/s10545-012-9560-4 [DOI] [PubMed] [Google Scholar]
- 5.Poser CM, Paty DW, Scheinberg L et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 1983;13:227–31. 10.1002/ana.410130302 [DOI] [PubMed] [Google Scholar]
- 6.McDonald WI, Compston A, Edan G et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol 2001;50:121–7. 10.1002/ana.1032 [DOI] [PubMed] [Google Scholar]
- 7.Tappino B, Biancheri R, Mort M et al. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum Mutat 2010;31:E1894–914. 10.1002/humu.21367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tokushige S, Sonoo T, Maekawa R et al. Isolated pyramidal tract impairment in the central nervous system of adult-onset Krabbe disease with novel mutations in the GALC gene. Brain Dev 2013;35:579–81. 10.1016/j.braindev.2012.08.004 [DOI] [PubMed] [Google Scholar]
- 9.Malandrini A, D'Eramo C, Palmeri S et al. Peripheral neuropathy in late-onset Krabbe disease: report of three cases. Neurol Sci 2013;34:79–83. 10.1007/s10072-012-0956-6 [DOI] [PubMed] [Google Scholar]
- 10.Ahmed RM, Murphy E, Davagnanam I et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry 2014;85:770–81. 10.1136/jnnp-2013-305888 [DOI] [PubMed] [Google Scholar]