

Abstract
The G protein-coupled receptor Smoothened (Smo) is the signal transducer of the developmentally- and therapeutically-relevant Hedgehog pathway. Although recent structural analyses have advanced our understanding of Smo biology, a number of questions remain. Chief among them are the identity of its natural ligand, the regulatory processes controlling its activation, and the mechanisms by which it signals to downstream effectors. This review will discuss recent discoveries from multiple model systems that have set the stage for solving these mysteries. We focus on the roles of distinct Smo functional domains, post-translational modifications and trafficking, and conclude by discussing their contributions to signal output.
Keywords: Smoothened, Hedgehog, GPCR, canonical signaling, noncanonical signaling
The Hedgehog Signaling Pathway
The evolutionarily conserved Hedgehog (Hh) signaling pathway provides instructional cues that drive morphogenesis during development and contribute to post-developmental tissue homeostasis and wound healing. Consistent with these important functionalities, corruption of Hh signaling can lead to developmental disorders and cancer [1, 2]. Ectopic Hh signaling is implicated in a range of tumor types including medulloblastoma, basal cell carcinoma (BCC), breast cancer and lung cancer and, as such, has become an attractive therapeutic target [3].
The Hh pathway is controlled by two membrane proteins, Patched (Ptch), the Hh receptor, and Smoothened (Smo), a Class F G protein-coupled receptor (GPCR) which transduces the activation signal to downstream pathway components (Figure 1A and [4-7]). In the absence of Hh ligand, Ptch inhibits signaling by Smo. Binding of Hh to Ptch triggers its internalization and degradation, thereby allowing Smo to accumulate and signal to intracellular effectors [8]. In most cases, signaling culminates in activation of the Gli family of transcription factors and induction of Hh target gene expression [9, 10] (Figure 1B). Many of these important discoveries were first revealed using the Drosophila model system, and subsequently found to be conserved to vertebrates. However, clear divergence exists, and is highlighted by i) the requirement for a sensory organelle termed the primary cilium as an organizing center for vertebrate signaling, and ii) the ability of vertebrate Smo to signal through distinct G protein-dependent and independent routes. Whereas Drosophila have single Hh, Ptch and Gli genes, vertebrates encode three ligands (Sonic Hh, Desert Hh and Indian Hh), two Ptch receptors (Ptch1 and Ptch2) and three Gli transcription factors (Gli1, 2 and 3) [11]. For the purposes of this review, we will not specify distinct ligands, receptors or Glis, and instead refer to them collectively as Hh, Ptch and Gli.
Figure 1. Regulation of the Smoothened GPCR.
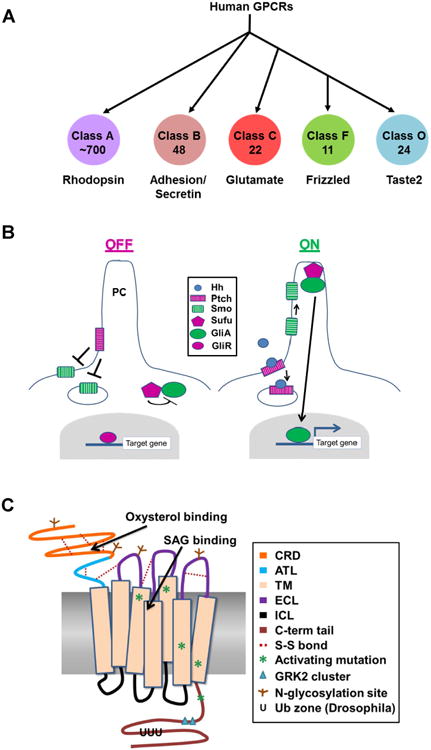
A GPCR classes. Proteins of the GPCR superfamily, which possess 7 TM domains, are grouped into classes based upon their functionality and homology. The diagram indicates human GPCR classes, and denotes the number of proteins found in each class. The largest amount of structural data is available for Class A, the Rhodopsin family. Much of what is known about GPCR topology and activation has been deduced from Class A structural analyses. Smo is a member of the Class F Frizzled family. Classes D (mating pheromone receptors) and E (cAMP receptors) are not found in humans. B. The core Hh pathway. In the absence of Hh ligand, the Hh receptor Ptch localizes to the base of the primary cilium (PC) and inhibits Smo ciliary entry and signaling. Sufu inhibits Gli activator while Gli repressor keeps target genes silenced. Hh binding triggers Ptch internalization, which allows Smo to traffic to the tip of the PC where it signals for Sufu to release Gli activator. Gli activator then translocates to the nucleus to activate target gene expression. Many additional proteins contribute to regulation of these processes. Only the core pathway components are shown here. C. Smoothened functional domains. The cartoon illustrates the distinct Smo functional domains, ligand binding pockets, post-translational modifications and location of known activating mutations. CRD – cysteine rich domain; ATL – amino terminal linker; TM – transmembrane helix; ECL – extracellular loop; ICL – intracellular loop; C-term tail – carboxyl-terminal tail.
Herein, we summarize recent discoveries that provide insight into how Smo signaling is regulated to control Hh pathway output. We focus on Smo protein functional domains, post-translational modifications and trafficking, which work together to control its signaling. We conclude by highlighting important open questions that, when answered, will significantly enhance understanding of Hh pathway biology.
Functional Domains and Post-translational Modifications
Smo possesses a set of stereotypical GPCR functional domains including an amino-terminal cysteine rich domain (CRD), three extracellular and three intracellular loops (ECL and ICL), seven transmembrane domains (TM) and an intracellular carboxyl-terminal tail which can undergo a range of post-translational modifications [6, 7] (Figure 1C). Until recently, the majority of studies interrogating Smo were focused on its carboxyl tail. However, recent structural insights have helped to reveal essential regulatory contributions by other functional domains.
Cysteine Rich Domain
The CRD is localized to the extracellular amino-terminal region, and shares homology with the CRD of the Wnt receptor Frizzled (Fz) [6, 12, 13]. The structure of the CRDs of zebrafish and Drosophila Smo proteins were recently solved, and revealed a conserved hydrophobic groove structurally similar to the pocket through which Fz binds the palmityl modification of Wnt [12-14]. Structural and mutagenesis studies revealed that the oxysterol 20(S)-OHC, an established positive regulator of Smo, could bind the vertebrate CRD pocket, and that the inhibitory glucocorticoid budesonide could bind the Drosophila pocket [12, 13, 15, 16]. Mutagenesis of residues predicted to coordinate the binding of these molecules to the CRD of both vertebrate and Drosophila proteins attenuated Smo-mediated pathway activation in response to 20(S)-OHC or Hh [12, 13, 15]. These studies support that the Smo CRD pocket plays an essential and conserved role in signaling, potentially through functioning as a physiologic ligand binding site (Box 1).
Text Box 1. Does Patched control an endogenous Smoothened ligand?
Although the series of events leading to Smo activation are becoming clearer, questions regarding the existence and identity of its natural ligand remain a roadblock in our understanding of its biology. The inherent druggability of Smo by both synthetic and naturally occurring compounds reveals its ability to be influenced by small molecule ligands [17, 18, 22, 85]. Although the essential endogenous ligand has yet to be identified, a significant amount of evidence supports that it exists, and that Ptch likely governs Smo through controlling availability of or access to it. Consistent with this notion, Ptch does not physically interact with Smo [86]. Instead, it inhibits Smo in a catalytic manner, with one molecule of Ptch estimated to be capable of regulating up to fifty molecules of Smo [87]. This evidence, combined with Ptch having a requisite sterol sensing domain and sharing homology with an ancient class of bacterial transporters, has led to a model whereby Ptch functions by pumping a sterol-like Smo regulator [88]. Whether the regulator is an agonist or antagonist has yet to be established, but based upon available information, either scenario is plausible. That Smo is constitutively active in the absence of Ptch suggests that the molecule could be an antagonist, and that removal of Ptch removes it from Smo [89]. However, the discovery of oxysterols as CRD-binding positive modulators of Smo suggests that the molecule in question could be an agonist, and that Ptch attenuates signaling by sequestering it away from Smo or diminishing its cellular concentration ([90, 91] and Figure 3).
Other data suggest that, rather than controlling a Smo-binding compound, Ptch may influence its activity by controlling the local membrane environment. This alternate model is supported by evidence that lipid kinases and phosphatases are capable of impacting Smo signal output [92]. Loss of the kinase controlling PI4P production in Drosophila prevents accumulation of Smo on the plasma membrane, thereby attenuating Hh pathway activity. Conversely, loss of Ptch results in accumulation of PI4P, leading to the hypothesis that it may function indirectly through lipid kinases to control Smo. In this scenario, a local membrane environment rich in PI4P would foster a conformation or localization of Smo permissive for signaling, and a membrane devoid of PI4P would not. Similar processes may be active in vertebrate signaling, as the lipid kinase PI3K-C2α has been found to impact Smo ciliary accumulation, an essential first step in signal induction [61].
Extracellular Loops and Transmembrane Domains
The CRD is connected to the extracellular loop (EC) domains through a disulfide bridge between an amino-terminal linker domain (ATL) and the first extracellular loop (EC1) [17]. Structures for the CRD and the EC loop/7TM domains were solved independently, and as such, a composite structure illustrating their precise interface is not available. However, the crystal structure of the 7TM region revealed multiple stabilizing interactions between the ATL and EC loops 1 and 3, suggesting that proper positioning of the CRD relative to the EC loops is important for Smo regulation [17]. Consistent with this notion, disruption of the ATL-EC1 disulfide bridge triggers ER retention and ablates signaling of the Drosophila protein [12]. Collaboration between the amino terminus and the EC loops is further supported by structural data demonstrating that the ATL assists the ECLs in coordinating binding of SAG1.5, a potent Smo agonist which docks into a second ligand binding pocket within the 7TM core [18]. Surprisingly, binding of this potent agonist to the ATL/7TM pocket did not induce pronounced TM shifts or remodeling of the intracellular domains of the protein, as would be predicted based upon activating shifts common to Class A GPCRs (Figure 1A and [18]). The most notable intracellular change between antagonist and agonist bound Smo occurred in intracellular loop 1 (IC1), which shifted inward towards W535 of TM7 in the SAG-bound conformation. IC1 communication with W535 may be a trigger for Smo activation, as W535L mutation is a known driver of oncogenic Smo signaling ([19, 20] and Box 2).
Text Box 2. Smoothened in cancer.
Inappropriate Hh signaling is implicated in cancer and can occur through Smo mutation [93]. Documented mutations fall into one of three categories depending upon their impact on signaling. The first are passenger mutations, which can occur in a number of tumor types, but have minimal impact on signaling or ligand binding [94, 95]. Conversely, activating mutations have profound effects on Smo activity, and can serve as driving mutations in basal cell carcinoma and medulloblastoma [94]. These mutations affect the TM domains or carboxyl-tail, and are thought to induce conformation shifts that drive ligand-independent signaling. Examples include V321M, L412F, F460L, W535L and R562Q [20, 95]. Of these, W535L (SmoM2) is most studied. This lesion is localized to the seventh transmembrane helix and impacts a region commonly considered to be a trigger point for activation of Class A GPCRs [17, 95]. V231M, L412 and F460L affect TM domains 3, 5 and 6, which are essential pivot regions for activation of stereotypical GPCRs, and likely activate Smo by mimicking its ligand-induced active conformation [94]. The carboxyl-terminal tail mutation R562Q is adjacent to the first conserved GRK2 phosphorylation cluster, and could potentially trigger ligand-independent signaling through altering conformation of this important regulatory domain [43].
The final category of Smo mutants associated with cancer affect its 7TM ligand binding pocket. These alterations are not sufficient to drive ligand-independent Smo signaling, but do impact binding of Smo inhibitors to promote drug resistance, and are therefore clinically relevant [96]. A number of amino acid changes in TM domains have been identified in relapsed BCCs following treatment with the Smo antagonist Vismodegib [96]. Recurrent tumors harboring drug-resistant mutations are thought to result primarily from outgrowth of drug-resistant subclones from the original tumor, but in some cases may occur through de novo mutation [96]. Avenues being explored to overcome or minimize such resistance are to target downstream of Smo, or to develop Smo inhibitors that function non-competitively with Vismodegib [97]. Candidates for the latter include Intraconazole, which targets an as yet unidentified domain, and select glucocorticoids and oxysterol binding inhibitors, which occlude the 20(S)-OHC binding pocket in the CRD [16, 98, 99]. Given the importance of trafficking in Smo signaling, another potential point of intervention could be to target processes influencing its loading into the secretory route or trafficking to the primary cilium. ER stress may be one such process, as stress modulators block exit of SmoM2 from the ER, leading to its elimination through the ER associated degradation pathway [100].
Structures of antagonist-bound Smo have been solved for a number of small molecule inhibitors, and show that Smo harbors multiple distinct docking sites at varying depths within its 7TM core [18]. Many of the 7TM binding compounds engage both EC2 and EC3, indicating an important role for EC loop conformation in coordinating Smo ligand binding [17, 18, 21, 22]. Accordingly, mutations that disrupt the conserved EC loop-stabilizing disulfide bridge between TM3 and EC2 have profound effects on Drosophila Smo, ranging from weak hypomorphic activity to robust ligand-independent signaling [23, 24]. Intriguingly, the outward positioning of EC2 relative to the 7TM core is reminiscent of Class A peptide binding GPCRs [17]. It is therefore tempting to speculate that Smo may be capable of binding larger peptide or lipid ligands that bridge the ligand binding pockets of the CRD and the EC/TM core ([12]and Figure 2). Notably, both the amino terminal domain and the EC loops of Smo harbor N-linked glycan modifications [25]. N-glycosylation can affect ligand binding and/or responsiveness for a number of GPCR superfamily members [26-29]. The close proximity of glycan acceptor sites to the CRD and 7TM pockets suggests that this may also be true for Smo.
Figure 2. A Model for the Stages of Smo Activation.
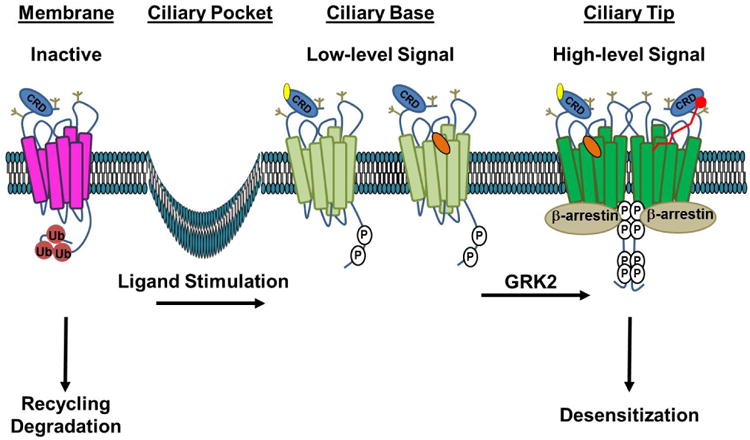
In the absence of ligand, inactive Smo (magenta) is ubiquitin modified (Ub, red), which signals for its internalization and membrane recycling or degradation. Stimulation with Hh ligand or a direct agonist such as a CRD-binding oxysterol (yellow) or the 7TM binding-compound SAG (orange) induces tail phosphorylation (P, white), leading to a conformation shift (light green) that promotes entry and accumulation in the primary cilium. Higher-order phosphorylation of the membrane-proximal GRK clusters leads to complete opening of the tail, which is permissive for Smo oligomerization and β-arrestin recruitment. This conformation (dark green) drives high-level signaling and correlates with increased accumulation of Smo in the tip of the primary cilium. We speculate that higher order signaling could be achieved by occupation of both ligand binding pockets by distinct ligands (left half of dimer) or by a larger peptide or fatty acid ligand bridging the two pockets (red ligand, right half of dimer). Recruitment of β-arrestin to the hyper-active conformation drives high-level signal propagation and eventual Smo desensitization. Extracellular N-linked glycans, which may contribute to extracellular conformation and/or ligand binding, are indicated in tan.
Intracellular Loops
Upon activation by an extracellular signal, GPCRs will typically interact with heterotrimeric G-proteins through their IC loop domains, and with arrestins through their carboxyl-terminal tails [30, 31]. Smo has been demonstrated to utilize Gαi heterotrimeric G proteins, but regulation of their direct binding has not yet been characterized [32, 33]. With the exception of the structural data discussed above, little is known about functionality of the Smo IC loops. However, modest functional data support a role for this domain in Smo signaling. An ethyl methanesulfonate mutagenesis screen in Drosophila identified a residue in IC3, that when mutated, attenuates Smo activity [24]. Of note, this residue is localized to the carboxyl-terminal end of IC3, which is a crucial domain for G protein coupling by stereotypical GPCRs [34]. The importance of the IC domain in regulating Smo signaling is further supported by an in vitro study in cultured cell lines demonstrating that peptide mimics of IC2 and IC3 suppress Smo-induced cancer cell proliferation [35]. Taken together, these studies suggest that the IC loops might influence Smo activity by interacting with essential regulatory or effector proteins. Additional studies are needed to identify these effectors and to determine how they are recruited to and activated by Smo.
Carboxyl Tail
Although the intracellular carboxyl-terminal tails of fly and vertebrate Smo proteins differ significantly in length and sequence homology, aspects of their functionality are conserved. For example, the tails of both fly and vertebrate proteins undergo hyper-phosphorylation in response to pathway activation. Multisite phosphorylation triggers a conformation change that enhances plasma membrane localization of the fly protein and prompts the vertebrate protein to enrich in the primary cilium, a sensory organelle that projects from the basal body [36, 37]. The extent of Smo phosphorylation and activity directly correlate with the level of Hh ligand stimulation a cell receives [38]. It has therefore been suggested that differential Smo tail phosphorylation is a mechanism by which the Hh morphogen gradient is interpreted by target cells exposed to different thresholds of ligand. This so-called “Hh phosphorylation code,” which is written by a number of different kinases and phosphatases was recently reviewed elsewhere. [38]. Only G protein-coupled receptor kinase (GRK) phosphorylation will be discussed here.
For most GPCRs, phosphorylation of the intracellular tail by GRK, serves to recruit arrestin molecules which can both initiate GPCR removal from the plasma membrane, and provide a scaffold upon which G protein-independent signaling is propagated or maintained [39]. Smo conforms to this model, as it is phosphorylated by GRK2 and engages β-arrestin in both flies and vertebrates [40-42]. The essential GRK2 phosphorylation sites are localized to two clusters in a well conserved membrane proximal region of the tail. Phosphorylation at these clusters is required for high-level Smo signaling, and in vertebrates, serves to recruit β-arrestin to initiate its internalization through clathrin-coated pits [43-45]. Although phosphorylation by the Drosophila GRK2 homolog does trigger the eventual internalization of Smo, it does not appear to be through recruitment of the Drosophila arrestin Kurtz [42]. Also, whereas vertebrate signaling appears to require β-arrestin recruitment to Smo for signal propagation, Kurtz loss does not have an obvious effect on Smo protein levels or signaling activity [40, 42, 46]. This could indicate that an additional Smo binding partner present in Drosophila functions semi-redundantly with Kurtz to control internalization and/or signaling by active Smo. Consistent with this hypothesis, the dynamin protein Shibire has been shown to contribute to membrane recycling of Drosophila Smo [47].
Whereas phosphorylation has a positive correlation with cell surface accumulation and signaling, ubiquitination has suppressive effects. The carboxyl tail of Drosophila Smo harbors multiple ubiquitin (Ub) acceptor sites [48, 49]. Mono- and poly-ubiquitination at these residues occurs in the absence of Hh, and contributes to recycling of the inactive protein. Removal of Ub occurs through direct engagement of the deubiquitinating enzyme USP8 by the carboxyl tail [48, 49]. Curiously, USP8 association with Smo is not affected by Hh stimulation or tail phosphorylation, suggesting that deubiquitination by this enzyme may not be directly regulated by Hh [49]. Further studies are needed to clarify whether changes in Ub modification represent an early step in Smo accumulation and signal induction, and to test whether these events are conserved in vertebrates.
Trafficking and Localization
Smo signaling is dependent upon its trafficking to an active subcellular localization, the plasma membrane in flies and the primary cilium (PC) in vertebrates (Figure 2 and [8, 50]). Relocalization to these sites typically occurs within ∼20 minutes of agonist stimulation, and represents an important regulatory event in initiation of Smo signal transduction. For the purposes of this review, we will focus on trafficking of vertebrate Smo, which must reach the PC to interact with a specific set of ciliary proteins to mount a robust signal.
Although it is clear that ciliary entry is tightly controlled, the precise mechanisms governing Smo ciliary trafficking are not yet fully established. The PC is separated from the plasma membrane through two checkpoints, the ciliary pockets and the transition zone, which function in tandem to gate movement of lipids and proteins into and out of the organelle [51, 52]. The Hh pathway has evolved to exploit this trafficking as a means of regulating Smo [52]. The Hh receptor Ptch contains a ciliary localization sequence (CLS) within its carboxyl-terminal tail, which unlocks its transport into the PC. Ciliary localization of Ptch is essential for Smo inhibition; deletion of its CLS prevents not only its PC localization, but also its ability to silence Smo [53]. Smo also contains a CLS, but unlike Ptch, accumulates in the PC only upon ligand stimulation [54]. For Smo to accumulate in the PC in a ligand-independent manner, Ptch must be disabled [55]. This suggests that enrichment of Smo within the PC is a key step in pathway activation, and that Ptch functions, at least in part, to regulate this essential trafficking event. How Ptch might target Smo to govern this event has not yet been established, but a number of models have been proposed (Figure 3).
Figure 3. Models of Ptch-mediated Smo Regulation.
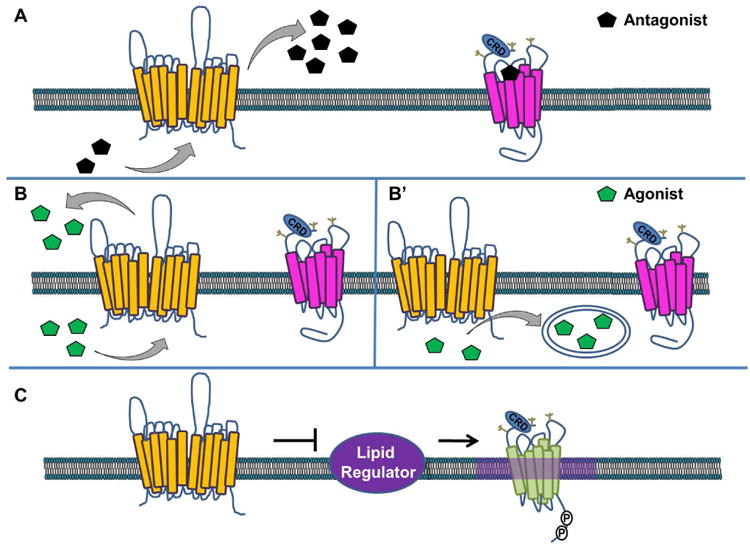
Several models for Ptch-mediated Smo regulation have been hypothesized. In A, Ptch (yellow) pumps an antagonist (black), which binds to cell-surface localized Smo to block its accumulation and signaling (magenta, inactive Smo). In B, Ptch pumps a Smo agonist (green) out of the cell (B) or sequesters it in vesicles (B′) where it is unable to bind and activate Smo. In C, Ptch controls activity of a lipid regulator to block accumulation of a specific lipid species in membrane microdomains that are permissive for Smo signaling (purple haze, active Smo shown in green).
Given the inverse ciliary relationship of Ptch and Smo, an early model postulated that Ptch functioned as a physical barrier for Smo PC entry [55]. However, the effects of chemical agonists (SAG or 20-OHC) and antagonists (cyclopamine) that directly bind Smo call this view into question. Binding of these small molecules induces Smo conformation shifts that are sufficient for its ciliary accumulation in a Hh- and Ptch-independent manner [56]. This, taken with the recent observation that removal of Ptch from the PC is not strictly required for Smo entry, suggests that while Ptch does present a barrier for Smo, it is not a physical one [53].
Smo ciliary entry is thought to occur through one of two routes, either laterally through the ciliary pockets or internally through recycling endosomes [57]. Once inside the PC Smo can diffuse freely within the ciliary membrane, but shows a preference for specific ciliary compartments depending upon its activation state. Inactive Smo spends more time confined to the base of the PC while active Smo enriches at the ciliary tip [58]. Although it is not yet known how Smo movement is controlled under these differing conditions, studies from Drosophila suggest that modifications of membrane lipids could have an effect. In flies, phosphatidylinositol-4-phosphate (PI4P) has been documented to influence Smo membrane trafficking and cell surface accumulation [59]. PIs can influence membrane trafficking by marking different cellular membranes; PI4P is found predominately in the Golgi and phosphatidylinositol 4,5-bisphosphate (PI4,5P2) at the plasma membrane [60]. It is therefore tempting to speculate that one mechanism by which Ptch controls Smo trafficking is through modulating the concentration or availability of specific phosphatidylinositol phosphates (PIP) in the membrane. Consistent with this hypothesis, the phosphatidylinositol 3-kinase PI3K-C2α was recently shown to produce an unidentified PI3P species at the base of the PC [61]. Deletion of PI3K-C2α prevented Smo ciliary accumulation, suggesting that a ciliary PI3P pool may be important for Smo stabilization or PC trafficking. Further support for lipids in Smo modulation was revealed by recent work demonstrating that Ptch utilizes endocannabinoids from lipoprotein particles to destabilize Smo [62-65]. This suggests that multiple lipid species could potentially be exploited by Ptch to govern Smo stabilization and activity. Whether the putative lipid modulator functions to alter the local Smo membrane environment, or serves as direct Smo agonist or antagonist is not yet known, and represents one of the most important unanswered questions in Smo biology.
Smo Signaling
The regulatory processes discussed above reveal that some aspects of Smo regulation are stereotypical and some unique. This duality is echoed by the signaling behavior of vertebrate Smo, which is capable of activating distinct canonical and noncanonical signaling routes. Whereas Drosophila Smo appears to govern a sole canonical signal that culminates in activation of a Gli transcription factor, vertebrate Smo can activate two independent signaling arms [11]. The canonical signal is dedicated to regulation of Hh target gene activation through Gli transcriptional effectors, and is thought to occur in a G protein-independent manner. Conversely, the noncanonical route utilizes Gαi to regulate cellular processes including metabolism, proliferation, Ca2+ flux and migration [66-69].
Canonical Signaling
Smo-mediated Gli activation is dependent upon Smo entering the PC in Hh-stimulated cells. However, how the activation signal is transmitted from ciliary Smo to the Gli effectors is not yet clear. It is, however, clear that regulation of Gli phosphorylation by cyclic AMP (cAMP)-dependent protein kinase (PKA) is paramount to controlling transcriptional repressor and activator formation [70-72]. Signaling from Smo to PKA and Sufu, a negative regulator of Gli activity, occurs in the PC and is dependent upon Smo interaction with the Ellis van Creveld Syndrome (Evc2) ciliary complex [73]. Evc2 associates with activated Smo in the EvC zone, which is situated adjacent to the transition zone at the ciliary base [73]. The complex is anchored to the EvC zone through association with EVCAB7 and IQ Motif Containing E (IQCE) ciliary proteins, which were recently revealed to be positive regulators of canonical signal output [74]. Canonical Smo signaling in the PC bifurcates to differentially regulate Gli activator and repressor activity, and correlates with Smo binding distinct functional partners in different ciliary localizations. Whereas Smo associated with the ciliary motor Kif7 can regulate Gli repressor from the ciliary tip, it is thought to signal from the Evc2 complex in the EvC zone to control ciliary entry and activation of Gli [74-78]. Consistent with this notion, the basal body localized protein Dlg5 was recently shown to assist Smo in Gli activation, but not in attenuating Gli repressor formation [75]. Despite all this new information, the precise mechanism(s) by which Smo signals through its multiple ciliary partners to regulate Gli remains enigmatic.
Noncanonical Smo signaling
Despite Smo having the 7TM topology that is characteristic of the GPCR superfamily, whether or not it couples with a G protein to induce downstream signaling has remained controversial. Early support for Smo-mediated Gαi activation was obtained in zebrafish, which showed phenotypes similar to Hh loss of function following pertussis toxin treatment [79]. Subsequent work in frog melanophores revealed that cAMP-regulated pigment distribution could be modulated by Smo over-expression, further supporting a Smo-Gαi link [80]. However, since those initial studies, a number of additional genetic and cell biologic studies failed to reach a consensus on Smo-Gαi coupling, and as such, Smo was commonly referred to as an “atypical” GPCR-like protein [33, 66, 67, 81-84].
The first biochemical evidence to support Smo-Gαi coupling was obtained by examining Gα-GTPγS binding in membranes isolated from Sf9 cells expressing murine Smo along with distinct classes of Gαβγ heterotrimeric G proteins [32]. This study revealed Smo to stimulate heterotrimeric G proteins of the Gαi family, but not to couple with Gαs, Gαq or G12/13 family members. Subsequent work in Drosophila provided the first genetic evidence supporting Smo-Gαi signaling, demonstrating that Gαi was required for low-threshold Hh target gene induction [33]. Genetic evidence connecting Gαi with Hh target gene expression in vertebrate systems has not yet been reported, but evidence supporting Gαi function in a vertebrate-specific noncanonical Smo signaling axis has begun to mount. This noncanonical signal does not regulate Gli, but instead modulates Ca2+ flux, RhoA and Rac activation and Warburg-like metabolism via Gαi (Box 1 and [66, 67, 69]).
How Smo selects between canonical and noncanonical routes is not known. However, the existence of at least two distinct Smo ligand binding pockets raises the possibility that selective bias may occur. We postulate that allosteric ligand engagement by the individual pockets could alter the Smo conformation or ciliary localization such that one effector route is favored over the other. The ability of sub-cellular localization to influence signal output is suggested by Smo mutants that lack their ciliary localization sequence. These mutants fail to activate Gli, but maintain their ability to couple with Gαi, suggesting that ciliary localization may not be strictly required for the noncanonical pathway [66]. It is also possible that differential use of the numerous phosphorylation, ubiquitination and/or N-linked glycosylation sites on Smo could modulate effector selection to achieve tissue or developmental context specific regulation of canonical or noncanonical signal output. This hypothesis is supported by a recent report demonstrating that a murine Smo mutant lacking its N-glycan modifications can signal potently to Gli, but cannot induce Gαi [25].
Concluding Remarks
Despite the new information regarding Smo regulation and signaling highlighted here, many important unanswered questions remain (see Outstanding Questions). Paramount among them is the identification of the natural Smo ligand, and an understanding of how Ptch controls Smo access or response to it. This knowledge will enhance our understanding of the canonical signal, and will likely also reveal novel aspects of noncanonical signaling. The physiological relevance of the noncanonical pathway is only beginning to be examined. As such, its contribution to developmental patterning has not been established, nor has its importance in disease. These open questions are likely to become the focus of new avenues of study as interest in Smo-Gαi signaling expands. Furthermore, the recent emergence of Smo inhibitors in the clinic necessitates an understanding of noncanonical signaling if we are to predict and mitigate on-target, Gli-independent effects that could lead to unfavorable outcomes.
Box 1. G proteins in Hedgehog Signaling at a Glance.
Canonical Signaling
Noncanonical Signaling
-
Smo couples with Gαi to govern:
-
– Cell migration
Hh-induces fibroblast migration by activating Rac1 and RhoA [66].
-
– Tubulogenesis
Hh-induces tubulogenesis of endothelial cells in 3D culture through activation of Rac1 and RhoA [84].
-
– Ca2+ flux
Hh-induces transient Ca2+ spikes in embryonic spinal neurons via Phospholipase C and inositol triphosphate [67].
-
– Proliferation
Gαi enhances Hh-induced proliferation of cerebellar granular neuronal precursors in culture [81].
-
– Warburg-like metabolism
Smo signals through Gαi, Ca2+ and AMP-activated protein kinase (Ampk) to reprogram metabolism in muscle and brown fat [69].
-
Trends Box.
Recent structural information reveals how ligands bind and regulate Smoothened.
Smoothened signaling is regulated by its trafficking within the primary cilium.
Smoothened can signal through G protein-dependent and independent mechanisms.
Outstanding Questions Box.
How does Ptch regulate Smo?
Does Smo have an endogenous ligand?
Do the CRD and 7TM ligand binding pockets collaborate in Smo activation?
How is Smo trafficking through the ciliary transition zone regulated?
How is selection between canonical and noncanonical signal output achieved?
What is the contribution of noncanonical Smo signaling to development?
Is noncanonical signaling corrupted in disease?
Acknowledgments
We thank members of the Ogden lab for thoughtful discussion and comments on the manuscript. This work was supported by 5R01GM101087 (S.K.O.) and by ALSAC of St. Jude Children's Research Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–87. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 2.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15(6):801–12. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins DJ, Goetz JA, Yuan Z, Stegman MA. Inhibitors of the Hedgehog Signal Transduction Pathway. Curr Cancer Ther Rev. 2005;1:227–288. [Google Scholar]
- 4.Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin JP, Stevens RC, Vriend G, Gloriam DE. Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol Sci. 2015;36(1):22–31. doi: 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, Noll M, Hooper JE, de Sauvage F, Rosenthal A. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384(6605):129–34. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 6.Alcedo J, Ayzenzon M, Von Ohlen T, Noll M, Hooper JE. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell. 1996;86(2):221–32. doi: 10.1016/s0092-8674(00)80094-x. [DOI] [PubMed] [Google Scholar]
- 7.van den Heuvel M, Ingham PW. smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature. 1996;382(6591):547–51. doi: 10.1038/382547a0. [DOI] [PubMed] [Google Scholar]
- 8.Denef N, Neubuser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell. 2000;102(4):521–31. doi: 10.1016/s0092-8674(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 9.Von Ohlen T, Hooper JE. Hedgehog signaling regulates transcription through Gli/Ci binding sites in the wingless enhancer. Mech Dev. 1997;68(1-2):149–56. doi: 10.1016/s0925-4773(97)00150-0. [DOI] [PubMed] [Google Scholar]
- 10.Alexandre C, Jacinto A, Ingham PW. Transcriptional activation of hedgehog target genes in Drosophila is mediated directly by the cubitus interruptus protein, a member of the GLI family of zinc finger DNA-binding proteins. Genes Dev. 1996;10(16):2003–13. doi: 10.1101/gad.10.16.2003. [DOI] [PubMed] [Google Scholar]
- 11.Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal. 2012;5(246):re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rana R, Carroll CE, Lee HJ, Bao J, Marada S, Grace CR, Guibao CD, Ogden SK, Zheng JJ. Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nat Commun. 2013;4:2965. doi: 10.1038/ncomms3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, Ingham PW, Covey DF, Siebold C, Rohatgi R. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. Elife. 2013;2:e01340. doi: 10.7554/eLife.01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science. 2012;337(6090):59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Myers BR, Sever N, Chong YC, Kim J, Belani JD, Rychnovsky S, Bazan JF, Beachy PA. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell. 2013;26(4):346–57. doi: 10.1016/j.devcel.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nedelcu D, Liu J, Xu Y, Jao C, Salic A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat Chem Biol. 2013;9(9):557–64. doi: 10.1038/nchembio.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497(7449):338–43. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Wu H, Evron T, Vardy E, Han GW, Huang XP, Hufeisen SJ, Mangano TJ, Urban DJ, Katritch V, Cherezov V, Caron MG, Roth BL, Stevens RC. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat Commun. 2014;5:4355. doi: 10.1038/ncomms5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam CW, Xie J, To KF, Ng HK, Lee KC, Yuen NW, Lim PL, Chan LY, Tong SF, McCormick F. A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene. 1999;18(3):833–6. doi: 10.1038/sj.onc.1202360. [DOI] [PubMed] [Google Scholar]
- 20.Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, Jr, de Sauvage FJ. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–2. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 21.McCabe JM, Leahy DJ. Smoothened goes molecular: new pieces in the hedgehog signaling puzzle. J Biol Chem. 2015;290(6):3500–7. doi: 10.1074/jbc.R114.617936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharpe HJ, Wang W, Hannoush RN, de Sauvage FJ. Regulation of the oncoprotein Smoothened by small molecules. Nat Chem Biol. 2015;11(4):246–55. doi: 10.1038/nchembio.1776. [DOI] [PubMed] [Google Scholar]
- 23.Carroll CE, Marada S, Stewart DP, Ouyang JX, Ogden SK. The extracellular loops of Smoothened play a regulatory role in control of Hedgehog pathway activation. Development. 2012;139(3):612–21. doi: 10.1242/dev.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakano Y, Nystedt S, Shivdasani AA, Strutt H, Thomas C, Ingham PW. Functional domains and sub-cellular distribution of the Hedgehog transducing protein Smoothened in Drosophila. Mech Dev. 2004;121(6):507–18. doi: 10.1016/j.mod.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 25.Marada S, Navarro G, Truong A, Stewart DP, Arensdorf AM, Nachtergaele S, Angelats E, Opferman JT, Rohatgi R, McCormick PJ, Ogden SK. Functional Divergence in the Role of N-Linked Glycosylation in Smoothened Signaling. PLoS Genet. 2015;11(8):e1005473. doi: 10.1371/journal.pgen.1005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soto AG, Trejo J. N-linked glycosylation of protease-activated receptor-1 second extracellular loop: a critical determinant for ligand-induced receptor activation and internalization. J Biol Chem. 2010;285(24):18781–93. doi: 10.1074/jbc.M110.111088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanctot PM, Leclerc PC, Clement M, Auger-Messier M, Escher E, Leduc R, Guillemette G. Importance of N-glycosylation positioning for cell-surface expression, targeting, affinity and quality control of the human AT1 receptor. Biochem J. 2005;390(Pt 1):367–76. doi: 10.1042/BJ20050189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He J, Xu J, Castleberry AM, Lau AG, Hall RA. Glycosylation of beta(1)-adrenergic receptors regulates receptor surface expression and dimerization. Biochem Biophys Res Commun. 2002;297(3):565–72. doi: 10.1016/s0006-291x(02)02259-3. [DOI] [PubMed] [Google Scholar]
- 29.Wheatley M, Hawtin SR. Glycosylation of G-protein-coupled receptors for hormones central to normal reproductive functioning: its occurrence and role. Hum Reprod Update. 1999;5(4):356–64. doi: 10.1093/humupd/5.4.356. [DOI] [PubMed] [Google Scholar]
- 30.Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang CR, Gu LL, Shan JM, Chen X, Hanna R, Choi M, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL, Jr, Kobilka BK, Skiniotis G, Lefkowitz RJ. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512(7513):218–22. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreira IS. Structural features of the G-protein/GPCR interactions. Biochim Biophys Acta. 2014;1840(1):16–33. doi: 10.1016/j.bbagen.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 32.Riobo NA, Saucy B, Dilizio C, Manning DR. Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci U S A. 2006;103(33):12607–12. doi: 10.1073/pnas.0600880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogden SK, Fei DL, Schilling NS, Ahmed YF, Hwa J, Robbins DJ. G protein Galphai functions immediately downstream of Smoothened in Hedgehog signalling. Nature. 2008;456(7224):967–70. doi: 10.1038/nature07459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang M, Zhao X, Chen HC, Catt KJ, Hunyady L. Activation of the AT1 angiotensin receptor is dependent on adjacent apolar residues in the carboxyl terminus of the third cytoplasmic loop. J Biol Chem. 2000;275(21):15782–8. doi: 10.1074/jbc.M000198200. [DOI] [PubMed] [Google Scholar]
- 35.Remsberg JR, Lou H, Tarasov SG, Dean M, Tarasova NI. Structural analogues of smoothened intracellular loops as potent inhibitors of Hedgehog pathway and cancer cell growth. J Med Chem. 2007;50(18):4534–8. doi: 10.1021/jm0705657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Tong C, Jiang J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature. 2007;450(7167):252–8. doi: 10.1038/nature06225. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Sasai N, Ma G, Yue T, Jia J, Briscoe J, Jiang J. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011;9(6):e1001083. doi: 10.1371/journal.pbio.1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Jiang J. Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. 2013;23(2):186–200. doi: 10.1038/cr.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shenoy SK, Lefkowitz RJ. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32(9):521–33. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng S, Maier D, Neubueser D, Hipfner DR. Regulation of smoothened by Drosophila G-protein-coupled receptor kinases. Dev Biol. 2010;337(1):99–109. doi: 10.1016/j.ydbio.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molnar C, Holguin H, Mayor F, Jr, Ruiz-Gomez A, de Celis JF. The G protein-coupled receptor regulatory kinase GPRK2 participates in Hedgehog signaling in Drosophila. Proc Natl Acad Sci U S A. 2007;104(19):7963–8. doi: 10.1073/pnas.0702374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molnar C, Ruiz-Gomez A, Martin M, Rojo-Berciano S, Mayor F, de Celis JF. Role of the Drosophila non-visual ss-arrestin kurtz in hedgehog signalling. PLoS Genet. 2011;7(3):e1001335. doi: 10.1371/journal.pgen.1001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maier D, Cheng S, Faubert D, Hipfner DR. A Broadly Conserved G-Protein-Coupled Receptor Kinase Phosphorylation Mechanism Controls Drosophila Smoothened Activity. PLoS Genet. 2014;10(7):e1004399. doi: 10.1371/journal.pgen.1004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meloni AR, Fralish GB, Kelly P, Salahpour A, Chen JK, Wechsler Reya RJ, Lefkowitz RJ, Caron MG. Smoothened signal transduction is promoted by G protein-coupled receptor kinase 2. Mol Cell Biol. 2006;26(20):7550–60. doi: 10.1128/MCB.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W, Ren XR, Nelson CD, Barak LS, Chen JK, Beachy PA, de Sauvage F, Lefkowitz RJ. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science. 2004;306(5705):2257–60. doi: 10.1126/science.1104135. [DOI] [PubMed] [Google Scholar]
- 46.Kovacs JJ, Whalen EJ, Liu R, Xiao K, Kim J, Chen M, Wang J, Chen W, Lefkowitz RJ. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science. 2008;320(5884):1777–81. doi: 10.1126/science.1157983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia R, Jia H, Fan J, Liu Y, Jia J. USP8 promotes smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PLoS Biol. 2012;10(1):e1001238. doi: 10.1371/journal.pbio.1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan J, Jiang K, Liu Y, Jia J. Hrs promotes ubiquitination and mediates endosomal trafficking of smoothened in Drosophila hedgehog signaling. PLoS One. 2013;8(11):e79021. doi: 10.1371/journal.pone.0079021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li S, Chen Y, Shi Q, Yue T, Wang B, Jiang J. Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLoS Biol. 2012;10(1):e1001239. doi: 10.1371/journal.pbio.1001239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102(32):11325–30. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohatgi R, Snell WJ. The ciliary membrane. Curr Opin Cell Biol. 2010;22(4):541–6. doi: 10.1016/j.ceb.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goetz SC, Ocbina PJ, Anderson KV. The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol. 2009;94:199–222. doi: 10.1016/S0091-679X(08)94010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J, EY CH, Brigui A, Plessis A, Beachy PA, Zheng X. The role of ciliary trafficking in Hedgehog receptor signaling. Sci Signal. 2015;8(379):ra55. doi: 10.1126/scisignal.aaa5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–21. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 55.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–6. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 56.Wilson CW, Chen MH, Chuang PT. Smoothened adopts multiple active and inactive conformations capable of trafficking to the primary cilium. PLoS One. 2009;4(4):e5182. doi: 10.1371/journal.pone.0005182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Milenkovic L, Scott MP, Rohatgi R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J Cell Biol. 2009;187(3):365–74. doi: 10.1083/jcb.200907126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Milenkovic L, Weiss LE, Yoon J, Roth TL, Su YS, Sahl SJ, Scott MP, Moerner WE. Single-molecule imaging of Hedgehog pathway protein Smoothened in primary cilia reveals binding events regulated by Patched1. Proc Natl Acad Sci U S A. 2015;112(27):8320–5. doi: 10.1073/pnas.1510094112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yavari A, Nagaraj R, Owusu-Ansah E, Folick A, Ngo K, Hillman T, Call G, Rohatgi R, Scott MP, Banerjee U. Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev Cell. 2010;19(1):54–65. doi: 10.1016/j.devcel.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santiago-Tirado FH, Bretscher A. Membrane-trafficking sorting hubs: cooperation between PI4P and small GTPases at the trans-Golgi network. Trends Cell Biol. 2011;21(9):515–25. doi: 10.1016/j.tcb.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franco I, Gulluni F, Campa CC, Costa C, Margaria JP, Ciraolo E, Martini M, Monteyne D, De Luca E, Germena G, Posor Y, Maffucci T, Marengo S, Haucke V, Falasca M, Perez-Morga D, Boletta A, Merlo GR, Hirsch E. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev Cell. 2014;28(6):647–58. doi: 10.1016/j.devcel.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khaliullina H, Bilgin M, Sampaio JL, Shevchenko A, Eaton S. Endocannabinoids are conserved inhibitors of the Hedgehog pathway. Proc Natl Acad Sci U S A. 2015;112(11):3415–20. doi: 10.1073/pnas.1416463112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khaliullina H, Panakova D, Eugster C, Riedel F, Carvalho M, Eaton S. Patched regulates Smoothened trafficking using lipoprotein-derived lipids. Development. 2009;136(24):4111–21. doi: 10.1242/dev.041392. [DOI] [PubMed] [Google Scholar]
- 64.Palm W, Sampaio JL, Brankatschk M, Carvalho M, Mahmoud A, Shevchenko A, Eaton S. Lipoproteins in Drosophila melanogaster--assembly, function, and influence on tissue lipid composition. PLoS Genet. 2012;8(7):e1002828. doi: 10.1371/journal.pgen.1002828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palm W, Swierczynska MM, Kumari V, Ehrhart-Bornstein M, Bornstein SR, Eaton S. Secretion and signaling activities of lipoprotein-associated hedgehog and non-sterol-modified hedgehog in flies and mammals. PLoS Biol. 2013;11(3):e1001505. doi: 10.1371/journal.pbio.1001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polizio AH, Chinchilla P, Chen L, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi Proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem. 2011 doi: 10.1074/jbc.M110.197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Belgacem YH, Borodinsky LN. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc Natl Acad Sci U S A. 2011;108(11):4482–7. doi: 10.1073/pnas.1018217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Teperino R, Aberger F, Esterbauer H, Riobo N, Pospisilik JA. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin Cell Dev Biol. 2014 doi: 10.1016/j.semcdb.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, Jaeger C, Kammerer B, Winter L, Wiche G, Dalgaard K, Selvaraj M, Gaster M, Lee-Young RS, Febbraio MA, Knauf C, Cani PD, Aberger F, Penninger JM, Pospisilik JA, Esterbauer H. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151(2):414–26. doi: 10.1016/j.cell.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 70.Niewiadomski P, Kong JH, Ahrends R, Ma Y, Humke EW, Khan S, Teruel MN, Novitch BG, Rohatgi R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014;6(1):168–81. doi: 10.1016/j.celrep.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010;24(7):670–82. doi: 10.1101/gad.1902910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191(2):415–28. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dorn KV, Hughes CE, Rohatgi R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev Cell. 2012;23(4):823–35. doi: 10.1016/j.devcel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pusapati GV, Hughes CE, Dorn KV, Zhang D, Sugianto P, Aravind L, Rohatgi R. EFCAB7 and IQCE regulate hedgehog signaling by tethering the EVC-EVC2 complex to the base of primary cilia. Dev Cell. 2014;28(5):483–96. doi: 10.1016/j.devcel.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chong YC, Mann RK, Zhao C, Kato M, Beachy PA. Bifurcating action of Smoothened in Hedgehog signaling is mediated by Dlg5. Genes Dev. 2015;29(3):262–76. doi: 10.1101/gad.252676.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Endoh-Yamagami S, Evangelista M, Wilson D, Wen X, Theunissen JW, Phamluong K, Davis M, Scales SJ, Solloway MJ, de Sauvage FJ, Peterson AS. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr Biol. 2009;19(15):1320–6. doi: 10.1016/j.cub.2009.06.046. [DOI] [PubMed] [Google Scholar]
- 77.Cheung HO, Zhang X, Ribeiro A, Mo R, Makino S, Puviindran V, Law KK, Briscoe J, Hui CC. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci Signal. 2009;2(76):ra29. doi: 10.1126/scisignal.2000405. [DOI] [PubMed] [Google Scholar]
- 78.Liem KF, Jr, He M, Ocbina PJ, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci U S A. 2009;106(32):13377–82. doi: 10.1073/pnas.0906944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hammerschmidt M, McMahon AP. The effect of pertussis toxin on zebrafish development: a possible role for inhibitory G-proteins in hedgehog signaling. Dev Biol. 1998;194(2):166–71. doi: 10.1006/dbio.1997.8796. [DOI] [PubMed] [Google Scholar]
- 80.DeCamp DL, Thompson TM, de Sauvage FJ, Lerner MR. Smoothened activates Galphai-mediated signaling in frog melanophores. J Biol Chem. 2000;275(34):26322–7. doi: 10.1074/jbc.M004055200. [DOI] [PubMed] [Google Scholar]
- 81.Barzi M, Kostrz D, Menendez A, Pons S. Sonic Hedgehog-induced proliferation requires specific Galpha inhibitory proteins. J Biol Chem. 2011;286(10):8067–74. doi: 10.1074/jbc.M110.178772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Low WC, Wang C, Pan Y, Huang XY, Chen JK, Wang B. The decoupling of Smoothened from Galphai proteins has little effect on Gli3 protein processing and Hedgehog-regulated chick neural tube patterning. Dev Biol. 2008;321(1):188–96. doi: 10.1016/j.ydbio.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carbe CJ, Cheng L, Addya S, Gold JI, Gao E, Koch WJ, Riobo NA. Gi Proteins Mediate Activation of the Canonical Hedgehog Pathway in the Myocardium. Am J Physiol Heart Circ Physiol. 2014 doi: 10.1152/ajpheart.00166.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chinchilla P, Xiao L, Kazanietz MG, Riobo NA. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle. 2010;9(3):570–79. doi: 10.4161/cc.9.3.10591. [DOI] [PubMed] [Google Scholar]
- 85.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002;99(22):14071–6. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Y, Struhl G. In vivo evidence that Patched and Smoothened constitute distinct binding and transducing components of a Hedgehog receptor complex. Development. 1998;125(24):4943–8. doi: 10.1242/dev.125.24.4943. [DOI] [PubMed] [Google Scholar]
- 87.Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418(6900):892–7. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- 88.Mukhopadhyay S, Rohatgi R. G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin Cell Dev Biol. 2014 doi: 10.1016/j.semcdb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rohatgi R, Scott MP. Patching the gaps in Hedgehog signalling. Nat Cell Biol. 2007;9(9):1005–9. doi: 10.1038/ncb435. [DOI] [PubMed] [Google Scholar]
- 90.Corcoran RB, Scott MP. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci U S A. 2006;103(22):8408–13. doi: 10.1073/pnas.0602852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, Rohatgi R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat Chem Biol. 2012;8(2):211–20. doi: 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yavari A, Nagaraj R, Owusu-Ansah E, Folick A, Ngo K, Hillman T, Call G, Rohatgi R, Scott MP, Banerjee U. Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev Cell. 19(1):54–65. doi: 10.1016/j.devcel.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barakat MT, Humke EW, Scott MP. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol Med. 2011;16(8):337–48. doi: 10.1016/j.molmed.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Atwood SX, Sarin KY, Li JR, Yao CY, Urman NM, Chang AL, Tang JY, Oro AE. Rolling the Genetic Dice: Neutral and Deleterious Smoothened Mutations in Drug-Resistant Basal Cell Carcinoma. J Invest Dermatol. 2015;135(8):2138–41. doi: 10.1038/jid.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang AL, Oro AE, Tang JY. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):342–53. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, Bourgon R, Tang JY, Sarin KY, Dirix L, Fisher DC, Rudin CM, Sofen H, Migden MR, Yauch RL, de Sauvage FJ. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):327–41. doi: 10.1016/j.ccell.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Long J, Li B, Rodriguez-Blanco J, Pastori C, Volmar CH, Wahlestedt C, Capobianco A, Bai F, Pei XH, Ayad NG, Robbins DJ. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J Biol Chem. 2014;289(51):35494–502. doi: 10.1074/jbc.M114.595348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Kim J, Chen B, King EM, Borodovsky A, Riggins GJ, Epstein EH, Jr, Beachy PA, Rudin CM. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23(1):23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang Y, Davidow L, Arvanites AC, Blanchard J, Lam K, Xu K, Oza V, Yoo JW, Ng JM, Curran T, Rubin LL, McMahon AP. Glucocorticoid compounds modify smoothened localization and hedgehog pathway activity. Chem Biol. 2012;19(8):972–82. doi: 10.1016/j.chembiol.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Marada S, Stewart DP, Bodeen WJ, Han YG, Ogden SK. The unfolded protein response selectively targets active smoothened mutants. Mol Cell Biol. 2013;33(12):2375–87. doi: 10.1128/MCB.01445-12. [DOI] [PMC free article] [PubMed] [Google Scholar]