

Abstract
Bmi-1 is a member of the Polycomb repressor complex 1 that mediates gene silencing by regulating chromatin structure and is indispensable for self-renewal of both normal and cancer stem cells. Despite three decades of research that have elucidated the transcriptional regulation, post-translational modifications and functions of Bmi-1 in regulating the DNA damage response, cellular bioenergetics, and pathologies, the entire potential of a protein with such varied functions remains to be realized. This review attempts to synthesize the current knowledge on Bmi-1 with an emphasis on its role in both normal physiology and cancer. Additionally, since cancer stem cells are emerging as a new paradigm for therapy resistance, the role of Bmi-1 in this perspective is also highlighted. The wide spectrum of malignancies that implicate Bmi-1 as a signature for stemness and oncogenesis also make it a suitable candidate for therapy. Nonetheless, new approaches are vitally needed to further characterize physiological roles of Bmi-1 with the long-term goal of using Bmi-1 as a prognostic marker and a therapeutic target.
Keywords: Aging, Bmi-1, Cancer, Post translational modification, Stem cell
Abbreviations: ASC, adult stem cell; ATM, Ataxia telangiectasia mutated; BASC, bronchioalveolar stem cell; Bmi-1, B-cell specific Moloney murine virus integration site 1; CIC, cancer initiating cell; CSC, cancer stem cell; EMT, epithelial–mesenchymal transition; ESC, embryonic stem cell; HCC, hepatocellular carcinoma; HDACi, histone deacetylase inhibitor; HSC, hematopoietic stem cell; hTERT, human telomerase reverse transcriptase; HTH, helix-turn-helix; ISC, intestinal stem cell; MEC, mammary epithelial cell; NSC, neural stem cell; PcG, polycomb group; PEST, domain rich in proline, glutamic acid, serine, and threonine; PRC, polycomb repressive complex; PTM, post-translational modification; RING, really interesting new gene; Rb, retinoblastoma; TSC, trophectodermal stem cell; UTR, untranslated region; XEN, extraembryonic stem cell
Introduction
In the Eμ-myc transgenic mice, Bmi-1 (B cell-specific Moloney murine leukemia virus integration site 1) was discovered as a frequent target of the Moloney virus insertion, resulting in virally accelerated B-lymphoid tumors, hence its name.1 Since its discovery, Bmi-1 has been implicated in a number of biological functions including development, cell cycle, DNA damage response (DDR), senescence, stem cell, self-renewal and cancer. Recently, Bmi-1 has proven to be of significant clinical interest as it has been noted to be overexpressed in a number of diseases and malignancies. This review will seek to give a basic overview of Bmi-1, its functions, and its potential research and clinical implications.
Bmi-1 protein
The Bmi-1 gene localizes on chromosome 10 (10p11.23) encodes for a 37 kDa protein composed of 326 amino acids.2, 3 Its protein structure is highly evolutionarily conserved, demonstrating considerable homology with the Mel-18 gene—a transcriptional repressor of Bmi-1—in humans and the Posterior Sex Combs (Psc) protein in Drosophila melanogaster.2, 4
The functionality of the Bmi-1 protein is primarily characterized by three regions: a central helix-turn-helix (HTH) domain, a conserved RING finger domain at the N-terminal end, and a carboxyl-terminal PEST-like domain.2, 5, 6 The RING domain of Bmi-1 comprises of a three-stranded β-sheet, two zinc binding loops, and an α-helix.5 The RING domain is required for Bmi-1 to localize to DNA strand breaks, thus critical for its role in DDR.7 Both the RING and HTH motifs are necessary for prevention of senescence in cells.8 Thus, these two regions aid in continuing the replicative life span of cells. The HTH domain is also important in binding Bmi-1 to DNA and providing a scaffold for subsequent protein binding.9 PEST domains are protein regions rich in proline (P), glutamic acid (E), serine (S), and threonine (T) bounded by basic residues that tend to act as proteolytic signals leading to protein degradation10. One study noted that the PEST-like domain of Bmi-1 primarily contained proline and serine residues and that the deletion of this region resulted in an increased half-life of Bmi-1 and promoted the proliferation of cells.11
In addition to the three primary functional regions, the Bmi-1 protein also contains two nuclear localization signals (NLS): NLS1 and NLS2. Of these two, only NLS2 appears to be functional in nuclear localization of Bmi-1 (Fig 1).12
Figure 1.

Bmi-1 Function and Protein Structure. (A) A visual representation of the known roles of Bmi-1. (B) Graphic of the Bmi-1 protein structure. Key protein motifs are indicated along with their relative sizes and locations. (HTH – helix-turn-helix; NLS – nuclear localization signal; PEST – motif rich in proline, glutamic acid, serine, and threonine; RING – really interesting new gene).
PcG functionality
As a polycomb group (PcG) protein, Bmi-1 associates with other PcG partners to function as an epigenetic repressor, remodeling chromatin through histone 2A (H2A) monoubiquitination and subsequent methylation at histones.3 PcG proteins were initially studied in Drosophila melanogaster and identified as transcriptional repressors of Hox genes—homeotic genes that regulate morphogenesis and tissue differentiation.13
Consequently, PcG proteins have been studied in their potential connection to cancer stem cells. Like stem cells in healthy tissues, tumors appear to contain a small subset of cells that have the potential to repopulate and affect transcriptional regulation patterns. Since PcG proteins play a role in transcriptional repression, it is hypothesized that they may be highly involved in stem cell renewal and cancer development.14
There are two multimeric PcG protein complexes: Polycomb repressor complex 1 (PRC1) and Polycomb repressor complex 2 (PRC2).3 As these complexes have been investigated, core functional components have been determined for both families of PcG proteins. In humans, the canonical PRC1 is comprised of Bmi-1, RING1A/B, PCGF, CBX, and HPH, while the core PRC2 is comprised of EZH, SUZ12, and EED.15 (summarized in Table 1). As a part of PRC1, Bmi-1 interacts with RING1B via its own RING domain and enhances the E3 ubiquitin ligase activity to ubiquitinate histone H2A.5 PRC2 operates as a histone transmethylase that mono-, di-, and trimethylates the Lys27 residue of histone H3.16 Traditionally, EED has only been associated with PRC2; however, a recent study suggests that EED plays an important role in both PRC1 and PRC2, and thus may potentially be a key coordinator in transcriptional regulation.17
Table 1.
Components of the PRC1 and PRC2 complexes.
| Protein motif | Function | |
|---|---|---|
| PRC1 | ||
| Bmi-1 Mel-18 CBX2, 6, 7, 8 CBX4 HPH1, 2 RING1A/B SCMH1, SCML2 L3MBTL |
RING domain5 RING domain28 Chromodomain144 Chromodomain144 Zinc finger and SEP domain145 RING domain5 Zinc finger, SAM and MBT domains146 MBT and SPM domains146 |
E3 ubiquitin ligase5 Undetermined Chromatin association,6, 147 Chromatin association and sumolyation E3 ubiquitin ligase5 |
| PRC2 | ||
| EED EZH1, 2 SUZ12 PCL1,2,3 JARID2 AEBP2 RBBP4, 7 YY1 |
WD40 domain17, 148 SET domain149 Zinc finger149 PHD finger, TUDOR147 Zinc finger, ARID domain, JmjC and JmjN147 Zinc finger,147,149 WD40 domain147 Zinc finger150 |
PcG binding and stabilization14 Histone transmethylase144 Histone transmethylase151 Hypothesized147 Hypothesized147 Increases histone Transmethylase activity149, Histone binding147 Recruits PcG proteins to DNA150 |
Listed are the most common proteins that associate as part of PRC1 and PRC2. Also included are the important functional protein motifs and how each protein contributes—if known—to the complex as a whole.
AEBP – adipocyte enhancer-binding protein; ARID – A–T interaction rich domain; CBX – chromobox; EED – embryonic ectoderm development; EZH – enhancer of zeste homolog; HPH – human polyhometic homolog; JARID – Jumonji/ARID containing; L3MBTL – lethal(3)malignant brain tumor-like protein; MBT – malignant brain tumor; PCL – polycomb-like; PHD – plant homodomain; RBBP – retinoblastoma-binding protein; RING – really interesting new gene; SAM – sterile alpha motif; SEP – serine-glutamine-proline; SET – Su(var)3–9, enhancer-of-zeste; SPM – SCM, Ph, and MBT domain; SCMH – sex comb on midleg homolog; SCML – sex comb on midleg-like domain; SUZ – supressor of zeste; TUDOR – domain originally described in Drosophila Tudor protein; WD40 – 40 residue tryptophan and aspartic acid repeat; YY – Yin–Yang.
Mouse models
Murine and human Bmi-1 display a high degree of similarity at the cDNA (92.4%) and at the protein level (98%), making mice the primary model organism for Bmi-1.2 A definitive study conducted by van der Lugt et al, demonstrated that Bmi-1 knockout mice are characterized by a survival rate of only ∼50% by the third day after birth.4 Additionally, knockout mice experienced increased frequency of illness, hematopoietic abnormalities in the liver and bone marrow, lymphoid abnormalities in the thymus and spleen, skeletal defects, ataxic gait, and reduced density in cerebellum and neural layers.4 Hematopoietic cell counts in the Bmi-1 knockout mice were reduced to roughly 30% of wild-type levels and continued to decrease as the mice aged. The majority of thymocytes in the knockout mice were immature, with total thymocyte levels being decreased to below 1%. In vitro, the fetal hematopoietic cells tend to demonstrate considerable growth defects, whereas in vivo the fetal cells developed relatively normally and only displayed severe defects with age. Despite the hematopoietic abnormalities, the red blood cell count and associated blood parameters did not significantly change in the knockout mice.18
A further study using Bmi-1 knockout mice found that reactive oxygen species (ROS) increased in various cell populations, especially thymocytes.19 In this study, the Bmi-1 knockout thymocytes demonstrated diminished oxidative capacity as well as reduced basal mitochondrial oxygen consumption—both of which contributed to an enhanced DNA damage response (DDR).19
An interesting Bmi-1 reporter study found that Bmi-1 is highly expressed in quiescent intestinal stem cells (ISCs). Self-renewal proteins Lgr5 and Bmi-1 were fluorescently tagged within mice ISCs and were studied before and after irradiation. Before irradiation, the Bmi-1 expressing ISCs were characterized as quiescent, while the Lgr5 ISCs were shown to be mitotically active. However, upon irradiation, the Lgr5 ISCs demonstrated susceptibility and died out quickly; in contrast, the Bmi-1 ISCs displayed high resistance to irradiation and proliferated to aid in regeneration. This ISC study suggests that Bmi-1 expression in vivo is highly important within injury response and regeneration of stem cells.20
Transcriptional regulation
Transcription factors
Bmi-1 has a number of transcriptional and post-transcriptional regulators. N-Myc, c-Myc, Sp1, Twist1, FOXM1, E2F-1 and SALL4 have been shown to positively regulate Bmi-1 expression, while KLF4, Mel-18 and histone deacetylase inhibitors (HDACi) have been shown to suppress expression at the transcriptional level.21, 22, 23, 24, 25, 26, 27, 28, 29, 30 N-Myc and c-Myc were shown to directly target and upregulate Bmi-1 mRNA in a dose-dependent manner.21 This mechanism may occur by c-Myc binding to the enhancer box sequence in the Bmi-1 promoter region.28 Zinc finger transcription factor Sp1 was shown to directly interact with and upregulate Bmi-1, possibly by binding to a G/C rich region.30 Twist1 has been shown to upregulate Bmi-1 in head and neck squamous cell carcinomas through binding PRC2 components like EZH2.22 Forkhead box transcriptional factor FOXM1 transcriptionally stimulates genes implicated in cell proliferation, and consequently positively regulates Bmi-1 transcription.23 E2F-1 directly targets both Bmi-1 and MYCN, upregulating both.24 E2F-1 was also shown to regulate PRC2 components and Bmi-1, but no other PRC1 components.24 Zinc finger transcription factor SALL4 positively regulated the Bmi-1 promoter in a dose-dependent manner, possibly through SALL4-induced hypermethylation at Lys4 on histone H3.25 Zinc finger protein KLF4 was shown to bind to and repress Bmi-1 expression in colon cancer.26 Mel-18 reduces Bmi-1 expression by repressing Myc, a positive regulator of Bmi-1.27, 28 HDACi have been shown to repress PRC2 components as well as Bmi-1 expression.29 While the correlations between Bmi-1 transcription and these transcription factors have been characterized, the exact mechanisms and interactions of many of these transcription factors still require further investigation.
Regulation by miRNA
MicroRNAs (miRNAs) are short 21–23 nucleotide RNA sequences that post-transcriptionally regulate gene expression. Bmi-1 expression is repressed by miRNAs in a number of malignancies. We showed that miR-15 and miR-16 directly downregulated expression of Bmi-1 in ovarian cancer.31 Within gastrointestinal cancer, miR-30e* has been identified to suppress Bmi-1.32 miR-135a suppresses Bmi-1 expression in pancreatic ductal adenocarcinoma.33 miR-141 induces senescence in diploid fibroblasts by repressing Bmi-1, EZH2, and consequently, PRC1 and PRC2 activities.34 In melanomas, miR-203 has been shown to suppress Bmi-1 expression.35 And finally, miR-320a was shown to directly target Bmi-1 and suppress its expression in nasopharyngeal carcinoma.36 Together, these reports indicate that depending on the cancer context a variety of miRNAs target the Bmi-1 mRNA, nonetheless suggesting the importance of Bmi-1 in various malignancies.
Post-translational modifications
Post-translational modification (PTM) of Bmi-1 is a relatively under-explored field and is directly associated with the functionality of the protein. Several PTMs of Bmi-1 have been reported, of which sumoylation, phosphorylation, acetylation and ubiquitination are of particular interest. Although the reports related to each such modification are limited and restricted to tissue and/or cancer types, it is interesting to note the diversity in the functional alteration that each such modification induces. The following paragraphs will further discuss how site-specific phosphorylation of Bmi-1 by different kinases, influence its tumorigenicity both positively and negatively.
Acetylation has been shown to influence the nuclear arrangement and function of the Bmi-1. Through time-lapse confocal microscopy, micro-irradiation by UV laser (355 nm), and GFP technology, the dynamics and recruitment of Bmi-1 to UV-damaged chromatin were studied. Bmi-1 was rapidly recruited (half-time t¼15 s), suggesting that Bmi-1 might be required for recognition of UV-damaged sites.37 Histone hyper-acetylation, stimulation by HDACi/TSA, suppression of transcription by actinomycin D, and ATP-depletion were all shown to prevent increased accumulation of Bmi-1 to γH2AX-positive irradiated chromatin.37 While there was no direct evidence of acetylated Bmi-1, this demonstrated that the nuclear arrangement of Bmi-1 was influenced by acetylation and occurred as an early event prior to the recruitment of HPβ to the UV-irradiated chromatin.
CBX4, a component of the PRC1, has also been shown to accumulate at sites of DNA damage as a part of the DDR pathway. A novel role for CBX4 as an early DDR protein involved SUMO conjugation at sites of DNA lesions.38 DNA damage stimulates sumoylation of Bmi-1 by CBX4 at Lys88, which is required for the accumulation of Bmi-1 at DNA damage sites.38 Moreover, CBX4 recruitment to the sites of laser micro-irradiation-induced DNA damage requires PARP activity but does not require H2AX, RNF8, Bmi-1 nor PI-3-related kinases.38 The importance of CBX4 in the DDR was confirmed by the depletion of CBX4, which resulted in decreased cellular resistance to ionizing radiation.38 Both the studies interestingly indicated that a PTM of Bmi-1—either acetylation or sumoylation—is an important prerequisite for recruitment of Bmi-1 to initiate the DDR pathway.
Several groups have simultaneously reported on phosphorylation of Bmi-1. Phosphorylated Bmi-1 at different serine residues modified both the Ink4a/Arf dependent and independent functions. As a PcG protein, Bmi-1 is phosphorylated by 3 pK (MAPKAP kinase 3), which is a convergence point downstream of activated ERK and p38 signaling pathways that have been implicated in differentiation and developmental processes.39In vitro and in vivo experiments—as well as yeast two-hybrid interaction and co-immunoprecipitation—established 3 pK as an interacting partner of PRC1 via Bmi-1.39 Gain of 3 pK activity—either through genetic or pharmacological intervention—resulted in phosphorylation of Bmi-1 and other PcG members, and in their dissociation from chromatin, culminating in de-repression of targets. One such reported Bmi-1 target is the Cdkn2a/Ink4A locus. Cells overexpressing 3 pK showed PcG complex/chromatin dissociation and concomitant de-repression of p14Arf, which was encoded by the Cdkn2a/Ink4A locus. Thus, 3 pK is a candidate regulator of phosphorylation-dependent PcG/chromatin interaction.39
Bmi-1 was also reported to be phosphorylated by PI3K/Akt pathway.40 Both Bmi-1 and Akt were coactivated in a substantial fraction of human high-grade tumors. Akt mediated Bmi-1 phosphorylation, enhanced its oncogenic potential in an Ink4a/Arf-independent manner.40 This process also modulated the DDR and affected genomic stability. These experiments placed Bmi-1 in a close niche of cellular signaling networks, opening up a world of possibilities as to how external signaling stimuli—in collaboration with chromosomal modifications through the Bmi-1/PRC1 complex—can dictate an oncogenic outcome.40 These data have direct bearing in providing new mechanistic insights into the modulation of Bmi-1 function by phosphorylation, either by 3 pK or by Akt.
Another independent study showed that phosphorylation of Bmi-1 by Akt could also modulate its function through the Ink/Arf pathway. Phosphorylation of human Bmi-1 at Ser316 by Akt impaired its function by triggering dissociation from the Ink4a/Arf locus, which resulted in decreased ubiquitination of histone H2A and the inability of Bmi-1 to promote cellular proliferation and tumor growth.41 Moreover, Akt-mediated phosphorylation of Bmi-1 also inhibited its ability to promote self-renewal of hematopoietic stem and progenitor cells. These studies provide a mechanism for the increased abundance of p16Ink4a and p14Arf observed in cancer cells with an activated phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, and identify crosstalk between phosphorylation events and chromatin structure. Together these reports point to the context dependent variability in functional outcome of phosphorylated Bmi-1.
Other important aspects of modulating Bmi-1 function are through the alteration in protein stability via ubiquitination. A recent study by Sahasrabuddhe et al, identified that Bmi-1 contains a functional recognition motif for the F box protein βTrCP, which regulates ubiquitination and proteasome-mediated degradation of various proteins. Overexpression of wild-type βTrCP—but not the ΔF mutant—promoted Bmi-1 ubiquitination and degradation. Also, compared to wild-type Bmi-1, the ΔF mutant protein exhibited increased pro-oncogenic activity.42 In summary, this finding establishes the importance of ubiquitination of Bmi-1 in regulating the turnover of Bmi-1 and its functional relevance in oncogenesis.
Regarding the existing data on PTMs of Bmi-1, it appears that the field is still at a nascent stage and further evidence needs to be accumulated to fully elucidate the diversified role of Bmi-1 in various physiological processes. However, it is not premature to speculate that PTMs of Bmi-1 are most likely cancer-tissue specific and that the specific site largely dictates the unique physiological role that this protein plays.
Bmi-1 in cellular physiology
Development
As previously noted, the PcG complexes were first identified as trans-acting regulators of homeotic gene function in Drosophila.43 Two clusters of homeotic genes, the antennapedia complex and the bithorax complex, collectively referred to as the homeotic complex are responsible for the determination of segmental identities in Drosophila. In vertebrates, PcG complexes regulate the self-renewal of various types of embryonic and adult stem cells44, 45 as well skeletal malformations.46, 47 PcG proteins are also evolutionarily conserved chromatin modifiers that can transcriptionally silence their targets through many rounds of cell division during development. Bmi-1 is arguably the PcG protein most strongly associated with neoplastic development.48
Bmi-1 plays an important role in morphogenesis during embryonic development and in hematopoiesis throughout pre- and post-natal life. In most tissues, low levels of the Bmi-1 mRNA are detected, whereas higher mRNA levels are found in thymus, heart, brain, and testes. Bmi-1 is also expressed in embryonic stem cells (ESCs) during embryonic development and in the placenta. Bmi-1 has an important role in anterior–posterior axis specification and in maintaining the correct spatial Hox gene expression pattern.49, 50 Loss of function of Bmi-1 has pleiotropic effects. Mice lacking Bmi-1 exhibit a homeotic posterior transformation coupled with strong proliferative defects during lymphocyte development, and also neurological abnormalities.4 In absence of Bmi-1, primary embryonic fibroblasts are unable to progress into S phase and to undergo premature senescence.51 Bmi-1 functions in normal development and stem cell maintenance have been attributed to its transcriptional repression of the Ink4a/Arf locus (Fig 2).45, 52, 53 In the central and peripheral nervous system, Bmi-1 deficiency is associated with increased expression of p16Ink4a and p19Arf, and inactivation of p16Ink4a in Bmi-1−/− mice partially rescued the self-renewal defect of Bmi-1−/− neural stem cells (NSCs).54 The PcG members are dynamically expressed throughout pre-implantation and development.55, 56 Recently Lavialy et al, reported a regulatory pathway that underlies cell fate allocation during early development. This process mechanistically links Bmi-1 to the lineage-specific transcription factors Nanog and Gata6.57 Bmi1 is repressed by Nanog in ESCs and highly expressed in the extraembryonic endoderm (XEN) and trophectodermal stem cells (TSCs) where Nanog is not present. Bmi-1 physically interacts with Gata6 in extraembryonic primitive endoderm derived XEN cells and controls its protein stability and resultant activity by inhibiting Gata6 ubiquitination and proteasome-mediated degradation. Interestingly, Bmi-1 also interacts with and maintains high Gata3 protein levels in TE-derived TSCs,58 suggesting a broader function for Bmi-1 in extraembryonic lineage formation and maintenance.
Figure 2.
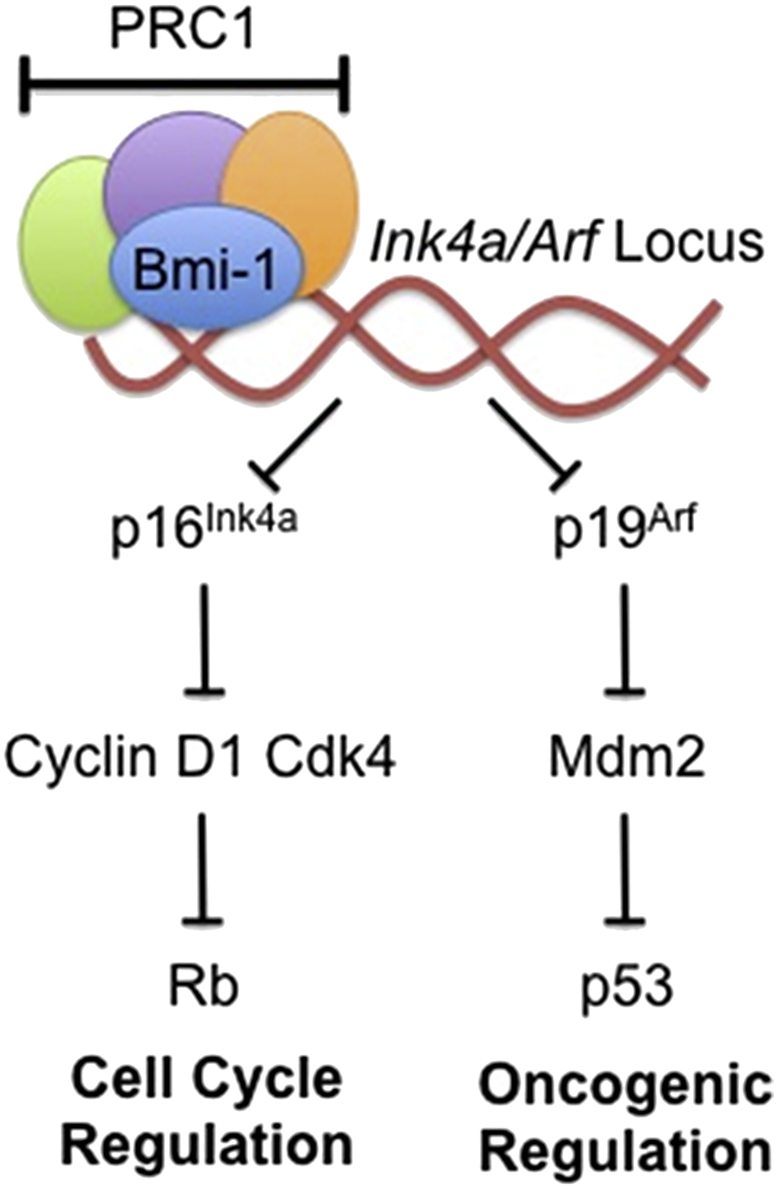
Role of Bmi-1 in Ink4a/Arf Pathway. This figure shows how PRC1 component Bmi-1 helps in the mediation of transcriptional repression at the Ink4a/Arf locus. This mechanism leads to downstream repression of cell cycle and oncogenic pathways.
Cell cycle
The normal cell cycle progression and regulation is tightly controlled by a variety of molecular checkpoints that supervise the various biological functions of the cell that occur within the different phases of the cell cycle.59 Bmi-1, being a transcriptional repressor and PcG protein, plays an important role in cell cycle regulation.4 Bmi-1 controls self-renewal and cell cycle by regulating the tumor suppressor proteins p16Ink4a and p14Arf.60, 61 Bmi-1 promotes CDK4 and CDK6 activity by repressing the Ink4a/Arf locus, which encodes the CDK4/6 inhibitor p16Ink4a and p19Arf (a homolog of human p14Arf) that activates p53 (Fig 2).51 Bmi-1 can also directly regulate p53 stability, further stressing its role in cellular proliferation and tumorigenesis by negatively acting through the pRb-p53 pathway.62, 63 The p16Ink4a protein inhibits binding of Cyclin D to CDK4/6, resulting in the suppression of retinoblastoma (Rb) activity and induction of cell cycle arrest.51, 60 p19Arf induces p53 and causes cell cycle arrest.51, 64 Bmi-1 promotes cell proliferation by suppressing p16Ink4a/Rb and/or p14Arf/MDM2/p53 tumor suppressor pathways.51 The absence of Bmi-1 is reported to relieve the repression of the Ink4a, resulting in the expression of p16Ink4a and p14Arf. Bmi-1 abolishes cell cycle checkpoints p16/p14 in various cell types.3 The cell cycle checkpoint that involves the Ink4a/Arf-p53-pRb axis has been described as the principle barrier to the initiation and maintenance of neoplastic transformation.65, 66
In contrast, a few studies have also reported that the proto-oncogene Bmi-1 is required for hepatocellular carcinoma (HCC) cell proliferation; however, the effect of Bmi-1 in promoting HCC cell growth was independent of the Ink4a/Arf status.67, 68, 69 The crosstalk among the proteins active in these pathways and in the epigenetic control of Ink4a/Arf expression has been widely investigated to characterize the role of proto-oncogenes that negatively affect this molecular checkpoint.70, 71 The down-regulation of Bmi-1 also inhibits HCC cell growth independent of Ink4a/Arf status. In mouse tumor cells induced by Bmi-1/RasV12, there is no down-regulation of p16Ink4a or p19Arf expression.72 A recent report evidenced that PRCs regulate cellular proliferation and transformation independently of the Ink4a/Arf-pRb-p53 pathway. PRCs localize at replication forks, and the loss of their function directly affects the progression and symmetry of DNA replication forks.73 Ink4a proteins function as antagonists of the Cyclin D–CDK4/6 complex, thereby hindering phosphorylation of Rb family members and consequent entry into S phase. p19Arf, in contrast, is not a CDK inhibitor and instead positively regulates p53.52 Deletion of both p16Ink4a and p19Arf through the disruption of the entire Ink4a locus has minimal consequences for HSC activity. Their limited expression is likely the result of transcriptional repression by Bmi-1, which is expressed preferentially in HSCs and actively represses the Ink4a locus.18, 74 Bmi-1 deficiency is lethal in adult mice due to hematopoietic failure caused by a progressive depletion of HSCs.18, 74 The escalation in p16Ink4a and p19Arf expression caused by Bmi-1 removal may completely inhibit the infrequent cell cycle entry of adult HSCs, which is crucial for HSC self-renewal and maintenance of blood homeostasis. Recent reports also suggested that Bmi-1 controls mouse glioma development in an Ink4a/Arf independent manner.69 Bmi-1 knockdown significantly inhibits cell growth in both wildtype and p16Ink4a null Ewing sarcoma67 and medulloblastoma cell lines.75 Thus, although inhibition of Ink/Arf tumor suppressor gene expression has been widely considered to be the key mechanism for the oncogenic activity of Bmi-1, more recent data suggest a critical role of Ink/Arf independent mechanisms for Bmi-1 during carcinogenesis.
DNA damage response
Genome integrity is constantly challenged by endogenous (metabolic) and exogenous (environmental) sources. To combat threats posed by DNA damage, cells have evolved mechanisms that are collectively termed as DDR. DDR involves a plethora of proteins whose sequential recruitment and function at DNA damage sites are modulated by numerous highly dynamic and reversible PTMs, including phosphorylation, ubiquitination, acetylation, methylation and sumoylation.
Bmi-1 is known to play a key role not only in preventing DNA damage, but also in DDR. Mitochondrial ROS has been known to be a prominent agent for oxidative DNA damage. Liu et al, has demonstrated that independent of p16Ink4a, Bmi-1 has a role in maintaining mitochondrial function and redox homeostasis.19 Further, cells lacking Bmi-1 have significant mitochondrial dysfunction accompanied by a sustained increase in ROS that is sufficient to engage the DDR pathway.19 We also have shown that silencing Bmi-1 in ovarian cancer model leads to increased ROS generation that induces PARP cleavage and apoptosis. Thus, Bmi-1 prevents oxidative DNA damage.76
Apart from protecting against oxidative DNA damage, Bmi-1 has been shown to be a key component in DDR, as it is required and sufficient to recruit the DDR machinery to DNA double-strand break (DSB) sites in response to radiation and can prolong NSC survival.77 Facchino et al, have shown that Bmi-1 is enriched at the chromatin after irradiation, localized and co-purified with ataxia telangiectasia mutated (ATM) protein and γH2AX. Bmi-1 was also shown to preferentially co-purify with non-homologous end joining proteins DNA-PK, PARP-1, hnRNP U, and histone H1 in CD133+ glioblastoma cells.77
Following DNA damage, Bmi-1 tethers RING finger protein 2 (RNF2 or RING1B) to DNA, and associates more stably with damaged compared to undamaged chromatin.78 Computational models based on a recently derived crystal structure of Bmi-1–RING1B–ubiquitin-conjugating enzyme H5c (UbcH5c) suggest that the complex binds to both nucleosomal DNA and histone H479, whereas initial recruitment of RING1B–Bmi-1 to DSBs is dependent on sumoylation of Bmi-1.38 As previously discussed, the PRC1 complex member CBX4 promotes sumoylation (SUMO-1) of Bmi-1 at Lys88 (K88), while the Bmi-1 K88R mutant failed to be recruited to the repair foci.38 RING1B–Bmi-1 catalyzes mono-ubiquitination of H2AX/H2A at Lys119 and Lys120 (Lys118 and Lys119 in H2A).7, 80, 81, 82 This modification is essential for the recruitment of ATM to damage site, and consequently, is necessary for efficient formation of γ-H2AX.81, 82 Although Bmi-1 is required for initial recruitment of ATM, ATM is required for sustained localization of Bmi-1 at breaks, which is important for efficient homologous recombination.7
Further experimentation will be required to dissect the mechanism by which sumoylation mediates RING1B–Bmi-1 assembly at DSBs, and how H2AX ubiquitination enables binding of ATM. Initial studies suggest that ubiquitination of H2A may weaken interaction with DNA, destabilizing the nucleosome,83 thus helping in binding of other DDR proteins.
Therefore, by inhibiting ROS induced oxidative DNA damage along with facilitating DDR, Bmi-1 contributes to maintaining genome integrity and resistance to genotoxic therapeutic reagents. Since the efficacy of cancer chemotherapy and radiotherapy relies on generation of DNA damage that is recognized and repaired by intrinsic DNA repair pathways, high expression of Bmi-1 correlates with therapy failure and could be a biomarker for resistance of DNA damaging therapy. Along these lines Sauvageau et al, showed that transformed human cell lines as well as primitive hematopoietic cells exhibited a high frequency of spontaneous chromosome breaks upon Bmi-1 depletion and were hypersensitive to genotoxic agents. Bmi-1 was recruited rapidly to the DNA damage foci where it blocked transcriptional elongation. In addition, Bmi-1 was required for homologous recombination repair and checkpoint recovery.84
Senescence and stem cells
Cellular senescence is characterized by an irreversible cell cycle arrest often in response to acute insults in an attempt to prevent damaged or mutated cells from proliferating uncontrollably.85 Histone deacetylase-associated Sin3B protein is implicated in cell cycle withdrawal, where it is transcriptionally upregulated.86 Sin3B has been identified as a novel direct target of Bmi-1, where Bmi-1-driven repression of Sin3B as an essential regulator of cellular senescence has been proposed.87 Bmi-1 has also been reported to prevent senescence and immortalize cells through the activation of telomerase in breast cancer cells61 and ovarian cancer cells, where elevation of Bmi-1 expression is closely correlated to the increased telomerase activity.88 Human telomerase reverse transcriptase (hTERT) expression, which leads to induction of telomerase activity is a direct target of c-Myc–induced transcription in mammary epithelial cells (MECs)89, 90 Apparently, Bmi-1, being a transcriptional repressor, acts independently from c-Myc. These data suggest that Bmi-1 regulates telomerase expression in MECs and might play a role in the development of human breast cancer. Deletion analysis of the Bmi-1 protein suggested that the RING finger, as well as a conserved HTH domain, were required for its ability to induce telomerase and immortalize MECs.61 However, Bmi-1 induction of telomerase is cell type specific; Bmi-1 fails to induce telomerase in fibroblasts.61 This is consistent with the observation that Bmi-1 overexpression did not immortalize human fibroblasts.8 It is not known whether Bmi-1 is involved in telomere function in normal breast stem cells. However, in the fetal liver, Bmi-1 was reported to play equally important roles both in the normal as well as progenitor stem cells.91 Hosen et al, showed that the expression of Bmi-1 is high in primitive HSCs, and is decreased when HSCs are differentiated into a particular lineage.92 The self-renewal and maintenance of HSCs and NSCs were reported to depend on the levels of Bmi-1.60, 64 These reports suggest a strong correlation between Bmi-1 and the differentiation and renewal of stem cells.
Bmi-1 is reported to play a crucial role during the self-renewal and maintenance of prostate, intestinal, lung epithelial and bronchioalveolar stem cells.93, 94, 95 It would not be out of place to underscore the commonality between stem cells and cancer cells to micromanage the bioenergetic needs for influencing epigenetic/genetic programs. Stem cells are characterized by well classified energetic and biosynthetic demands compared to quiescent differentiated cells.96 Changing gears between the glycolytic and mitochondrial oxphos pathways triggers differentiation or reprogramming to pluripotency that are furthermore accompanied by consequent changes in cell cycle, biomass, metabolite levels, and redox state. So either a direct or indirect role of Bmi-1 in regulating the cellular bioenergetics may be well conceived as a strategy to answer how Bmi-1 integrates with epigenetic and genetic programs to coordinately regulate stem cell lineage and/or fate.
Stem cells are of two types: ESCs and adult stem cells (ASCs). ESCs are pluripotent stem cells capable of developing into different cells; while ASCs maintain and repair their resident tissues in adult organisms. Thus, self-renewal, differentiation, and prevention of senescence of ASCs are critical for tissue homeostasis. Aging is the progressive decline in physiology and function of adult tissues often attributable to the loss of regenerative capacity of ASCs.97 ASCs play key roles in overall tissue homeostasis and repair. The function of ASCs declines with age, which may contribute to the physiological decline in tissue homeostasis and the increased risk of neoplasm during aging. Control of gene expression by chromatin remodeling is critical for ASC function.98
Bmi-1 plays a crucial role in self-renewal and differentiation of leukemic stem and progenitor cells. In breast cancer cells, gain of Bmi-1 function resulted in increased self-renewal and promoted epithelial–mesenchymal transition (EMT), while contrasting phenotypes were reported with Bmi-1 knockdown through regulation of Nanog expression via the NFκB pathway.99
In the nervous system, Bmi-1 is also required for the self-renewal of adult NSCs. Both constitutive deletion and acute knockdown of Bmi-1 result in impaired self-renewal of cultured NSCs isolated from young adult mice. The effect of Bmi-1 knockdown on NSCs is aggravated if NSCs are isolated from adult as opposed to embryonic and postnatal mice. In vivo, Bmi-1 deficiency causes a decrease in the numbers of proliferating, bromodeoxyuridine positive SVZ cells (neural progenitors) without affecting apoptosis.54, 100
In addition to modulating the self-renewal of stem cells, Bmi-1 regulates stem cell differentiation potential in both HSCs and NSCs. Loss of Bmi-1 does not block the differentiation of more committed hematopoietic progenitors,51, 101 but affects the ability of stem cells and early progenitors to retain all cell fate choices. In culture, HSCs from young adult Bmi-1-deficient mice have reduced multi-lineage potential compared with wild-type HSCs when assessed at early passage.101 The effects of Bmi-1 on HSC differentiation have been linked to its effects on chromatin state. In a mixed population of HSCs and multipotent progenitors (IL7Rα−/KLS), Bmi-1 binds at genomic loci that are marked by both repressive H3K27me3 and active H3–K4me3,102 a ‘bivalent’ chromatin state associated with genes that are poised to be expressed during differentiation.103 Constitutive loss of Bmi-1 in the HSC/multipotent progenitor population results in a reduction in H3K27me3 binding, de-repression of B-cell lineage factors and consequent increase in B-lymphopoiesis.102 Thus, Bmi-1 is a promising candidate for the regulation of HSC differentiation potential during aging. Bmi-1 function in young adult HSC and NSC self-renewal is mediated, in large part, through its transcriptional repression of the p16Ink4a/p19Arf aging locus.51 p16Ink4a inhibits Cyclin-D/CDK4/6 complexes to control cell cycle and senescence, whereas p19Arf contributes to cell cycle control, senescence and apoptosis through the regulation of p53.104
Genetic experiments suggest that in HSCs, p16Ink4a is the dominant mediator of the effects of Bmi-1 on stem cell proliferation. Deletion of the entire p16Ink4a/p19Arf locus, but not that of p19Arf alone, can mostly rescue the effect of Bmi-1 deficiency on HSC self-renewal in long-term competitive repopulation assays.105 p19Arf may be a more critical target in adult NSCs, as p19Arf deletion partially rescues self-renewal defects caused by Bmi-1 deficiency, although to a lesser extent than deletion of the entire p16Ink4a/p19Arf locus.64, 106 In contrast to chronic Bmi-1 loss, acute RNA interference-mediated knockdown of Bmi-1 in NSC cultures from young adult mice does not lead to an increase in p16Ink4a or p19Arf expression, but instead results in altered expression of another cell cycle inhibitor, p21CIP1, which can rescue the anti-proliferative phenotype of Bmi-1 knockdown.107 Thus, acute loss of Bmi-1 is likely insufficient for bringing about quick changes to the chromatin state of the p16Ink4a/p19Arf locus; however, constitutive deletion of Bmi-1 may result in gradually accumulating and stably maintained activating chromatin marks, such as H3K4me3 or histone acetylation, at the p16Ink4a/p19Arf locus.
The expression of Bmi-1 itself does not change significantly in isolated HSC and NSC populations during aging.108, 109 By contrast, the role of Bmi-1 in maintaining self-renewal and multipotency notably declines during aging, arguing for altered activity of Bmi-1 at yet unidentified targets. Indeed, growing evidence proposes additional age-related targets for Bmi-1 in addition to p16Ink4a/p19Arf. Overexpression of Bmi-1 in HSCs isolated from p19Arf mutant mice and p16Ink4a/p19Arf compound mutant mice can still enhance multipotency of HSCs in vitro.101, 105 Furthermore, Bmi-1 plays a non-cell autonomous role in the bone marrow microenvironment that does not depend on p16Ink4a or p19Arf.105 Similarly, deletion of the entire p16Ink4a/p19Arf locus in Bmi-1−/− mice does not completely rescue NSC defects in self-renewal capacity.64, 106
The p16Ink4a/p19Arf-independent requirement for Bmi-1 in ASC populations may be due to the ability of Bmi-1 to regulate the DDR pathway via repression of the cell cycle checkpoint protein Chk2.19 Deletion of Chk2 in Bmi-1−/− mice restores hematopoietic stem and progenitor cell function and enhances progenitor cell proliferation.19 These reports suggest that modulators of chromatin state, such as Bmi-1, are crucial for maintaining the ability of ASCs to integrate and respond to environmental stresses during aging.
Overexpression of p16Ink4a and p19Arf in adult HSCs induced cell cycle arrest and apoptosis via the pRb and the p53-dependent pathway, respectively. Double deletion of the Bmi-1 and p16Ink4a/p19Arf genes partially rescued the phenotypes observed in Bmi-1-deficient mice,51 suggesting that p16Ink4a, p19Arf, and p53 are downstream effectors of Bmi-1 that are involved in the control of the proliferation and survival of HSCs during self-renewing cell divisions.
In cancer, recurrence after optimal treatment has often been a critical clinical limitation indicative of the existence of another emerging stem cell category that evaded the existing therapy, classified as cancer initiating cells (CIC) or cancer stem cells (CSC). Evidence of CSCs from xenograft models and surviving fraction of treated tumors further consolidate this theory. Therefore, the stemness properties of CICs pose a new challenge to existing cancer therapy and from different studies, Bmi-1 surfaces as a bio-signature of these CIC/CSC.110, 111, 112 The complex dynamics of Bmi-1 function ranging from cell cycle regulation, stem cell maintenance to DDR extends beyond its capacity as a transcriptional repressor of the Ink/Arf pathway. It can be speculated that as future research brings into light new interactors of Bmi-1, many other cellular processes would be discovered in which Bmi-1 plays an important role.
Cancer
As previously noted, Bmi-1 was first identified as a Myc-cooperating oncogene in murine B- and T-cell lymphomas.61, 74, 113, 114 Several recent studies have suggested that in human cancers, Bmi-1 is often overexpressed and acts as an oncogene to promote carcinogenesis including prostate, lung, ovarian, urinary bladder, lymphoma, mesothelioma, medulloblastoma, glioma, acute myeloid leukemia and breast cancer.115 Bmi-1 overexpression drives stem-like properties associated with induction of EMT that promotes invasion, metastasis, and poor prognosis.51, 61 This section summarizes (Table 2) the critical roles of Bmi-1 in all reported types of cancer to date.
Table 2.
Known roles of Bmi-1 in cancer.
| Cancer | Role of Bmi-1 | Reference |
|---|---|---|
| Breast | Bmi-1 plays a crucial role in invasion and metastasis by modulating the Akt/GSK-3b/Snail pathway and the expression of EMT markers in breast cancer | 130,131 |
| Colorectal | Bmi-1 regulates histone ubiquitination for colon cancer proliferation in vitro and in vivo | 132,26 |
| Glioblastoma | Bmi-1 overexpressed in human glioblastoma tumors and highly enriched in tumor-initiating CD133+ stem cells | 120 |
| Head and neck | Bmi-1 directly regulates the EMT regulator, Twist1 | 128 |
| Hepatic | Bmi-1 is upregulated in human hepatocellular carcinoma in an Ink4a/Arf-independent manner in liver cancer development | 72 |
| Intestine | Bmi-1 deficiency impairs the progression and maintenance of small intestinal tumors in a cell autonomous and highly Arf-dependent manner. | 134 |
| Leukemia | Bmi-1 is highly expressed in M0-subtype acute myeloid leukemia | 135, 136 |
| Lung | Bmi-1 is required for the self-renewal of the lung epithelial stem cells and bronchioalveolar stem cells | 138, 139 |
| Lymphoma | Bmi-1 can contribute to lymphomagenesis in the T and B cell lineages and collaborate with the Myc gene in tumor development | 152 |
| Medullo-blastomas | Bmi-1 is strongly expressed in proliferating cerebellar precursor cells in mice and humans. | 117 |
| Naso-pharyngeal carcinoma | Bmi-1 is highly overexpressed which lead to the induction of telomerase activity, reduction of p16Ink4a expression, and immortalization of nasopharyngeal epithelial cells | 125 |
| Ovary | Bmi-1 protein is highly expressed in ovarian epithelial cancer tissues | 31, 88 |
| Pancreatic cancer | Bmi-1 is overexpressed in human pancreatic cancer | 153 |
| Prostate | Bmi-1 is a crucial regulator of self-renewal in adult prostate cells and plays important roles in prostate cancer initiation and progression | 93 |
| Skin | Bmi-1 levels are markedly elevated in epidermis-derived cell lines and in epidermal squamous cell carcinoma | 140 |
| Tongue | Bmi-1 is abnormally overexpressed in tongue cancers, which is associated with cervical node metastasis. | 141 |
This table gives a brief overview of the reported functions of Bmi-1 in progression and maintenance of human cancers.
EMT – epithelial–mesenchymal transition.
Glioblastoma: Bmi-1 has been shown to be essential for the proliferation and self-renewal of the NSCs of the sub-ventricular zone.18, 54, 64, 100, 106, 116, 117 Also, Bmi-1 is necessary for the propagation of leukemic stem cells and is expressed in glioblastoma stem cells.74, 118 Häyry et al, have shown that Bmi-1 is expressed in all histological types of gliomas and that relative protein expression is a novel independent prognostic marker in oligodendroglial tumors.119 Bmi-1 is overexpressed in human glioblastomas and is highly enriched in the tumor-initiating CD133+ stem cells. Knockdown of Bmi-1 completely prevented brain tumor formation in mice.120 miR128 has also been shown to negatively regulate Bmi-1 expression and inhibits self-renewal of glioma.121
Medulloblastomas: Bmi-1 is strongly expressed in proliferating cerebellar precursor cells in mice and humans.117 Overexpression of Bmi-1 promotes cell proliferation and is required for the hedgehog pathway-driven tumorigenesis in medulloblastomas. Sonic hedgehog (Shh) modulates Bmi-1 in childhood medulloblastoma brain tumor-initiating cells. Bmi-1 expression positively correlates with increasing Shh ligand concentrations.122
Ovarian cancer: Bmi-1 is highly expressed in ovarian epithelial cancer tissues, and its expression level correlates with histological grade and clinical phase of the patients.31 We had previously found that inhibition of the Shh pathway by cyclopamine inhibited sub-cutaneous ovarian tumor growth and potently downregulated Bmi-1 at the protein level.123 Elevation of Bmi-1 expression is closely correlated to the increased telomerase activity.88 miR-15a and miR-16, which are under expressed in ovarian cell lines and in primary ovarian tissues, directly target the 3′ untranslated region (UTR) and significantly correlate with Bmi-1 protein levels in ovarian cancer patients and cell lines. Furthermore, Bmi-1 protein levels are downregulated in response to miR-15a or miR-16 expression and cause significant reduction in ovarian cancer cell proliferation and clonal growth.31
Prostate cancer:Bmi-1 is overexpressed in prostate cancer with adverse pathologic and clinical features. Tumors with higher Gleason scores have a significant upregulation of Bmi-1, while the presence of Bmi-1 in lower grade prostate cancer samples is highly predictive for prostate-specific antigen recurrence.124 Microarray meta-analyses have found that the presence of Bmi-1 in prostate cancer specimens often indicates metastatic disease and a high probability of unfavorable therapeutic outcome.115 Bmi-1 has been shown enriched in a population of prostate cancer cells with higher tumor initiating capacities.115 Bmi-1 inhibition protects prostate cells from FGF10 driven hyperplasia and slows the growth of aggressive PTEN-deletion induced prostate cancer.93
Nasopharyngeal carcinoma: Bmi-1 is overexpressed in nasopharyngeal carcinoma cell lines at both mRNA and protein levels. Overexpression of Bmi-1 was also observed in a significant number of nasopharyngeal carcinoma tumors, which correlated with advanced invasive stage of the tumor progression and poor prognosis. Overexpression of Bmi-1 led to induction of telomerase activity, reduction of p16Ink4a expression and immortalization of nasopharyngeal epithelial cells.125 Bmi-1 plays an important role in the pathogenesis of nasopharyngeal carcinoma by inducing EMT partially by targeting the tumor suppressor PTEN, thus activating the PI3K/Akt pathway. Upregulation of Bmi-1 led to the stabilization of Snail, a transcriptional repressor associated with EMT, via modulation of PI3K/Akt/GSK-3β signaling.126
Head and neck cancer: Bmi-1 mediates its diverse effects through upregulation of the mitotic kinase Aurora A, which is encoded by the AURKA gene. During Bmi-1-overexpression, the β-catenin/TCF4 complex regulates the transcription of AURKA.127 In patients with head and neck cancers, increased levels of both Twist1 and Bmi-1 correlate with downregulation of E-cadherin and p16Ink4a, and are associated with worse prognosis.128 In addition, in head and neck squamous cell carcinoma (HNSCC) a CD44+ fraction comprising of less than 10% of the tumor significantly expresses nuclear Bmi-1 and demonstrates stem-like properties by giving rise to tumor in vivo.129
Breast cancer: Breast cancer shows a high prevalence of Bmi-1 expression, which is significantly correlated with aggressive features and unfavorable prognosis. Bmi-1 plays a crucial role in invasion and metastasis by modulating the Akt/GSK-3b/Snail pathway and the expression of EMT markers in breast cancer. Bmi-1 promotes the invasion and metastasis of human breast cancer.130 Bmi-1 is critical to the oncogenic behavior of several established breast cancer cell lines in vitro and in vivo. Most importantly, the collaboration of Bmi-1 and H-RAS in human mammary epithelial cells leads to a highly aggressive phenotype that includes increased spontaneous metastasis to the liver and spleen, and novel metastasis to the brain.131
Colorectal cancer: Bmi-1 regulates histone ubiquitination in primary human colorectal cancer cells which plays an important role in colon cancer proliferation in vitro and in vivo.26 Kreso et al, recently reported for the first time that the stemness function of CICs could be targeted with a small-molecule inhibitor of Bmi-1.132 In human colorectal cancer, CIC function is dependent on the canonical self-renewal regulator Bmi-1. Downregulation of Bmi-1 inhibits the self-renewal property of colorectal CICs, and thus abrogates their tumorigenic potential. Bmi-1 inhibitor treatment in primary colorectal cancer xenografts also resulted in colorectal CIC loss with long-term and irreversible impairment of tumor growth. Targeting the Bmi-1-related self-renewal machinery provides the framework for clinical evaluation of anti-Bmi-1 therapy not only in colorectal cancer but other cancers such as ovarian cancer which are characterized by frequent relapse after chemotherapy.
Hepatocellular carcinoma: Bmi-1 is upregulated in HCC.133 Bmi-1 can cooperate with other oncogenic signals to promote hepatic carcinogenesis in vivo. Bmi-1 functions independent of Ink4A/Arf repression in liver cancer development.72
Intestinal cancer: Bmi-1 deficiency impairs the progression and maintenance of small intestinal tumors in a cell autonomous and highly Arf-dependent manner. Bmi-1 loss reduces both the number and size of small intestinal adenomas.134
Leukemia: High expression of Bmi-1 in acute myeloid leukemia cells is associated with an unfavorable prognosis.135 Expression of Bmi-1 is required for maintenance and self-renewal of normal and leukemic stem and progenitor cells, and protects cells against oxidative stress.136 Also, Bmi-1 has an essential role in maintaining the self-renewing capacity of leukemic stem cells.137
Lung cancer: Bmi-1 is required for the self-renewal of ASCs including the lung epithelial stem cells, bronchioalveolar stem cells (BASCs).138, 139Bmi-1 knockout mice exhibited an impaired ability to repair Clara cell injury that was associated with failure of BASC expansion in vivo.14, 138
Skin cancer: Bmi-1 plays an important role in maintaining proliferation of basal keratinocytes and in maintaining the viability. Bmi-1 levels are distinctly elevated in immortalized and transformed epidermis-derived cell lines and in epidermal squamous cell carcinoma of suprabasal differentiating keratinocytes.140
Tongue cancer: Bmi-1 is aberrantly overexpressed in a significant portion of tongue cancers. Elevated Bmi-1 is associated with cervical node metastasis. Bmi-1 serves as a key driver with multiple oncogenic functions during tongue cancer progression and as a novel biomarker for diagnosis and prognostic prediction for patients.141
The exhaustive above list of malignancies where Bmi-1 acts as an oncoprotein can be expected to be expanded as research progresses. Given the wide variety of cancers ranging from solid tumors to glioblastoma to leukemia, where Bmi-1 is implicated and its involvement in maintenance of CSC's that are chemoresistant makes Bmi-1 a potential candidate for drug therapy.
Conclusion and future directions
Bmi-1 is at an important crossroad in at least 16 different types of cancer and stands out as a promising target in the small list of genes known to regulate CSC function. Thus, Bmi-1 is an important new target for therapy in malignancies characterized by overexpression of Bmi-1. However, since no direct enzymatic activity of Bmi-1 has yet been established, developing a small molecule inhibitor that directly targets Bmi-1 poses a pharmacological challenge.
However in principle, several independent groups have investigated the effect of reducing Bmi-1 levels in vitro and in vivo in different cancers ranging from ovarian to prostrate and these studies have validated Bmi-1 as a therapeutic target. Our previous study on ovarian cancer has argued in favor of anti-Bmi-1 therapy in clinical settings due to enhanced cisplatin sensitivity upon Bmi-1 silencing.76 Also downregulation of Bmi-1 leads to de-repression of Ink4a, which encodes tumor suppressors p16Ink and p19Arf that regulate senescence and apoptosis.18 Activation of these pathways has been invoked as important tumor suppressors, because these pathways act as potent inhibitors of proliferation or propagation of damaged cells. In addition, we posit that cisplatin and Bmi-1 act on similar pathways affecting mitochondrial function and/or through increased ROS generation, causing DNA damage augmenting the signal initiating apoptosis.76 As noted above in the context of various cancers, different miRs target the Bmi-1 mRNA, and we have shown that miR15a/16 significantly reduce Bmi-1 levels and subsequent clonal growth in ovarian cancer cells.31In vivo silencing of Bmi-1 by siRNA delivered using DOPC (1,2-dioleoyl-sn-glycero-3-phosphatidylc holine) nanoliposomes in the orthotopic A2780-CP20 mouse model resulted in significant reduction in tumor weight and nodules compared to the control siRNA group. Combination therapy with Bmi-1 siRNA and cisplatin resulted in even greater reduction in tumor weight and nodule.76 Therefore, though siRNA or miRNA approaches against Bmi-1 might prove useful in therapy, delivery and off-target effects remain of considerable concern. In this context, several known drugs have been reported to reduce Bmi-1 expression. For example, Bommi et al, and Jung et al, independently investigated the effects of HDACi on Bmi-1 expression and downstream targets.29 HDACi treatment downregulated PcG genes such as Bmi-1, EZH2 and SUZ12 in senescent mesenchymal stem cells.142 This was associated with hypophosphorylation of Rb, which causes Rb to bind and decrease the transcriptional activity of E2F and cellular senescence by p16Ink4a regulation.142 Blocking Bmi-1 expression by HDACi in immortal non-transformed breast epithelial cells and breast cancer cells was accompanied by a decrease in histone-2A Lys119 ubiquitination (H2AK119Ub), de-repression of growth inhibitory genes and putative tumor suppressors, which are known to be silenced by PcG proteins and PRCs.29
Recently, the drug Artemisinin was also shown to inhibit the expression of Bmi-1. Artemisinin and its derivatives are particularly useful for the treatment of resistant Plasmodium falciparum malarial parasites. This drug has an endoperoxide group that is activated by intraparasitic heme–iron to form free radicals, which kill malarial parasites by alkylating biomolecules. Artemisinin inhibited Bmi-1 at both protein and transcriptional levels. Bmi-1 knockdown made the cells more sensitive to Artemisinin with an increase in accumulation at the G1-phase of the cell cycle.143 However, none of these drugs can be claimed to be a specific Bmi-1 inhibitor and therefore run the risk of bystander effect when evaluated from a clinical perspective.
In this regard PTC Therapeutics (NJ) has attempted to exploit the possible post-transcriptional regulation of Bmi-1 in a bid to screen for Bmi-1 specific drugs. The Bmi-1 3′-UTR contains multiple A–U rich elements that can significantly upregulate reporter gene expression, while its 5′-UTR contains a strong IRES activity. High-throughput screening using compounds that selectively inhibited 5′- and 3′- UTR-mediated Bmi-1 reporter expression identified PTC-209, which subsequently inhibited endogenous Bmi-1 expression in human colorectal HCT116 and human fibrosarcoma HT1080 tumor cells in a dose- and time- dependent manner (IC50 at about 0.5 μM).132 Cytotoxicity was ruled out since PTC-209 did not inhibit cell growth or viability in HEK293 human embryonic kidney cells after overnight treatment and had limited effects on cell proliferation in HT1080 cells after a 48-h treatment. That PTC-209 modulated the Bmi-1-PRC1 complex activity was evident from reduced global ubiquitinated histone H2A with no effect on total H2A or RING1A levels. PTC-209 selectively lowered Bmi-1 levels and decreased colorectal tumor cell growth. Further, unlike the genetic manipulation, PTC-209 provided the control to modulate the length and extent of Bmi-1 inhibition through the addition or removal of the inhibitor, a critical tool to further investigate the effects of Bmi-1 function in various cancers.132 Thus the future of anti-Bmi-1 therapy holds promise and development of kinase specific inhibitors that affect specific phosphorylation sites on Bmi-1 can be envisioned.
Conflicts of interest
None to declare.
Acknowledgements
This study was supported by the National Institutes of Health (NIH)CA157481 awarded to RB.
Footnotes
Peer review under responsibility of Chongqing Medical University.
Contributor Information
Resham Bhattacharya, Email: Resham-Bhattacharya@ouhsc.edu.
Soumyajit Banerjee Mustafi, Email: Soumyajit-BanerjeeMustafi@ouhsc.edu.
Mark Street, Email: mark.street@eagles.oc.edu.
Anindya Dey, Email: Anindya-Dey@ouhsc.edu.
Shailendra Kumar Dhar Dwivedi, Email: Shailendra-Dwivedi@ouhsc.edu.
References
- 1.van Lohuizen M., Verbeek S., Scheijen B., Wientjens E., van der Gulden H., Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- 2.Alkema M.J., Wiegant J., Raap A.K., Berns A., van Lohuizen M. Characterization and chromosomal localization of the human proto-oncogene BMI-1. Hum Mol Gene. 1993;2:1597–1603. doi: 10.1093/hmg/2.10.1597. [DOI] [PubMed] [Google Scholar]
- 3.Raaphorst F.M. Deregulated expression of Polycomb-group oncogenes in human malignant lymphomas and epithelial tumors. Hum Mol Genet. 2005;14(Spec No 1):R93–R100. doi: 10.1093/hmg/ddi111. [DOI] [PubMed] [Google Scholar]
- 4.van der Lugt N.M., Domen J., Linders K. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994;8:757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- 5.Li Z., Cao R., Wang M., Myers M.P., Zhang Y., Xu R.M. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J Biol Chem. 2006;281:20643–20649. doi: 10.1074/jbc.M602461200. [DOI] [PubMed] [Google Scholar]
- 6.Cao L., Bombard J., Cintron K., Sheedy J., Weetall M.L., Davis T.W. BMI1 as a novel target for drug discovery in cancer. J Cell Biochem. 2011;112:2729–2741. doi: 10.1002/jcb.23234. [DOI] [PubMed] [Google Scholar]
- 7.Ginjala V., Nacerddine K., Kulkarni A. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol. 2011;31:1972–1982. doi: 10.1128/MCB.00981-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itahana K., Zou Y., Itahana Y. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23:389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brennan R.G., Matthews B.W. The helix-turn-helix DNA binding motif. J Biol Chem. 1989;264:1903–1906. [PubMed] [Google Scholar]
- 10.Rechsteiner M., Rogers S.W. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- 11.Yadav A.K., Sahasrabuddhe A.A., Dimri M., Bommi P.V., Sainger R., Dimri G.P. Deletion analysis of BMI1 oncoprotein identifies its negative regulatory domain. Mol Cancer. 2010;9:158. doi: 10.1186/1476-4598-9-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen K.J., Hanna J.S., Prescott J.E., Dang C.V. Transformation by the Bmi-1 oncoprotein correlates with its subnuclear localization but not its transcriptional suppression activity. Mol Cell Biol. 1996;16:5527–5535. doi: 10.1128/mcb.16.10.5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ringrose L., Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–443. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 14.Sauvageau M., Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Croce L., Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20:1147–1155. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- 16.Davidovich C., Goodrich K.J., Gooding A.R., Cech T.R. A dimeric state for PRC2. Nucleic Acids Res. 2014;42:9236–9248. doi: 10.1093/nar/gku540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao Q., Wang X., Zhao M. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. 2014;5:3127. doi: 10.1038/ncomms4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park I.K., Qian D., Kiel M. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 19.Liu J., Cao L., Chen J. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan K.S., Chia L.A., Li X. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci U S A. 2012;109:466–471. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang R., Cheung N.K., Vider J. MYCN and MYC regulate tumor proliferation and tumorigenesis directly through BMI1 in human neuroblastomas. FASEB J. 2011;25:4138–4149. doi: 10.1096/fj.11-185033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin A., Cano A. Tumorigenesis: twist1 links EMT to self-renewal. Nat Cell Biol. 2010;12:924–925. doi: 10.1038/ncb1010-924. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z., Zheng Y., Park H.J. Targeting FoxM1 effectively retards p53-null lymphoma and sarcoma. Mol Cancer Ther. 2013;12:759–767. doi: 10.1158/1535-7163.MCT-12-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowak K., Kerl K., Fehr D. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 2006;34:1745–1754. doi: 10.1093/nar/gkl119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J., Chai L., Liu F. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci U S A. 2007;104:10494–10499. doi: 10.1073/pnas.0704001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu T., Chen X., Zhang W. Regulation of the potential marker for intestinal cells, Bmi1, by beta-catenin and the zinc finger protein KLF4: implications for colon cancer. J Biol Chem. 2012;287:3760–3768. doi: 10.1074/jbc.M111.316349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo W.J., Zeng M.S., Yadav A. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res. 2007;67:5083–5089. doi: 10.1158/0008-5472.CAN-06-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo W.J., Datta S., Band V., Dimri G.P. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol Biol Cell. 2007;18:536–546. doi: 10.1091/mbc.E06-05-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bommi P.V., Dimri M., Sahasrabuddhe A.A., Khandekar J., Dimri G.P. The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle. 2010;9:2663–2673. doi: 10.4161/cc.9.13.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H.B., Liu G.H., Zhang H. Sp1 and c-Myc regulate transcription of BMI1 in nasopharyngeal carcinoma. FEBS J. 2013;280:2929–2944. doi: 10.1111/febs.12299. [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharya R., Nicoloso M., Arvizo R. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009;69:9090–9095. doi: 10.1158/0008-5472.CAN-09-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugihara H., Ishimoto T., Watanabe M. Identification of miR-30e* regulation of Bmi1 expression mediated by tumor-associated macrophages in gastrointestinal cancer. PloS One. 2013;8:e81839. doi: 10.1371/journal.pone.0081839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang Z., Xu W.H., Lu P. MicroRNA-135a inhibits cell proliferation by targeting Bmi1 in pancreatic ductal adenocarcinoma. Int J Biol Sci. 2014;10:733–745. doi: 10.7150/ijbs.8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimri M., Carroll J.D., Cho J.H., Dimri G.P. microRNA-141 regulates BMI1 expression and induces senescence in human diploid fibroblasts. Cell Cycle. 2013;12:3537–3546. doi: 10.4161/cc.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang X., Sun Y., Han S., Zhu W., Zhang H., Lian S. MiR-203 inhibits melanoma invasive and proliferative abilities by targeting the polycomb group gene BMI1. Biochem Biophys Res Commun. 2015;456:361–366. doi: 10.1016/j.bbrc.2014.11.087. [DOI] [PubMed] [Google Scholar]
- 36.Qi X., Li J., Zhou C., Lv C., Tian M. MicroRNA-320a inhibits cell proliferation, migration and invasion by targeting BMI-1 in nasopharyngeal carcinoma. FEBS Lett. 2014;588:3732–3738. doi: 10.1016/j.febslet.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 37.Sustackova G., Kozubek S., Stixova L. Acetylation-dependent nuclear arrangement and recruitment of BMI1 protein to UV-damaged chromatin. J Cell Physiol. 2012;227:1838–1850. doi: 10.1002/jcp.22912. [DOI] [PubMed] [Google Scholar]
- 38.Ismail I.H., Gagne J.P., Caron M.C. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012;40:5497–5510. doi: 10.1093/nar/gks222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voncken J.W., Niessen H., Neufeld B. MAPKAP kinase 3pK phosphorylates and regulates chromatin association of the polycomb group protein Bmi1. J Biol Chem. 2005;280:5178–5187. doi: 10.1074/jbc.M407155200. [DOI] [PubMed] [Google Scholar]
- 40.Nacerddine K., Beaudry J.B., Ginjala V. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin Invest. 2012;122:1920–1932. doi: 10.1172/JCI57477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y., Liu F., Yu H. Akt phosphorylates the transcriptional repressor bmi1 to block its effects on the tumor-suppressing ink4a-arf locus. Sci Signal. 2012;5 doi: 10.1126/scisignal.2003199. ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sahasrabuddhe A.A., Dimri M., Bommi P.V., Dimri G.P. BetaTrCP regulates BMI1 protein turnover via ubiquitination and degradation. Cell Cycle. 2011;10:1322–1330. doi: 10.4161/cc.10.8.15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kennison J.A. The Polycomb and trithorax group proteins of Drosophila: trans-regulators of homeotic gene function. Annu Rev Genet. 1995;29:289–303. doi: 10.1146/annurev.ge.29.120195.001445. [DOI] [PubMed] [Google Scholar]
- 44.Lund A.H., van Lohuizen M. Epigenetics and cancer. Genes Dev. 2004;18:2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 45.Valk-Lingbeek M.E., Bruggeman S.W., van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Akasaka T., Kanno M., Balling R., Mieza M.A., Taniguchi M., Koseki H. A role for mel-18, a Polycomb group-related vertebrate gene, during theanteroposterior specification of the axial skeleton. Development. 1996;122:1513–1522. doi: 10.1242/dev.122.5.1513. [DOI] [PubMed] [Google Scholar]
- 47.Core N., Bel S., Gaunt S.J. Altered cellular proliferation and mesoderm patterning in Polycomb-M33-deficient mice. Development. 1997;124:721–729. doi: 10.1242/dev.124.3.721. [DOI] [PubMed] [Google Scholar]
- 48.Sparmann A., van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 49.del Mar Lorente M., Marcos-Gutierrez C., Perez C. Loss- and gain-of-function mutations show a polycomb group function for Ring1A in mice. Development. 2000;127:5093–5100. doi: 10.1242/dev.127.23.5093. [DOI] [PubMed] [Google Scholar]
- 50.van der Lugt N.M., Alkema M., Berns A., Deschamps J. The Polycomb-group homolog Bmi-1 is a regulator of murine Hox gene expression. Mech Dev. 1996;58:153–164. doi: 10.1016/s0925-4773(96)00570-9. [DOI] [PubMed] [Google Scholar]
- 51.Jacobs J.J., Kieboom K., Marino S., DePinho R.A., van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 52.Sherr C.J. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 53.Sharpless N.E. Ink4a/Arf links senescence and aging. Exp Gerontol. 2004;39:1751–1759. doi: 10.1016/j.exger.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 54.Molofsky A.V., Pardal R., Iwashita T., Park I.K., Clarke M.F., Morrison S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Carroll D., Erhardt S., Pagani M., Barton S.C., Surani M.A., Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21:4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puschendorf M., Terranova R., Boutsma E. PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat Genet. 2008;40:411–420. doi: 10.1038/ng.99. [DOI] [PubMed] [Google Scholar]
- 57.Dupressoir A., Lavialle C., Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta. 2012;33:663–671. doi: 10.1016/j.placenta.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka S., Kunath T., Hadjantonakis A.K., Nagy A., Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science. 1998;282:2072–2075. doi: 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- 59.Medema R.H., Macurek L. Checkpoint control and cancer. Oncogene. 2012;31:2601–2613. doi: 10.1038/onc.2011.451. [DOI] [PubMed] [Google Scholar]
- 60.Park I.K., Morrison S.J., Clarke M.F. Bmi1, stem cells, and senescence regulation. J Clin Investi. 2004;113:175–179. doi: 10.1172/JCI20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dimri G.P., Martinez J.L., Jacobs J.J. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002;62:4736–4745. [PubMed] [Google Scholar]
- 62.Calao M., Sekyere E.O., Cui H.J. Direct effects of Bmi1 on p53 protein stability inactivates oncoprotein stress responses in embryonal cancer precursor cells at tumor initiation. Oncogene. 2013;32:3616–3626. doi: 10.1038/onc.2012.368. [DOI] [PubMed] [Google Scholar]
- 63.Su W.J., Fang J.S., Cheng F., Liu C., Zhou F., Zhang J. RNF2/Ring1b negatively regulates p53 expression in selective cancer cell types to promote tumor development. Proc Natl Acad Sci U S A. 2013;110:1720–1725. doi: 10.1073/pnas.1211604110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Molofsky A.V., He S., Bydon M., Morrison S.J., Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–1437. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hahn W.C., Counter C.M., Lundberg A.S., Beijersbergen R.L., Brooks M.W., Weinberg R.A. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 66.Ventura A., Kirsch D.G., McLaughlin M.E. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 67.Douglas D., Hsu J.H., Hung L. BMI-1 promotes ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res. 2008;68:6507–6515. doi: 10.1158/0008-5472.CAN-07-6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Datta S., Hoenerhoff M.J., Bommi P. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007;67:10286–10295. doi: 10.1158/0008-5472.CAN-07-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bruggeman S.W., Hulsman D., Tanger E. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell. 2007;12:328–341. doi: 10.1016/j.ccr.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 70.Sperka T., Wang J., Rudolph K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13:579–590. doi: 10.1038/nrm3420. [DOI] [PubMed] [Google Scholar]
- 71.Kim W.Y., Sharpless N.E. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 72.Xu C.R., Lee S., Ho C. Bmi1 functions as an oncogene independent of Ink4A/Arf repression in hepatic carcinogenesis. Mol Cancer Res. 2009;7:1937–1945. doi: 10.1158/1541-7786.MCR-09-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Piunti A., Rossi A., Cerutti A. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat Commun. 2014;5:3649. doi: 10.1038/ncomms4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lessard J., Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 75.Wiederschain D., Chen L., Johnson B. Contribution of polycomb homologues Bmi-1 and Mel-18 to medulloblastoma pathogenesis. Mol Cell Biol. 2007;27:4968–4979. doi: 10.1128/MCB.02244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang E., Bhattacharyya S., Szabolcs A. Enhancing chemotherapy response with Bmi-1 silencing in ovarian cancer. PloS One. 2011;6:e17918. doi: 10.1371/journal.pone.0017918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Facchino S., Abdouh M., Chatoo W., Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci. 2010;30:10096–10111. doi: 10.1523/JNEUROSCI.1634-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ismail I.H., Andrin C., McDonald D., Hendzel M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol. 2010;191:45–60. doi: 10.1083/jcb.201003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bentley M.L., Corn J.E., Dong K.C., Phung Q., Cheung T.K., Cochran A.G. Recognition of UbcH5c and the nucleosome by the Bmi1/Ring1b ubiquitin ligase complex. EMBO J. 2011;30:3285–3297. doi: 10.1038/emboj.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bergink S., Salomons F.A., Hoogstraten D. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev. 2006;20:1343–1352. doi: 10.1101/gad.373706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan M.R., Peng G., Hung W.C., Lin S.Y. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J Biol Chem. 2011;286:28599–28607. doi: 10.1074/jbc.M111.256297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu C.Y., Kang H.Y., Yang W.L. Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J Biol Chem. 2011;286:30806–30815. doi: 10.1074/jbc.M111.257469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li W., Nagaraja S., Delcuve G.P., Hendzel M.J., Davie J.R. Effects of histone acetylation, ubiquitination and variants on nucleosome stability. Biochem J. 1993;296:737–744. doi: 10.1042/bj2960737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chagraoui J., Hebert J., Girard S., Sauvageau G. An anticlastogenic function for the Polycomb Group gene Bmi1. Proc Natl Acad Sci U S A. 2011;108:5284–5289. doi: 10.1073/pnas.1014263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campisi J., d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 86.Grandinetti K.B., David G. Sin3B: an essential regulator of chromatin modifications at E2F target promoters during cell cycle withdrawal. Cell Cycle. 2008;7:1550–1554. doi: 10.4161/cc.7.11.6052. [DOI] [PubMed] [Google Scholar]
- 87.DiMauro T., Cantor D.J., Bainor A.J., David G. Transcriptional repression of Sin3B by Bmi-1 prevents cellular senescence and is relieved by oncogene activation. Oncogene. 2015;34:4011–4017. doi: 10.1038/onc.2014.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang F., Sui L., Xin T. Correlations of BMI-1 expression and telomerase activity in ovarian cancer tissues. Exp Oncol. 2008;30:70–74. [PubMed] [Google Scholar]
- 89.Greenberg R.A., O'Hagan R.C., Deng H. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene. 1999;18:1219–1226. doi: 10.1038/sj.onc.1202669. [DOI] [PubMed] [Google Scholar]
- 90.Wu K.J., Grandori C., Amacker M. Direct activation of TERT transcription by c-MYC. Nat Genet. 1999;21:220–224. doi: 10.1038/6010. [DOI] [PubMed] [Google Scholar]
- 91.Chiba T., Seki A., Aoki R. Bmi1 promotes hepatic stem cell expansion and tumorigenicity in both Ink4a/Arf-dependent and -independent manners in mice. Hepatology. 2010;52:1111–1123. doi: 10.1002/hep.23793. [DOI] [PubMed] [Google Scholar]
- 92.Hosen N., Yamane T., Muijtjens M., Pham K., Clarke M.F., Weissman I.L. Bmi-1-green fluorescent protein-knock-in mice reveal the dynamic regulation of bmi-1 expression in normal and leukemic hematopoietic cells. Stem Cells. 2007;25:1635–1644. doi: 10.1634/stemcells.2006-0229. [DOI] [PubMed] [Google Scholar]
- 93.Lukacs R.U., Memarzadeh S., Wu H., Witte O.N. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell. 2010;7:682–693. doi: 10.1016/j.stem.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tian H., Biehs B., Warming S. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zacharek S.J., Fillmore C.M., Lau A.N. Lung stem cell self-renewal relies on BMI1-dependent control of expression at imprinted loci. Cell Stem Cell. 2011;9:272–281. doi: 10.1016/j.stem.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ito K., Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Conboy I.M., Rando T.A. Aging, stem cells and tissue regeneration: lessons from muscle. Cell Cycle. 2005;4:407–410. doi: 10.4161/cc.4.3.1518. [DOI] [PubMed] [Google Scholar]
- 98.Pekovic V., Hutchison C.J. Adult stem cell maintenance and tissue regeneration in the ageing context: the role for A-type lamins as intrinsic modulators of ageing in adult stem cells and their niches. J Anat. 2008;213:5–25. doi: 10.1111/j.1469-7580.2008.00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Paranjape A.N., Balaji S.A., Mandal T. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC Cancer. 2014;14:785. doi: 10.1186/1471-2407-14-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zencak D., Lingbeek M., Kostic C. Bmi1 loss produces an increase in astroglial cells and a decrease in neural stem cell population and proliferation. J Neurosci. 2005;25:5774–5783. doi: 10.1523/JNEUROSCI.3452-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iwama A., Oguro H., Negishi M. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–851. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 102.Oguro H., Yuan J., Ichikawa H. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell. 2010;6:279–286. doi: 10.1016/j.stem.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 103.Bernstein B.E., Mikkelsen T.S., Xie X. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 104.Lowe S.W., Sherr C.J. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 105.Oguro H., Iwama A., Morita Y., Kamijo T., van Lohuizen M., Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med. 2006;203:2247–2253. doi: 10.1084/jem.20052477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bruggeman S.W., Valk-Lingbeek M.E., van der Stoop P.P. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005;19:1438–1443. doi: 10.1101/gad.1299305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fasano C.A., Dimos J.T., Ivanova N.B., Lowry N., Lemischka I.R., Temple S. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell. 2007;1:87–99. doi: 10.1016/j.stem.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 108.Janzen V., Forkert R., Fleming H.E. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 109.Molofsky A.V., Slutsky S.G., Joseph N.M. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen K., Huang Y.H., Chen J.L. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–740. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reya T., Morrison S.J., Clarke M.F., Weissman I.L. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 112.Szotek P.P., Pieretti-Vanmarcke R., Masiakos P.T. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154–11159. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Haupt Y., Alexander W.S., Barri G., Klinken S.P., Adams J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- 114.Jacobs J.J., Scheijen B., Voncken J.W., Kieboom K., Berns A., van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13:2678–2690. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Glinsky G.V., Berezovska O., Glinskii A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cui H., Hu B., Li T. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am J Pathol. 2007;170:1370–1378. doi: 10.2353/ajpath.2007.060754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leung C., Lingbeek M., Shakhova O. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 118.Hemmati H.D., Nakano I., Lazareff J.A. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hayry V., Tynninen O., Haapasalo H.K. Stem cell protein BMI-1 is an independent marker for poor prognosis in oligodendroglial tumours. Neuropathol Appl Neurobiol. 2008;34:555–563. doi: 10.1111/j.1365-2990.2008.00949.x. [DOI] [PubMed] [Google Scholar]
- 120.Abdouh M., Facchino S., Chatoo W., Balasingam V., Ferreira J., Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. 2009;29:8884–8896. doi: 10.1523/JNEUROSCI.0968-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Godlewski J., Nowicki M.O., Bronisz A. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 122.Wang X., Venugopal C., Manoranjan B. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene. 2012;31:187–199. doi: 10.1038/onc.2011.232. [DOI] [PubMed] [Google Scholar]
- 123.Bhattacharya R., Kwon J., Ali B. Role of hedgehog signaling in ovarian cancer. Clin Cancer Res. 2008;14:7659–7666. doi: 10.1158/1078-0432.CCR-08-1414. [DOI] [PubMed] [Google Scholar]
- 124.van Leenders G.J., Dukers D., Hessels D. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol. 2007;52:455–463. doi: 10.1016/j.eururo.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 125.Song L.B., Zeng M.S., Liao W.T. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res. 2006;66:6225–6232. doi: 10.1158/0008-5472.CAN-06-0094. [DOI] [PubMed] [Google Scholar]
- 126.Song L.B., Li J., Liao W.T. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009;119:3626–3636. doi: 10.1172/JCI39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chou C.H., Yang N.K., Liu T.Y. Chromosome instability modulated by BMI1-AURKA signaling drives progression in head and neck cancer. Cancer Res. 2013;73:953–966. doi: 10.1158/0008-5472.CAN-12-2397. [DOI] [PubMed] [Google Scholar]
- 128.Yang M.H., Hsu D.S., Wang H.W. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12:982–992. doi: 10.1038/ncb2099. [DOI] [PubMed] [Google Scholar]
- 129.Prince M.E., Sivanandan R., Kaczorowski A. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guo B.H., Feng Y., Zhang R. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer. 2011;10:10. doi: 10.1186/1476-4598-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hoenerhoff M.J., Chu I., Barkan D. BMI1 cooperates with H-RAS to induce an aggressive breast cancer phenotype with brain metastases. Oncogene. 2009;28:3022–3032. doi: 10.1038/onc.2009.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kreso A., van Galen P., Pedley N.M. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20:29–36. doi: 10.1038/nm.3418. [DOI] [PubMed] [Google Scholar]
- 133.Wang H., Pan K., Zhang H.K. Increased polycomb-group oncogene Bmi-1 expression correlates with poor prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134:535–541. doi: 10.1007/s00432-007-0316-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Maynard M.A., Ferretti R., Hilgendorf K.I., Perret C., Whyte P., Lees J.A. Bmi1 is required for tumorigenesis in a mouse model of intestinal cancer. Oncogene. 2014;33:3742–3747. doi: 10.1038/onc.2013.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sawa M., Yamamoto K., Yokozawa T. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int J Hematol. 2005;82:42–47. doi: 10.1532/IJH97.05013. [DOI] [PubMed] [Google Scholar]
- 136.Rizo A., Olthof S., Han L., Vellenga E., de Haan G., Schuringa J.J. Repression of BMI1 in normal and leukemic human CD34(+) cells impairs self-renewal and induces apoptosis. Blood. 2009;114:1498–1505. doi: 10.1182/blood-2009-03-209734. [DOI] [PubMed] [Google Scholar]
- 137.Yuan J., Takeuchi M., Negishi M., Oguro H., Ichikawa H., Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia. 2011;25:1335–1343. doi: 10.1038/leu.2011.85. [DOI] [PubMed] [Google Scholar]
- 138.Dovey J.S., Zacharek S.J., Kim C.F., Lees J.A. Bmi1 is critical for lung tumorigenesis and bronchioalveolar stem cell expansion. Proc Natl Acad Sci U S A. 2008;105:11857–11862. doi: 10.1073/pnas.0803574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim C.F., Jackson E.L., Woolfenden A.E. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 140.Lee K., Adhikary G., Balasubramanian S. Expression of Bmi-1 in epidermis enhances cell survival by altering cell cycle regulatory protein expression and inhibiting apoptosis. J Invest Dermatol. 2008;128:9–17. doi: 10.1038/sj.jid.5700949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li Z., Wang Y., Yuan C. Oncogenic roles of Bmi1 and its therapeutic inhibition by histone deacetylase inhibitor in tongue cancer. Lab Invest. 2014;94:1431–1445. doi: 10.1038/labinvest.2014.123. [DOI] [PubMed] [Google Scholar]
- 142.Jung J.W., Lee S., Seo M.S. Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cell Mol Life Sci. 2010;67:1165–1176. doi: 10.1007/s00018-009-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu J., Hu D., Yang G. Down-regulation of BMI-1 cooperates with artemisinin on growth inhibition of nasopharyngeal carcinoma cells. J Cell Biochem. 2011;112:1938–1948. doi: 10.1002/jcb.23114. [DOI] [PubMed] [Google Scholar]
- 144.Chou D.M., Adamson B., Dephoure N.E. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Levine S.S., Weiss A., Erdjument-Bromage H., Shao Z., Tempst P., Kingston R.E. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol. 2002;22:6070–6078. doi: 10.1128/MCB.22.17.6070-6078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Boccuni P., MacGrogan D., Scandura J.M., Nimer S.D. The human L(3)MBT polycomb group protein is a transcriptional repressor and interacts physically and functionally with TEL (ETV6) J Biol Chem. 2003;278:15412–15420. doi: 10.1074/jbc.M300592200. [DOI] [PubMed] [Google Scholar]
- 147.Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Denisenko O., Shnyreva M., Suzuki H., Bomsztyk K. Point mutations in the WD40 domain of Eed block its interaction with Ezh2. Mol Cell Biol. 1998;18:5634–5642. doi: 10.1128/mcb.18.10.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cao R., Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 150.Wilkinson F.H., Park K., Atchison M.L. Polycomb recruitment to DNA in vivo by the YY1 REPO domain. Proc Natl Acad Sci U S A. 2006;103:19296–19301. doi: 10.1073/pnas.0603564103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kirmizis A., Bartley S.M., Kuzmichev A. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004;18:1592–1605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Haupt Y., Bath M.L., Harris A.W., Adams J.M. bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene. 1993;8:3161–3164. [PubMed] [Google Scholar]
- 153.Proctor E., Waghray M., Lee C.J. Bmi1 enhances tumorigenicity and cancer stem cell function in pancreatic adenocarcinoma. PloS One. 2013;8:e55820. doi: 10.1371/journal.pone.0055820. [DOI] [PMC free article] [PubMed] [Google Scholar]