

Abstract
The molecular characterization of various cancers has shown that cancers with the same origins, histopathologic diagnoses, and clinical stages can be highly heterogeneous in their genetic and epigenetic alterations that cause tumorigenesis. A number of cancer driver genes with functional abnormalities that trigger malignant transformation and that are required for the survival of cancer cells have been identified. Therapeutic agents targeting some of these cancer drivers have been successfully developed, resulting in substantial improvements in clinical symptom amelioration and outcomes in a subset of cancer patients. However, because such therapeutic drugs often benefit only a limited number of patients, the successes of clinical development and applications rely on the ability to identify those patients who are sensitive to the targeted therapies. Thus, biomarkers that can predict treatment responses are critical for the success of precision therapy for cancer patients and of anticancer drug development. This review discusses the molecular heterogeneity of lung cancer pathogenesis; predictive biomarkers for precision medicine in lung cancer therapy with drugs targeting epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), c-ros oncogene 1 receptor tyrosine kinase (ROS1), and immune checkpoints; biomarkers associated with resistance to these therapeutics; and approaches to identify predictive biomarkers in anticancer drug development. The identification of predictive biomarkers during anticancer drug development is expected to greatly facilitate such development because it will increase the chance of success or reduce the attrition rate. Additionally, such identification will accelerate the drug approval process by providing effective patient stratification strategies in clinical trials to reduce the sample size required to demonstrate clinical benefits.
Keywords: Precision medicine, Personalized therapy, Epidermal growth factor receptor (EGFR), Anaplastic lymphoma kinase (ALK), Immunotherapy, Biomarkers, Drug development
Background
A recent study analyzing the global disease burden revealed that lung cancer is the second leading cause of death in the United States and in most other high-income countries [1]. In China, lung cancer is the fifth leading cause of death, following stroke, ischemic heart disease, road injuries, and chronic obstructive pulmonary disease [1, 2]. Globally, the annual incidence of lung cancer is approximately 1.8 million, with an annual mortality of approximately 1.6 million [3, 4]. The 5-year overall survival (OS) rate for lung cancer patients in the United States has improved only moderately over the past four decades from 12.2% in 1975 to 17.8% in 2010, despite the use of many therapeutic modalities [5–7]. The 5-year OS rate of lung cancer patients in China is approximately 16.1% [8], which is close to that observed in the United States. Histologically, lung cancer is classified as small cell lung cancer (SCLC, approximately 15%) and non-small cell lung cancer (NSCLC, approximately 85%); the latter includes adenocarcinoma, squamous cell carcinoma, and large cell carcinoma. Tobacco smoking is the major cause for all types of lung cancer, particularly SCLC and squamous cell carcinoma [9]. Air pollution is another major cause of lung cancer, as a substantial number of lung cancer deaths in China and other East Asian countries may be attributed to fine particles in the air [10–12].
Small cell lung cancer is highly aggressive, and most (95%) SCLC patients are current or former smokers [13, 14]. Although the initial response rate to platinum-based chemotherapy and radiotherapy is high, recurrence and resistance occur inevitably in most SCLC patients, leading to 5-year OS rates of approximately 24% for patients with limited-stage SCLC and of less than 3% for those with metastases [5]. Fortunately, the incidence of SCLC in the United States has been declining over the past decades because of a decreasing prevalence of tobacco smoking [15]. For NSCLC, surgical resection is the standard care for patients with stage I disease, whereas surgical resection plus adjuvant therapy is used for patients with stages II–III disease. NSCLC patients at the advanced stages (stages IIIB–IV) are usually treated with chemotherapy, pathway-targeted therapies, and/or supportive medicine. Early diagnosis is the major factor proven to improve outcomes [16]; the 5-year OS rate is approximately 70% for patients with stage I NSCLC and drops to approximately 5% for patients with stage IV lung cancer [17]. Unfortunately, most lung cancers are diagnosed at an advanced stage and thus have a poor prognosis [9]. Early detection by low-dose computed tomography (CT) screening significantly reduces lung cancer mortality; nevertheless, approximately 96% of the positive results detected by such screening are false positives [18]. Moreover, the current estimated cost of screening to avoid one premature lung cancer death is approximately $240,000, thereby adding substantial expenditures to the health care system [19].
Advances in molecular profiling and targeted therapy have shown that subgroups of lung cancer patients are highly sensitive to some small-molecule inhibitors targeting key molecular nodes that drive carcinogenesis in those patients, such as epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK). These breakthrough discoveries have not only led to a new paradigm of biomarker-directed precision or personalized therapy but also greatly accelerated the development of novel anticancer drugs. It is now clear that mutations in cancer driver genes and alterations in gene expression and/or posttranscriptional modifications can all drastically affect treatment responses or clinical outcomes [20–22]. Moreover, the success of targeted anticancer therapy largely depends on biomarkers that can identify patient subgroups that may respond to specific therapeutic agents because alterations in a particular cancer driver usually exist in only a small subset of patients. This review discusses recent advances in predictive biomarker-directed precision medicine and drug development for lung cancers.
Molecular heterogeneity in lung cancer
Genome-wide gene sequencing analyses have found that each lung cancer may have an average of approximately 150 somatic mutations that are expected to alter their protein products; this number is much higher than the average of 30–60 mutations observed in other solid tumors [23]. The high somatic mutation rates observed in lung cancer reflect the mutagenic roles of cigarette smoking in the pathogenesis of lung cancer. Indeed, the total number of point mutations in coding regions identified in lung cancer is approximately 10 times higher in smokers than in non-smokers [24]. Smoking also causes distinct changes in gene mutation and gene expression signatures in cancer tissues and normal lung tissues [25, 26]. Smoking-induced gene expression alterations observed in normal lung tissues can be transient because the majority of changes at the expression level can revert to non-smoker levels following smoking cessation [26]. Nevertheless, some of these expression changes are irreversible and permanent [26].
The results from genome-wide sequencing analyses for primary lung adenocarcinoma [24, 25, 27, 28], squamous cell cancer [29], SCLC [30, 31], and carcinoids or neuroendocrine tumors [32] have been reported recently by several groups. Information regarding genetic alterations (mutations and copy number changes), mRNA expression data, and protein/protein phosphorylation levels in lung adenocarcinoma, squamous cell cancer, and SCLC can be retrieved from The Cancer Genome Atlas (TCGA) databases and publically available datasets at the website http://www.cbioportal.org. Figure 1 shows the frequencies of cancer driver genes that are frequently mutated (including copy number changes) in lung adenocarcinoma, squamous cell cancer, and SCLC. Based on data retrieved from cBioPortal, tumor protein p53 (TP53) is the most frequently mutated cancer driver gene for all three histological types of lung cancer, with frequencies varying from 46% in adenocarcinoma to 86% in SCLC. Additionally, Kirsten rat sarcoma viral oncogene homolog (KRAS), cyclin-dependent kinase inhibitor 2A (CDKN2A), mixed-lineage leukemia 3 (MLL3), serine/threonine kinase 11 (STK11), kelch-like ECH-associated protein 1 (KEAP1), and EGFR are the top frequently mutated genes detected in lung adenocarcinomas. Phosphatidylinositol-4,5-biphosphate 3-kinase, catalytic subunit alpha (PIK3CA), sex-determining region Y-related gene family 2 (SOX2), CDKN2A, TP63, fibroblast growth factor receptor 1(FGFR1), and MLL2 are the top mutant genes in squamous cell cancer; retinoblastoma 1 (RB1), E1A-binding protein p300 (EP300), MLL2, smoothened (SMO), and PIK3CA are the top mutant genes in SCLC. Our recent sequencing analysis of the exomes of 200 cancer-related genes found that mutations in TP53, KRAS, MLL3, SET domain-containing 2 (SETD2), AT rich interactive domain 1A (ARID1A), PIK3CA, and ALK were frequently detected in primary tumors and corresponding patient-derived xenografts of NSCLC [33]. Most of the mutations (93%) detected in the primary tumors were also detected in their corresponding patient-derived xenografts. Nevertheless, the numbers of mutations detected in each primary tumor varied greatly, and most tumors had mutations in more than two genes. The results from these molecular profiling studies clearly demonstrated heterogeneity in the molecular pathogenesis of lung cancer. As shown in Figure 2a, the mutational status of the top seven frequently mutated cancer driver genes varied greatly in 230 lung adenocarcinomas [28]. Moreover, mutations in those genes, particularly for tumor suppressor genes such as TP53 (Figure 2b), are often widely distributed in the entire coding region. Evidence has shown that mutations in TP53 can lead to either loss or gain of functions [34–38], and both may promote tumorigenesis through different mechanisms. Similarly, different mutations in KRAS, even if in the same codon such as G12C, G12V, and G12D mutations in the KRAS gene, may lead to different conformational changes in KRAS and have different effects on clinical outcomes and molecular pathway activation [39]. Ultimately, this molecular heterogeneity may affect treatment responses to therapeutics targeting different pathways.
Figure 1.
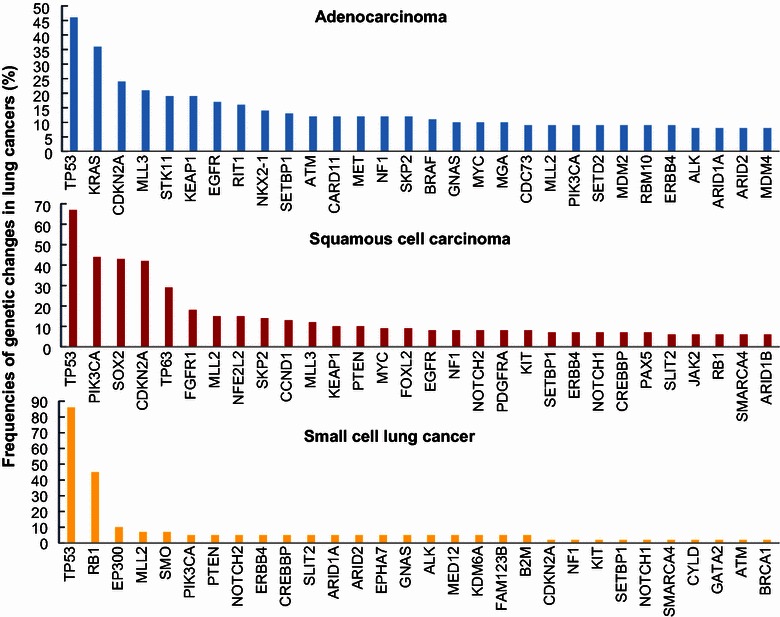
Frequencies of genetic alternations (mutations and copy number changes) in cancer driver genes. The frequencies for the top 30 mutated genes in lung adenocarcinoma, squamous cell carcinoma, and small cell lung cancer were retrieved from http://www.cbioportal.org. TP53 tumor protein p53; KRAS Kirsten rat sarcoma viral oncogene homolog; CDKN2A cyclin-dependent kinase inhibitor 2A; MLL3 mixed-lineage leukemia 3; STK11 serine/threonine kinase 11; KEAP1 kelch-like ECH-associated protein 1; EGFR epidermal growth factor receptor; RIT1 Ras-like without CAAX 1; NKX2-1 NK2 homeobox 1; SETBP1 SET-binding protein 1; ATM ataxia telangiectasia-mutated; CARD11 caspase recruitment domain family, member 11; MET MET proto-oncogene; NF1 neurofibromin1; SKP2 S-phase kinase-associated protein 2; BRAF v-Raf murine sarcoma viral oncogene homolog B1; GNAS GNAS complex locus; MYC v-myc avian myelocytomatosis viral oncogene homolog; MGA MAX dimerization protein; CDC73 cell division cycle 73; PIK3CA phosphatidylinositol-4,5-biphosphate 3-kinase, catalytic subunit alpha; SETD2 SET domain-containing 2; MDM2 mouse double minute 2 homolog; RBM10 RNA-binding motif protein 10; ERBB4 erb-b2 receptor tyrosine kinase 4; ALK anaplastic lymphoma kinase; ARID1A AT rich interactive domain 1A; SOX2 sex-determining region Y-related gene family 2; FGFR1 fibroblast growth factor receptor 1; NFE2L2 nuclear factor, erythroid 2-like 2; CCND1 cyclin D1; PTEN phosphatase and tensin homolog; FOXL2 forkhead box L2; NOTCH2 notch 2; PDGFRA platelet-derived growth factor receptor, alpha polypeptide; KIT v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; CREBBP CREB-binding protein; PAX5 paired box 5; SLIT2 slit homolog 2; JAK2 Janus kinase 2; RB1 retinoblastoma 1; SMARCA4 SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4; EP300 E1A-binding protein p300; SMO smoothened; EPHA7 EPH receptor A7; MED12 mediator complex subunit 12; KDM6A lysine (K)-specific demethylase 6A; FAM123B family with sequence similarity 123B; B2M beta-2-microglobulin; CYLD cyclin dromatosis (turban tumor syndrome); GATA2 GATA-binding protein 2; BRCA1 breast cancer early onset.
Figure 2.
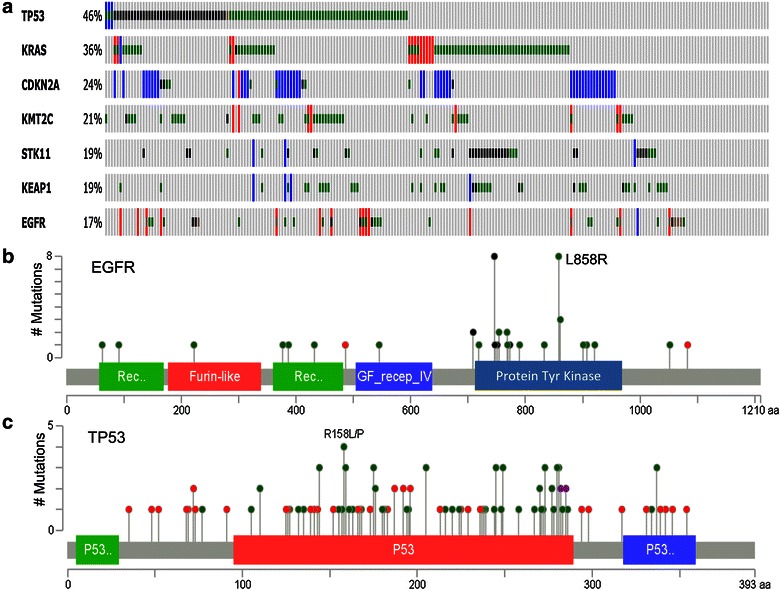
Molecular heterogeneity in lung adenocarcinoma. a The status of genetic alterations in 230 lung adenocarcinomas retrieved from The Cancer Genome Atlas (TCGA) database [28]. The mutation frequencies (%) are shown on the left of graph. Each vertical line represents a tumor. The graph shows mutations in the top seven cancer driver genes in lung adenocarcinoma. Red amplification, blue homozygous deletion, green missense mutation, black truncating mutation, brown in-frame mutation. b, c Mutations in EGFR and TP53 in the same 230 adenocarcinomas. The gray bars represent the entire lengths of the EGFR and TP53 proteins, with the number of amino acids indicated at the bottom of each gray bar. The green, red, and blue boxes indicate protein domains. The lines and dots indicate the locations and frequencies of mutations detected in the EGFR and TP53 genes. Green missense mutations, red nonsense or frameshift mutations, black in-frame deletions. The data were retrieved from http://www.cbioportal.org.
Notably, however, current TCGA data are derived mostly from patients in Western countries. The mutational status in other ethnic populations may differ [40–42]. For example, activated EGFR mutations are detected in approximately 10–17% of lung adenocarcinoma patients in the United States and Europe [27, 43–46] but in approximately 30–65% of lung adenocarcinoma patients in Asia [47–50]. In contrast, KRAS mutations were detected in 35–50% of lung adenocarcinomas in Caucasian patients [28, 51] but in less than 5% of lung adenocarcinomas in Chinese patients [48, 49, 52]. Nevertheless, changes in molecular signaling can occur in the absence of gene mutations. Indeed, the activation of RAS signaling pathways can be detected in substantial numbers of primary tumors with wild-type RAS genes [53]. Increased expression of the RAS genes [54, 55], increased upstream signaling from tyrosine kinase growth factor receptors such as v-erbB2 avian erythroblastic leukemia viral oncogene homolog 2 (HER2) and EGFR [56, 57], and altered expression of microRNAs such as let-7 [58, 59] may all contributes to the activation of RAS signaling pathways.
Biomarker-directed precision medicine for lung cancer
Anti-EGFR therapy
Epidermal growth factor receptor, a major driver in lung tumorigenesis, has been extensively investigated as a target for anticancer therapy. As noted above, activating EGFR mutations are detected in substantial numbers of lung cancer patients and are more commonly observed in women and non-smokers [27, 43–46]. The finding that EGFR mutations in primary lung cancer are associated with sensitivity to EGFR inhibitors gefitinib and erlotinib [44, 60, 61] contributed greatly to the final approval of these therapeutics for the treatment of lung cancer. Subsequently, EGFR gene mutations have served as biomarkers in the clinic for the identification of responders to EGFR inhibitors. Both gefitinib and erlotinib (Figure 3) are the first choice for therapy in lung cancer patients whose tumors harbor EGFR mutations and are reported to significantly prolong progression-free survival (PFS) in patients with EGFR-mutant lung cancer [62–64]. Icotinib, which has a clinical efficacy similar to that of gefitinib but less adverse effects [65], is approved for the treatment of EGFR-mutant lung cancer in China.
Figure 3.
Chemical structures of anti-EGFR therapeutic agents.
As shown in Figure 2, mutations in EGFR are also heterogeneous. There are multiple locations in the EGFR gene where driver mutations can occur, and these mutations associate with differential sensitivity to EGFR inhibition. The most common EGFR mutations are in-frame deletions in exon 19 and L858R point mutations in exon 21. Together, these mutations account for approximately 85% of the EGFR mutations observed in lung cancer [46, 48, 66]; tumors with these mutations are highly sensitive to treatment with erlotinib, gefitinib, and the second-generation EGFR inhibitor afatinib [63, 67–69]. Tumors with other less common mutations, such as exon 18 mutations at G719 and exon 21 mutation L861Q, are also susceptible to treatment with EGFR inhibitors [60, 70]. Nevertheless, tumors with exon 20 in-frame insertions, which account for approximately 4–10% of all EGFR mutations, usually do not respond to treatment with EGFR inhibitors [71–73]. Although these insertional mutations activate EGFR, they do not have increased affinity for EGFR inhibitors, as is observed for other EGFR mutations [74].
However, despite initial dramatic responses to gefitinib and erlotinib in patients with EGFR-mutant lung cancers, acquired resistance inevitably occurs at a median of 10–13 months after treatment initiation [69, 75, 76]. A number of mechanisms for this acquired resistance have been identified, including a secondary T790M mutation at exon 20 of the EGFR gene [77–79], amplification of the MET proto-oncogene (MET) [80–82] or HER2 gene [83, 84], mutations of the KRAS gene [85], activation of AXL receptor tyrosine kinase (AXL), V-Srcavian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (SRC) kinases [86–88], and extracellular signal-regulated kinase (ERK) [89], loss of phosphatase and tensin homolog (PTEN) [90], and activation of the nuclear factor-κB (NF-κB) [91] or interleukin-6 receptor (IL-6R)/signal transducer and activator of transcription 3 (STAT3) pathway [92, 93]. Among these mechanisms, the most common cause of resistance observed clinically is acquisition of the EGFRT790M mutation, which is found in approximately 50% of patients [77, 80, 94]. A number of second-generation EGFR inhibitors, most of which are irreversible, inhibit two or more receptors in the EGFR family, and may have some activity in T790M-mutant cancers, have been evaluated in clinical trials [95–98]. Among those evaluated in clinical trials, only afatinib [96, 99] has been approved (in July 2013) by the United States Food and Drug Administration (FDA) for the treatment of metastatic NSCLC with EGFR mutations. Unfortunately, diarrhea and rash, the two most common adverse effects associated with a good response to the therapy, appear to be more severe in patients treated with afatinib than those treated with erlotinib or gefitinib [100]. More recently, the third-generation EGFR inhibitors AZD9291 [101] and CO-1686 [102, 103] have shown promising anticancer activity in T790M-mutant tumors. Our recent studies have revealed that ibrutinib, an irreversible inhibitor of Bruton tyrosine kinase (BTK) that was recently approved by the FDA for the treatment of mantle cell lymphoma and chronic lymphocytic leukemia [104, 105], can function as an EGFR inhibitor to selectively inhibit growth and induce apoptosis in EGFR-mutant NSCLC cells in vitro and in vivo, including erlotinib-resistant cells that harbor a T790M mutation [106]. Because ibrutinib was recently reported to be well tolerated when used in combination with other targeted therapeutics [107, 108], it may have advantages in combination therapy to overcome resistance caused by alternative or bypass survival signals from cooperative or parallel pathways, as is observed in T790M-negative resistant cells.
Anti-ALK/ROS1 therapy
The discovery of gene rearrangements between the echinoderm microtubule-associated protein-like 4 (EML4) gene and the ALK gene that lead to constitutive activation of ALK in NSCLC [109, 110] greatly contributed to the subsequent development and approval of crizotinib, a small-molecule inhibitor of ALK, c-ros oncogene 1 receptor tyrosine kinase (ROS1), and MET. EML4/ALK rearrangements are detected in approximately 4–5% of patients with NSCLC, mostly in young patients with adenocarcinoma who are non-smokers or light smokers [111–114]. ALK rearrangements were found to be mutually exclusive with EGFR and KRAS mutations, although they share common downstream signaling pathways with EGFR such as the RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, and JAK/STAT pathways. Crizotinib (Figure 4) was originally developed as an MET inhibitor; however, crizotinib also inhibits ALK and ROS1 [115]. Soon after the first reports of EML4/ALK rearrangement and its role in lung cancer oncogenesis [109, 110], a diagnostic method for detecting EML4/ALK rearrangement by fluorescence in situ hybridization (FISH) was developed, and patients with EML4/ALK rearrangement-positive lung cancer were enrolled in clinical trials with crizotinib [114, 116]. Most of the patients with advanced ALK-positive NSCLC responded to crizotinib treatment, resulting in tumor shrinkage or disease stabilization. The objective response rate was approximately 60%, and the median PFS was 8–10 months [117, 118]. In comparison, conventional chemotherapy for the same type of patients achieved a response rate of approximately 20% and a PFS of 2–3 months [117, 118]. Subsequently, crizotinib was approved for the treatment of ALK-positive NSCLC in conjunction with an FDA-approved diagnostic assay, the Vysis ALK BreakApart FISH Probe Kit.
Figure 4.

Chemical structures of anti-ALK/ROS1 therapeutic agents. ROS1 c-ros oncogene 1 receptor tyrosine kinase.
ROS1 gene rearrangement and activation are detected in approximately 1–1.5% of NSCLC, predominantly in adenocarcinoma and in non-smokers, as is observed with ALK rearrangement [119–121]. However, ROS1 and ALK rearrangements rarely occur in the same tumors. Because the kinase domains of ALK and ROS1 share 77% homology at the protein sequence level and crizotinib is highly active for both ALK and ROS1 [115], clinical investigation into the treatment of NSCLC patients with ROS1 rearrangement has been pursued. The results indicated that crizotinib also has marked antitumor activity in patients with ROS1-rearranged advanced NSCLC, with an objective response rate of 72% and a median response duration of approximately 17.6 months [122]. The response duration in ROS1 rearrangement-positive patients appears to be longer than that in ALK-positive patients (10–11 months). Similar to the diagnosis of ALK rearrangement, FISH and/or reverse transcription-polymerase chain reaction (RT-PCR) are used to identify patients with ROS1 rearrangement [122]. Nevertheless, resistance to crizotinib eventually occurs in both ALK- and ROS1-positive patients.
Among the patients with ALK-positive NSCLC who acquired resistance to crizotinib, one-third had various secondary mutations in the ALK kinase domain and/or had ALK gene amplification [111]. Resistance can also be caused by aberrant activation of alternative or bypass pathways, such as marked v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) amplification and EGFR activation [123]. In some patients, multiple resistance mechanisms may exist simultaneously, indicating that combination therapy will be required to overcome resistance [124, 125]. Second-generation ALK inhibitors have been developed to overcome crizotinib resistance caused by mutations in the ALK kinase domain. Among these ALK inhibitors, ceritinib has been approved by the FDA for the treatment of ALK-positive NSCLC in the United States, and alectinib has been approved in Japan [126, 127] (Figure 4). Ceritinib is reported to be effective in patients with various resistant mutations. Of the patients who had received crizotinib previously, 56% responded to ceritinib treatment, including patients with central nervous system metastases [126, 128]. Nevertheless, ceritinib resistance has been observed in the clinic. Resistant cells obtained from patients treated with ceritinib revealed activation of the EGFR, mitogen-activated protein (MAP) kinase, and SRC pathways, and the cells were sensitive to combination therapies with inhibitors for EGFR, MAP kinase-ERK kinase (MEK), and SRC [129]. MET activation has been reported to cause resistance to alectinib because, as an ALK selective inhibitor, alectinib does not inhibit MET [130].
Immunotherapy
The presence of tumor antigens derived from mutant proteins, dysregulated or overexpressed proteins, and viral oncogenic proteins has inspired intensive investigation into anticancer immunotherapy through various approaches [131]. The high mutation rates observed in lung cancers suggest that lung cancer cells may possess high levels of tumor antigens from mutant proteins and may therefore be vulnerable to immunotherapy. However, during malignant evolution, cancer cells obtain the ability to escape immune system recognition and destruction through various mechanisms, including inhibiting tumor antigen presentation by the down-regulation of class I major histocompatibility complex (MHC) molecule expression on the surface of tumor cells, recruiting immunosuppressive cells (such as regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages) to the tumor microenvironment, secreting immunosuppressive cytokines [such as IL-10 and tumor growth factor-β (TGF-β)], activating immune co-inhibitory checkpoint receptors [such as cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and programmed death-1 (PD-1)], and inhibiting immune co-stimulatory receptors (CD40, OX40, CD137, and CD28) [132–134]. Advances in the mechanistic characterization of tumor cell evasion of immunosurveillance have led to the development of various approaches that aim to mobilize the immune system for the elimination of malignant cells. Currently, the most advanced available therapeutics developed for lung cancer are monoclonal antibodies that target checkpoints of the immune regulation cascade.
Immune checkpoints regulate the amplitude and duration of immune responses, preventing damage to normal tissues from overactivation of the immune system [132–134]. The key players in the checkpoints include CTLA-4, lymphocyte activation gene-3 (LAG-3), and PD-1, as well as the PD-1 ligands PD-L1 and PD-L2. CTLA-4 is constitutively expressed in regulatory T cells but only transiently expressed in activated cytotoxic T cells. It shares common ligands, CD80 and CD86, with the co-stimulatory receptor CD28. The interaction of CD28 with these ligands leads to the full activation of cytotoxic T cells, whereas the interaction of CTLA-4 with the ligands prevents T cell activation. The up-regulation of CTLA-4 was detected in anergic lymphocytes, and the blockade of CTLA-4 with anti-CTLA-4 antibodies resulted in antitumor activity [135]. The anti-CTLA-4 antibody ipilimumab is approved for the treatment of melanoma [136] and is currently in clinical trials for the treatment of NSCLC. PD-1 (also known as PDCD1 or CD279) is expressed in T, B, and natural killer (NK) cells. The interaction of PD-1 with its ligands, PD-L1 (also called B7-H1 or CD274) and PD-L2 (B7-DC), suppresses T cell activation and promotes T cell apoptosis [137–139], leading to immune evasion. The expression of PD-L1 in lung cancer is detected in 25–50% of NSCLC cases, depending on the assays used to detect its protein or mRNA [140], and is associated with the presence of EGFR mutations in NSCLC [141]. EGFR activation induced PD-L1 expression in bronchial epithelial cells, whereas treatment with EGFR inhibitors reduced PD-L1 expression [142]. PD-1 monoclonal antibodies (nivolumab and pembrolizumab) have been approved for the treatment of melanoma [143–146]. The objective response rates in melanoma patients are approximately 26–40% [143, 144, 146]. Nivolumab was approved for the treatment of NSCLC in the United States in March 2015 [147] and was found to be highly active in treating Hodgkin’s lymphoma [148]. Monoclonal antibodies for PD-L1 are also under intensive clinical evaluation for the treatment of NSCLC and other solid tumors [149, 150]. The objective response rates in patients with advanced NSCLC are approximately 17–20% in patients receiving ≥1 mg/kg of anti-PD1 or anti-PD-L1 antibodies every 2–3 weeks, and the responses may last 1 year or longer [147, 149–151]. The response rates to both anti-PD1 and anti-PD-L1 antibodies were significantly higher in PD-L1-positive patients than in PD-L1-negative patients [150, 151]. In a trial with an anti-PD1 antibody, 9 (36%) of 25 PD-L1-positive patients responded to the treatment, whereas none of 17 PD-L1-negative patients had an objective response [151]. The study with the anti-PD-L1 antibody also found that responses were often observed in patients with tumors expressing high levels of PD-L1, especially if PD-L1 was expressed by tumor-infiltrating immune cells. The expression of CTLA-4, the absence of fractalkine (CX3CL1) [150], and the presence of pre-existing CD8+ T cells distinctly located at the invasive tumor margin [145] were also found to be associated with treatment responses to the PD1/PD-L1 blockage antibodies. These results demonstrated the feasibility of using the expression of PD-L1 and the presence of immune cells in tumor sites for patient stratification for PD1/PD-L1 blockade therapy. Nevertheless, larger clinical trials will be required to further validate the associations between PD-L1 expression and improved clinical outcomes.
Predictive biomarkers in anticancer drug development
Anticancer drug development has a high failure or attrition rate, primarily because of a lack of efficacy, inability to identify responders, and intolerable toxic effects [152, 153]. Only approximately 5% of anticancer agents evaluated in human studies between 1991 and 2000 were successfully approved for clinical applications [152, 154]. The majority (80%) of phase III failures were caused by a lack of efficacy [152, 154–156] that resulted from a lack of efficacy proof-of-concept in humans and a lack of objective biomarkers capable of reporting such efficacy [152, 154]. The development of the anti-EGFR drugs gefitinib and erlotinib and the anti-ALK/ROS1 drugs crizotinib and ceritinib has provided excellent examples of using predictive biomarkers and patient stratifications in clinical trials to increase the chance of success or to reduce the attrition rate in anticancer drug development. Notably, both gefitinib [64, 157, 158] and erlotinib [159, 160] failed to show a benefit in randomized phase III trials with unselected patient populations. Similarly, the successes of the v-Raf murine sarcoma viral oncogene homolog B1 (BRAF) inhibitor vemurafenib [161, 162] and the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib [163–165] in anticancer therapy relied on biomarker-based patient stratifications in clinical trials. In fact, the inability to identify cancers that are most likely to respond to a treatment is a major challenge in anticancer drug development, not only leading to failure to demonstrate the potential benefit of a promising anticancer agent [64, 166] but also exposing patients to the risks of ineffective treatments. Indeed, a lack of predictive biomarkers for patient stratification has halted further development of many other investigational drugs [167–170]. Thus, reliable predictive biomarkers are crucial to the success of anticancer drug development, and the simultaneous development of predictive biomarkers will be highly desirable for promising anticancer drug candidates.
The identification of predictive biomarkers can be relatively straightforward if the therapeutic targets are themselves the cancer drivers and if the cancer cells are addicted to the presence of those targets for survival. As in the cases of EGFR, ALK, and ROS1, the constitutive activation of such targets is the driving cause of a subset of NSCLC and is required for the survival of those cancer cells; therefore, the presence of constitutive activating mutations or gene rearrangement can be explored as predictive biomarkers for patient stratification. Nevertheless, cancer cells can be killed indirectly through synthetic lethality between the cancer drivers and their synthetic lethal partners, as has been demonstrated using the PARP inhibitor olaparib to eliminate breast cancer early onset (BRCA)-mutant ovarian and breast cancer cells [165, 171, 172]. In this case, loss-of-function mutations in the BRCA1 and BRCA2 genes or other functional deficiencies in the DNA repair pathways will serve as predictive biomarkers for response to olaparib and other PARP inhibitors [173]. Synthetic lethality is a lethal phenotype caused by the combined effects of mutations in two or more genes. This concept has recently been exploited for the development of novel genotype-selective anticancer agents and/or the identification of novel therapeutic targets [173–176]. This approach could be useful for the indirect targeting of cancer drivers such as genetic changes in TP53, STK11, RB1, and RAS genes that are nondruggable or difficult to target using small molecules. Notably, a study conducting short hairpin RNA (shRNA) library screening for genes that may have synthetic lethal interactions in the oncogenic KRAS gene in the colon cancer cell line DLD-1 and its isogenic derivatives led to the identification of approximately 370–1,600 KRAS synthetic lethal genes, depending on the stringencies of statistical analyses [177], demonstrating the diversity of biological processes or pathways regulated by KRAS. The molecular pathology of RAS-mediated oncogenesis is further complicated by the fact that cancer cells with different mutations in KRAS, even if in the same codon (such as G12C, G12V, and G12D mutations), may lead to different conformational changes in KRAS, have different effects on clinical outcomes, and activate different molecular and metabolic pathways [39, 178]. Moreover, a substantial number of KRAS-mutant tumors have mutations in other cancer driver genes such as TP53, CDKN2A, and STK11. Studies in transgenic mice have found that activating mutations in KRAS predispose mice to early-onset lung adenocarcinoma [179, 180]. Nevertheless, such tumors do not have an invasive or metastatic phenotype [181]. Metastasis will occur only when additional genetic changes, such as Tp53 mutations or Stk11 deletion, are introduced [182, 183]. Finally, the RAS activation signature is observed in approximately 25% of KRAS wild-type cancers [53] because increased RAS activity in human cancers can result from RAS gene amplification [184, 185] or overexpression [55], an increase in upstream signals from tyrosine kinase growth factor receptors such as HER2 and EGFR [56, 57], or a decrease in the expression of microRNAs such as let-7 family members [58].
The above-discussed situations impose considerable challenges on the identification of predictive biomarkers for anti-RAS therapeutics developed through synthetic lethality screening. Substantial efforts will be required to identify predictive biomarkers for such therapeutic agents. Through synthetic lethality screening using a chemical library for isogenic cell lines with and without oncogenic KRAS, we previously reported a small molecule, designated oncrasin-1, that induces apoptosis in several KRAS-mutant lung cancer cell lines [186]. The in vitro and in vivo activities were optimized by the synthesis and evaluation of analogues [187–190]. The mechanistic characterization revealed that these compounds induce oxidative stress, activate c-Jun N-terminal kinase (JNK), and inhibit RNA polymerase II, STAT3, and cyclin D1 [187, 188, 191–194]. However, a predictive biomarker for oncrasin compounds remained elusive until our recent discovery that such compounds require the expression of a sulfotransferase (SULT), SULT1A1, in cancer cells for their anticancer activity and that the expression of SULT1A1 is capable of predicting the responses to oncrasin compounds [195]. SULT1A1 is a biotransformation enzyme that bioactivates several pro-carcinogens [196–202] and some anticancer drugs such as tamoxifen [203]. The identification of SULT1A1 as a predictive biomarker for oncrasin compounds shed light on mechanisms of the selectivity and, possibly, the toxicity of these compounds [204]. The process of identifying this predictive biomarker underscores the importance of activity characterization in a large set of molecularly annotated cancer cell lines and of collaborations between biostatisticians and molecular biologists. Statistical analysis of associations between the activity of NSC-743380 (oncrasin-72) and the gene expression profile in the NCI-60 cell line panel led to the identification of a number of candidate genes, and subsequent molecular characterizations demonstrated the causal relationship between the expression of SULT1A1 and the anticancer activity of NSC-743380, validating the feasibility of using SULT1A1 as a predictive marker for those compounds [195]. Larger panels of molecularly annotated cancer cell lines have also been used by other groups for the identification of predictive biomarkers for larger sets of anticancer drugs [150, 205–209]. Ultimately, rigorous validation of causal relationships between the sensitivity and the biomarker will be essential for successful clinical translations.
Conclusion
The diversity found in the molecular pathogenesis of lung cancer demonstrates that conventional histology-based classifications of the disease are not sufficient to provide guidance for treatment decisions in the clinic. Classifications based on molecular pathology are required for precision therapy. The successful development of anti-EGFR and anti-ALK/ROS1 therapeutics has proven the critical role of predictive biomarkers in anticancer drug development because the ability to identify patient subpopulations who may benefit from a drug candidate will directly affect the design and patient selection of clinical trials, dramatically reduce the sample size required to reveal potential benefits, and increase the chance of success. It is expected that more efforts will be devoted to the identification of predictive biomarkers and the development of effective assays for biomarkers during drug development. These efforts will likely be rewarded by increased success rates when the drug candidates enter clinical trials. Advances in technologies, such as next-generation sequencing and digital polymerase chain reaction, will also facilitate the detection of changes in genotypes and/or in DNA or RNA levels and the development of effective assays for biomarkers. On the other hand, biomarker-directed patient stratification has changed the clinical practice of anticancer therapy and has substantially improved treatment efficacy by providing guidance for the selection of optimal treatment for some lung cancer patients. However, biomarker-directed personalized therapy can currently benefit only a limited number of patients because most of the currently available predictive biomarkers occur infrequently [24, 27, 210] and because response-predictive biomarkers are not available for most first-line anticancer therapeutics. Thus, alternative approaches will be required to identify the appropriate therapeutics for a particular patient. Such patient-oriented approaches may require direct testing of the drug sensitivity of a patient’s cancer cells to various available anticancer therapeutics rather than predicting drug sensitivity based on the presence or absence of predictive biomarkers. Some recent studies have shown the feasibility of testing for the treatment responses of patient-derived tumor xenografts [211, 212] and/or primary cell cultures [213] to identify effective treatment regimens. Nevertheless, technology improvements and rigorous validations are required before such techniques can be used commonly in clinical practice.
Authors’ contribution
BF, RJM, JVH, and SGS conceived the concept and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Michael Worley and the Department of Scientific Publications of The University of Texas MD Anderson Cancer Center for editorial review of the manuscript. This work was supported in part by the National Institutes of Health Specialized Program of Research Excellence (SPORE) Grant CA070907 developmental award and The University of Texas MD Anderson Cancer Center support Grant CA-016672, and by endowed funds to The University of Texas MD Anderson Cancer Center, including the Moon Shots Program, the Homer Flower Research Fund, and the Goldman Sachs Philanthropy Fund.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Contributor Information
Bingliang Fang, Email: bfang@mdanderson.org.
Reza J Mehran, Email: rjmehran@mdanderson.org.
John V Heymach, Email: jheymach@mdanderson.org.
Stephen G Swisher, Email: swisher@mdanderson.org.
References
- 1.GBD 2013 Mortality and Causes of Death Collaborators Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen WQ, Zheng RS, Zhang SW, Zeng HM, Zou XN. The incidences and mortalities of major cancers in China, 2010. Chin J Cancer. 2014;33:402–405. doi: 10.5732/cjc.014.10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 4.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 5.Surveillance Epidemiology and End Results Program. Cancer of the lung and bronchus. National Cancer Institute. http://seer.cancer.gov/archive/csr/1975_2011/results_merged/sect_15_lung_bronchus.pdf (2014).
- 6.Edwards BK, Noone AM, Mariotto AB, Simard EP, Boscoe FP, Henley SJ, et al. Annual report to the Nation on the status of cancer, 1975–2010, featuring prevalence of comorbidity and impact on survival among persons with lung, colorectal, breast, or prostate cancer. Cancer. 2014;120:1290–1314. doi: 10.1002/cncr.28509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–271. doi: 10.3322/caac.21235. [DOI] [PubMed] [Google Scholar]
- 8.Zeng H, Zheng R, Guo Y, Zhang S, Zou X, Wang N, et al. Cancer survival in China, 2003–2005: a population-based study. Int J Cancer. 2015;136:1921–1930. doi: 10.1002/ijc.29227. [DOI] [PubMed] [Google Scholar]
- 9.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seow WJ, Hu W, Vermeulen R, Hosgood III HD, Downward GS, Chapman RS, et al. Household air pollution and lung cancer in China: a review of studies in Xuanwei. Chin J Cancer. 2014;33:471–475. doi: 10.5732/cjc.014.10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoek G, Raaschou-Nielsen O. Impact of fine particles in ambient air on lung cancer. Chin J Cancer. 2014;33:197–203. doi: 10.5732/cjc.014.10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loomis D, Huang W, Chen G. The International Agency for Research on Cancer (IARC) evaluation of the carcinogenicity of outdoor air pollution: focus on China. Chin J Cancer. 2014;33:189–196. doi: 10.5732/cjc.014.10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amini A. Progress in the management of limited-stage small cell lung cancer. Cancer. 2014;120:790–798. doi: 10.1002/cncr.28505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalemkerian GP. Advances in pharmacotherapy of small cell lung cancer. Exp Opin Pharmacother. 2014;15:2385–2396. doi: 10.1517/14656566.2014.957180. [DOI] [PubMed] [Google Scholar]
- 15.Govindan R. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24:4539–4544. doi: 10.1200/JCO.2005.04.4859. [DOI] [PubMed] [Google Scholar]
- 16.Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–573. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Unger M. A pause, progress, and reassessment in lung cancer screening. N Engl J Med. 2006;355:1822–1824. doi: 10.1056/NEJMe068207. [DOI] [PubMed] [Google Scholar]
- 18.National Lung Screening Trial Research Team. Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goulart BH, Bensink ME, Mummy DG, Ramsey SD. Lung cancer screening with low-dose computed tomography: costs, national expenditures, and cost-effectiveness. J Natl Compr Cancer Netw. 2012;10:267–275. doi: 10.6004/jnccn.2012.0023. [DOI] [PubMed] [Google Scholar]
- 20.Lu H, Wang L, Gao W, Meng J, Dai B, Wu S, et al. IGFBP2/FAK pathway is causally associated with dasatinib resistance in non-small cell lung cancer cells. Mol Cancer Ther. 2013;12:2864–2873. doi: 10.1158/1535-7163.MCT-13-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma. Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013;19:6322–6328. doi: 10.1158/1078-0432.CCR-13-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosse Y, Postma DS, Sin DD, Lamontagne M, Couture C, Gaudreault N, et al. Molecular signature of smoking in human lung tissues. Cancer Res. 2012;72:3753–3763. doi: 10.1158/0008-5472.CAN-12-1160. [DOI] [PubMed] [Google Scholar]
- 27.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research Network. Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez-Cuesta L. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat Commun. 2014;5:3518. doi: 10.1038/ncomms4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao C, Wang L, Peng S, Cao M, Li H, Hu J, et al. Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett. 2015;357:179–185. doi: 10.1016/j.canlet.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carnero A. Loss-of-function genetics in mammalian cells: the p53 tumor suppressor model. Nucleic Acids Res. 2000;28:2234–2241. doi: 10.1093/nar/28.11.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blagosklonny MV. Loss of function and p53 protein stabilization. Oncogene. 1997;15:1889–1893. doi: 10.1038/sj.onc.1201374. [DOI] [PubMed] [Google Scholar]
- 36.Dittmer D. Gain of function mutations in p53. Nat Genet. 1993;4:42–46. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 37.Pugacheva EN, Ivanov AV, Kravchenko JE, Kopnin BP, Levine AJ, Chumakov PM. Novel gain of function activity of p53 mutants: activation of the dUTPase gene expression leading to resistance to 5-fluorouracil. Oncogene. 2002;21:4595–4600. doi: 10.1038/sj.onc.1205704. [DOI] [PubMed] [Google Scholar]
- 38.Zhou G. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell. 2014;54:960–974. doi: 10.1016/j.molcel.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–239. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou W, Christiani DC. East meets West: ethnic differences in epidemiology and clinical behaviors of lung cancer between East Asians and Caucasians. Chin J Cancer. 2011;30:287–292. doi: 10.5732/cjc.011.10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Telbany A, Ma PC. Cancer genes in lung cancer: racial disparities: are there any? Genes Cancer. 2012;3:467–480. doi: 10.1177/1947601912465177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Broet P. Genomic profiles specific to patient ethnicity in lung adenocarcinoma. Clin Cancer Res. 2011;17:3542–3550. doi: 10.1158/1078-0432.CCR-10-2185. [DOI] [PubMed] [Google Scholar]
- 43.Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–967. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 44.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marchetti A, Martella C, Felicioni L, Barassi F, Salvatore S, Chella A, et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol. 2005;23:857–865. doi: 10.1200/JCO.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 46.Gahr S, Stoehr R, Geissinger E, Ficker JH, Brueckl WM, Gschwendtner A, et al. EGFR mutational status in a large series of Caucasian European NSCLC patients: data from daily practice. Br J Cancer. 2013;109:1821–1828. doi: 10.1038/bjc.2013.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi YL, Sun JM, Cho J, Rampal S, Han J, Parasuraman B, et al. EGFR mutation testing in patients with advanced non-small cell lung cancer: a comprehensive evaluation of real-world practice in an East Asian tertiary hospital. PLoS One. 2013;8:e56011. doi: 10.1371/journal.pone.0056011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li C, Fang R, Sun Y, Han X, Li F, Gao B, et al. Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS One. 2011;6:e28204. doi: 10.1371/journal.pone.0028204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao B, Sun Y, Zhang J, Ren Y, Fang R, Han X, et al. Spectrum of LKB1, EGFR, and KRAS mutations in Chinese lung adenocarcinomas. J Thorac Oncol. 2010;5:1130–1135. doi: 10.1097/JTO.0b013e3181e05016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka T, Matsuoka M, Sutani A, Gemma A, Maemondo M, Inoue A, et al. Frequency of and variables associated with the EGFR mutation and its subtypes. Int J Cancer. 2010;126:651–655. doi: 10.1002/ijc.24746. [DOI] [PubMed] [Google Scholar]
- 51.Mills NE, Fishman CL, Rom WN, Dubin N, Jacobson DR. Increased prevalence of K-ras oncogene mutations in lung adenocarcinoma. Cancer Res. 1995;55:1444–1447. [PubMed] [Google Scholar]
- 52.Sun Y. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28:4616–4620. doi: 10.1200/JCO.2010.29.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Algarra I, Perez M, Serrano MJ, Garrido F, Gaforio JJ. c-K-ras overexpression is characteristic for metastases derived from a methylcholanthrene-induced fibrosarcoma. Invasion Metastasis. 1998;18:261–270. doi: 10.1159/000024519. [DOI] [PubMed] [Google Scholar]
- 55.Coleman WB, Throneburg DB, Grisham JW, Smith GJ. Overexpression of c-K-ras, c-N-ras and transforming growth factor beta co-segregate with tumorigenicity in morphologically transformed C3H 10T1/2 cell lines. Carcinogenesis. 1994;15:1005–1012. doi: 10.1093/carcin/15.5.1005. [DOI] [PubMed] [Google Scholar]
- 56.Ehrhardt A, David MD, Ehrhardt GR, Schrader JW. Distinct mechanisms determine the patterns of differential activation of H-Ras, N-Ras, K-Ras 4B, and M-Ras by receptors for growth factors or antigen. Mol Cell Biol. 2004;24:6311–6323. doi: 10.1128/MCB.24.14.6311-6323.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell. 1993;73:611–620. doi: 10.1016/0092-8674(93)90146-H. [DOI] [PubMed] [Google Scholar]
- 58.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 59.Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 61.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 62.Cappuzzo F, Ciuleanu T, Stelmakh L, Cicenas S, Szczesna A, Juhasz E, et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010;11:521–529. doi: 10.1016/S1470-2045(10)70112-1. [DOI] [PubMed] [Google Scholar]
- 63.Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS) J Clin Oncol. 2011;29:2866–2874. doi: 10.1200/JCO.2010.33.4235. [DOI] [PubMed] [Google Scholar]
- 64.Thatcher N, Chang A, Parikh P, Rodrigues PJ, Ciuleanu T, von Pawel J, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 65.Shi Y, Zhang L, Liu X, Zhou C, Zhang L, Zhang S, et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013;14:953–961. doi: 10.1016/S1470-2045(13)70355-3. [DOI] [PubMed] [Google Scholar]
- 66.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25:587–595. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 67.Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 68.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 69.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 70.Yeh P. DNA-Mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): a catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapy. Clin Cancer Res. 2013;19:1894–1901. doi: 10.1158/1078-0432.CCR-12-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013;12:220–229. doi: 10.1158/1535-7163.MCT-12-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu JY, Wu SG, Yang CH, Gow CH, Chang YL, Yu CJ, et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res. 2008;14:4877–4882. doi: 10.1158/1078-0432.CCR-07-5123. [DOI] [PubMed] [Google Scholar]
- 73.Yasuda H. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012;13:e23–e31. doi: 10.1016/S1470-2045(11)70129-2. [DOI] [PubMed] [Google Scholar]
- 74.Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5:216ra177. doi: 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 76.Wykosky J, Fenton T, Furnari F, Cavenee WK. Therapeutic targeting of epidermal growth factor receptor in human cancer: successes and limitations. Chin J Cancer. 2011;30:5–12. doi: 10.5732/cjc.010.10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 79.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cappuzzo F, Janne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20:298–304. doi: 10.1093/annonc/mdn635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 83.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina SE, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 85.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 86.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stabile LP, He G, Lui VW, Henry C, Gubish CT, Joyce S, et al. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin Cancer Res. 2013;19:380–392. doi: 10.1158/1078-0432.CCR-12-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2:934–947. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi: 10.1158/0008-5472.CAN-08-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-kB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, Ha SJ, et al. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther. 2012;11:2254–2264. doi: 10.1158/1535-7163.MCT-12-0311. [DOI] [PubMed] [Google Scholar]
- 93.Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, et al. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res. 2012;18:4986–4996. doi: 10.1158/1078-0432.CCR-12-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12:5764–5769. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 95.Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2012;30:3337–3344. doi: 10.1200/JCO.2011.40.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 97.Hirsch FR, Janne PA, Eberhardt WE, Cappuzzo F, Thatcher N, Pirker R, et al. Epidermal growth factor receptor inhibition in lung cancer: status 2012. J Thorac Oncol. 2013;8:373–384. doi: 10.1097/JTO.0b013e31827ed0ff. [DOI] [PubMed] [Google Scholar]
- 98.Yu HA, Riely GJ. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J Natl Compr Cancer Netw. 2013;11:161–169. doi: 10.6004/jnccn.2013.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Langer CJ. Epidermal growth factor receptor inhibition in mutation-positive non-small-cell lung cancer: is afatinib better or simply newer? J Clin Oncol. 2013;31:3303–3306. doi: 10.1200/JCO.2013.49.8782. [DOI] [PubMed] [Google Scholar]
- 101.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790 M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tjin Tham Sjin R, Lee K, Walter AO, Dubrovskiy A, Sheets M, Martin TS, et al. In vitro and in vivo characterization of irreversible mutant-selective EGFR inhibitors that are wild-type sparing. Mol Cancer Ther. 2014;13:1468–1479. doi: 10.1158/1535-7163.MCT-13-0966. [DOI] [PubMed] [Google Scholar]
- 103.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gao W, Wang M, Wang L, Lu H, Wu S, Dai B, et al. Selective antitumor activity of ibrutinib in EGFR-mutant non-small cell lung cancer cells. J Natl Cancer Inst. 2014;106:dju204. doi: 10.1093/jnci/dju204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burger JA, Keating MJ, Wierda WG, Hartmann E, Hoellenriegel J, Rosin NY, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15:1090–1099. doi: 10.1016/S1470-2045(14)70335-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Younes A, Thieblemont C, Morschhauser F, Flinn I, Friedberg JW, Amorim S, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol. 2014;15:1019–1026. doi: 10.1016/S1470-2045(14)70311-0. [DOI] [PubMed] [Google Scholar]
- 109.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 110.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 111.Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19:4273–4281. doi: 10.1158/1078-0432.CCR-13-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Solomon B. ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J Thorac Oncol. 2009;4:1450–1454. doi: 10.1097/JTO.0b013e3181c4dedb. [DOI] [PubMed] [Google Scholar]
- 113.Rodig SJ. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clinl Cancer Res. 2009;15:5216–5223. doi: 10.1158/1078-0432.CCR-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cui JJ, Tran-Dube M, Shen H, Nambu M, Kung PP, Pairish M, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54:6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 116.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31:1105–1111. doi: 10.1200/JCO.2012.44.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 119.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pan Y, Zhang Y, Li Y, Hu H, Wang L, Li H, et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer. 2014;84:121–126. doi: 10.1016/j.lungcan.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 121.Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18:4570–4579. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci USA. 2011;108:7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15:1119–1128. doi: 10.1016/S1470-2045(14)70362-6. [DOI] [PubMed] [Google Scholar]
- 128.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kogita A, Togashi Y, Hayashi H, Banno E, Terashima M, De Velasco MA, et al. Activated MET acts as a salvage signal after treatment with alectinib, a selective ALK inhibitor, in ALK-positive non-small cell lung cancer. Int J Oncol. 2015;46:1025–1030. doi: 10.3892/ijo.2014.2797. [DOI] [PubMed] [Google Scholar]
- 131.Hu W, Davis JJ, Zhu H, Dong F, Guo W, Ang J, et al. Redirecting adaptive immunity against foreign antigens to tumors for cancer therapy. Cancer Biol Ther. 2007;6:1773–1779. doi: 10.4161/cbt.6.11.4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res. 2013;19:1021–1034. doi: 10.1158/1078-0432.CCR-12-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- 135.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm0902-1039c. [DOI] [PubMed] [Google Scholar]
- 138.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 139.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Velcheti V, Schalper KAC, Anagnostou VK, Syrigos KN, Sznol M, Herbst RS, et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014;94:107–116. doi: 10.1038/labinvest.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.D’Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer. 2015;112:95–102. doi: 10.1038/bjc.2014.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 144.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 147.Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16:257–265. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 153.Moreno L, Pearson AD. How can attrition rates be reduced in cancer drug discovery? Exp Opin Drug Discov. 2013;8:363–368. doi: 10.1517/17460441.2013.768984. [DOI] [PubMed] [Google Scholar]
- 154.Kola I. The state of innovation in drug development. Clin Pharmacol Ther. 2008;83:227–230. doi: 10.1038/sj.clpt.6100479. [DOI] [PubMed] [Google Scholar]
- 155.Arrowsmith J. Trial watch: phase III and submission failures: 2007–2010. Nat Rev Drug Discov. 2011;10:87. doi: 10.1038/nrd3375. [DOI] [PubMed] [Google Scholar]
- 156.Arrowsmith J. Trial watch: phase II failures: 2008–2010. Nat Rev Drug Discov. 2011;10:328–329. doi: 10.1038/nrd3439. [DOI] [PubMed] [Google Scholar]
- 157.Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial—INTACT 2. J Clin Oncol. 2004;22:785–794. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 158.Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial–INTACT 1. J Clin Oncol. 2004;22:777–784. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 159.Gatzemeier U, Pluzanska A, Szczesna A, Kaukel E, Roubec J, De Rosa F, et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol. 2007;25:1545–1552. doi: 10.1200/JCO.2005.05.1474. [DOI] [PubMed] [Google Scholar]
- 160.Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005;23:5892–5899. doi: 10.1200/JCO.2005.02.840. [DOI] [PubMed] [Google Scholar]
- 161.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 164.Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30:372–379. doi: 10.1200/JCO.2011.36.9215. [DOI] [PubMed] [Google Scholar]
- 165.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852–861. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- 166.Takano T, Fukui T, Ohe Y, Tsuta K, Yamamoto S, Nokihara H, et al. EGFR mutations predict survival benefit from gefitinib in patients with advanced lung adenocarcinoma: a historical comparison of patients treated before and after gefitinib approval in Japan. J Clin Oncol. 2008;26:5589–5595. doi: 10.1200/JCO.2008.16.7254. [DOI] [PubMed] [Google Scholar]
- 167.Allen SL, Lundberg AS. Amonafide: a potential role in treating acute myeloid leukemia. Exp Opin Invest Drugs. 2011;20:995–1003. doi: 10.1517/13543784.2011.585756. [DOI] [PubMed] [Google Scholar]
- 168.Innocenti F, Iyer L, Ratain MJ. Pharmacogenetics of anticancer agents: lessons from amonafide and irinotecan. Drug Metab Dispos. 2001;29:596–600. [PubMed] [Google Scholar]
- 169.Kreis W, Chan K, Budman DR, Allen SL, Fusco D, Mittelman A, et al. Clinical pharmacokinetics of amonafide (NSC 308847) in 62 patients. Cancer Invest. 1996;14:320–327. doi: 10.3109/07357909609012158. [DOI] [PubMed] [Google Scholar]
- 170.Ratain MJ, Mick R, Janisch L, Berezin F, Schilsky RL, Vogelzang NJ, et al. Individualized dosing of amonafide based on a pharmacodynamic model incorporating acetylator phenotype and gender. Pharmacogenetics. 1996;6:93–101. doi: 10.1097/00008571-199602000-00008. [DOI] [PubMed] [Google Scholar]
- 171.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 172.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 173.Fang B. Development of synthetic lethality anticancer therapeutics. J Med Chem. 2014;57:7859–7873. doi: 10.1021/jm500415t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fang B. Genetic interactions in translational research on cancer. World J Med Genet. 2011;1:14–22. doi: 10.5496/wjmg.v1.i1.14. [DOI] [Google Scholar]
- 175.Guo Z, Fang B. Synthetic lethality in anticancer drug discovery and target identification. Pharmacologia. 2012;3:352–361. doi: 10.5567/pharmacologia.2012.352.361. [DOI] [Google Scholar]
- 176.Fang B. Genetic interactions of STAT3 and anticancer drug development. Cancers. 2014;6:494–525. doi: 10.3390/cancers6010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Brunelli L, Caiola E, Marabese M, Broggini M, Pastorelli R. Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget. 2014;5:4722–4731. doi: 10.18632/oncotarget.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Meuwissen R, Linn SC, Van der Valk M, Mooi WJ, Berns A. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 2001;20:6551–6558. doi: 10.1038/sj.onc.1204837. [DOI] [PubMed] [Google Scholar]
- 181.Kim CF, Jackson EL, Kirsch DG, Grimm J, Shaw AT, Lane K, et al. Mouse models of human non-small-cell lung cancer: raising the bar. Cold Spring Harb Symp Quant Biol. 2005;70:241–250. doi: 10.1101/sqb.2005.70.037. [DOI] [PubMed] [Google Scholar]
- 182.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 183.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–559. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hoa M, Davis SL, Ames SJ, Spanjaard RA. Amplification of wild-type K-ras promotes growth of head and neck squamous cell carcinoma. Cancer Res. 2002;62:7154–7156. [PubMed] [Google Scholar]
- 185.Filmus JE, Buick RN. Stability of c-K-ras amplification during progression in a patient with adenocarcinoma of the ovary. Cancer Res. 1985;45:4468–4472. [PubMed] [Google Scholar]
- 186.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–7408. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Guo W, Wu S, Wang L, Wei X, Liu X, Wang J, et al. Antitumor activity of a novel oncrasin analogue is mediated by JNK activation and STAT3 inhibition. PLoS One. 2011;6:e28487. doi: 10.1371/journal.pone.0028487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wei X, Guo W, Wu S, Wang L, Lu Y, Xu B, et al. Inhibiting JNK dephosphorylation and induction of apoptosis by novel anticancer agent NSC-741909 in cancer cells. J Biol Chem. 2009;284:16948–16955. doi: 10.1074/jbc.M109.010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wu S, Wang L, Guo W, Liu X, Liu J, Wei X, et al. Analogues and derivatives of oncrasin-1, a novel inhibitor of the C-terminal domain of RNA polymerase II and their antitumor activities. J Med Chem. 2011;54:2668–2679. doi: 10.1021/jm101417n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wu S, Wang L, Huang X, Cao M, Hu J, Li H, et al. Prodrug oncrasin-266 improves the stability, pharmacokinetics, and safety of NSC-743380. Bioorg Med Chem. 2014;22:5234–5240. doi: 10.1016/j.bmc.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Guo W, Wu S, Wang L, Wang R, Wei L, Liu J, et al. Interruption of RNA processing machinery by a small compound 1-[(4-chlorophenyl) methyl]-1H-indole-3-carboxaldehyde (oncrasin-1) Mol Cancer Ther. 2009;8:441–448. doi: 10.1158/1535-7163.MCT-08-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Guo W, Wei X, Wu S, Wang L, Peng H, Wang J, et al. Antagonistic effect of flavonoids on NSC-741909-mediated antitumor activity via scavenging of reactive oxygen species. Eur J Pharmacol. 2010;649:51–58. doi: 10.1016/j.ejphar.2010.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu X, Guo W, Wu S, Wang L, Wang J, Dai B, et al. Antitumor activity of a novel STAT3 inhibitor and redox modulator in non-small cell lung cancer cells. Biochem Pharmacol. 2012;83:1456–1464. doi: 10.1016/j.bcp.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wei X, Guo W, Wu S, Wang L, Huang P, Liu J, et al. Oxidative stress in NSC-741909-induced apoptosis of cancer cells. J Transl Med. 2010;8:37. doi: 10.1186/1479-5876-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Huang X, Cao M, Wang L, Wu S, Liu X, Li H, et al. Expression of sulfotransferase SULT1A1 in cancer cells predicts susceptibility to the novel anticancer agent NSC-743380. Oncotarget. 2015;6:345–354. doi: 10.18632/oncotarget.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Svendsen C, Meinl W, Glatt H, Alexander J, Knutsen HK, Hjertholm H, et al. Intestinal carcinogenesis of two food processing contaminants, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and 5-hydroxymethylfurfural, in transgenic FVB min mice expressing human sulfotransferases. Mol Carcinog. 2012;51:984–992. doi: 10.1002/mc.20869. [DOI] [PubMed] [Google Scholar]
- 197.Suzuki Y, Umemura T, Ishii Y, Hibi D, Inoue T, Jin M, et al. Possible involvement of sulfotransferase 1A1 in estragole-induced DNA modification and carcinogenesis in the livers of female mice. Mutation Res. 2012;749:23–28. doi: 10.1016/j.mrgentox.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 198.Gamage NU, Duggleby RG, Barnett AC, Tresillian M, Latham CF, Liyou NE, et al. Structure of a human carcinogen-converting enzyme, SULT1A1. Structural and kinetic implications of substrate inhibition. J Biol Chem. 2003;278:7655–7662. doi: 10.1074/jbc.M207246200. [DOI] [PubMed] [Google Scholar]
- 199.Arlt VM, Glatt H, Muckel E, Pabel U, Sorg BL, Schmeiser HH, et al. Metabolic activation of the environmental contaminant 3-nitrobenzanthrone by human acetyltransferases and sulfotransferase. Carcinogenesis. 2002;23:1937–1945. doi: 10.1093/carcin/23.11.1937. [DOI] [PubMed] [Google Scholar]
- 200.Banoglu E. Current status of the cytosolic sulfotransferases in the metabolic activation of promutagens and procarcinogens. Curr Drug Metab. 2000;1:1–30. doi: 10.2174/1389200003339234. [DOI] [PubMed] [Google Scholar]
- 201.Michejda CJ, Kroeger Koepke MB. Carcinogen activation by sulfate conjugate formation. Adv Pharmacol. 1994;27:331–363. doi: 10.1016/S1054-3589(08)61038-8. [DOI] [PubMed] [Google Scholar]
- 202.Kroeger-Koepke MB, Koepke SR, Hernandez L, Michejda CJ. Activation of a beta-hydroxyalkylnitrosamine to alkylating agents: evidence for the involvement of a sulfotransferase. Cancer Res. 1992;52:3300–3305. [PubMed] [Google Scholar]
- 203.Mercer KE, Apostolov EO, da Costa GG, Yu X, Lang P, Roberts DW, et al. Expression of sulfotransferase isoform 1A1 (SULT1A1) in breast cancer cells significantly increases 4-hydroxytamoxifen-induced apoptosis. Int J Mol Epidemiol Genet. 2010;1:92–103. [PMC free article] [PubMed] [Google Scholar]
- 204.Eldridge SR, Cover J, Morris J, Fang B, Horn TL, Elsass KE, et al. Characterization of acute biliary hyperplasia in Fisher 344 rats administered the indole-3-carbinol analog, NSC-743380. Toxicol Appl Pharmacol. 2014;2014:303–309. doi: 10.1016/j.taap.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–4382. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bracht K, Nicholls AM, Liu Y, Bodmer WF. 5-Fluorouracil response in a large panel of colorectal cancer cell lines is associated with mismatch repair deficiency. Br J Cancer. 2010;103:340–346. doi: 10.1038/sj.bjc.6605780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mazumdar T, Byers LA, Ng PK, Mills GB, Peng S, Diao L, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13:2738–2750. doi: 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Soda M, Isobe K, Inoue A, Maemondo M, Oizumi S, Fujita Y, et al. A prospective PCR-based screening for the EML4-ALK oncogene in non-small cell lung cancer. Clin Cancer Res. 2012;18:5682–5689. doi: 10.1158/1078-0432.CCR-11-2947. [DOI] [PubMed] [Google Scholar]
- 211.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De OE, Rubio-Viqueira B, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10:1311–1316. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Morelli MP, Calvo E, Ordonez E, Wick MJ, Viqueira BR, Lopez-Casas PP, et al. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J Clin Oncol. 2012;30:e45–e48. doi: 10.1200/JCO.2011.36.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yuan H, Myers S, Wang J, Zhou D, Woo JA, Kallakury B, et al. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. N Engl J Med. 2012;367:1220–1227. doi: 10.1056/NEJMoa1203055. [DOI] [PMC free article] [PubMed] [Google Scholar]