

Abstract
Background
Human myxovirus resistant protein A (MxA), encoded by the myxovirus resistance 1 (Mx1) gene, is an interferon (IFN)-triggered dynamin-like multi-domain GTPase involved in innate immune responses against viral infections. Recent studies suggest that MxA is associated with several human cancers and may be a tumor suppressor and a promising biomarker for IFN therapy. Mx1 gene mutations in the coding region for MxA have been discovered in many types of cancer, suggesting potential biological associations between mutations in MxA protein and corresponding cancers. In this study, we performed a systematic analysis based on the crystal structures of MxA and elucidated how these mutations specifically affect the structure and therefore the function of MxA protein.
Methods
Cancer-associated Mx1 mutations were collected and screened from the COSMIC database. Twenty-two unique mutations that cause single amino acid alterations in the MxA protein were chosen for the analysis. Amino acid sequence alignment was performed using Clustal W to check the conservation level of mutation sites in Mx proteins and dynamins. Structural analysis of the mutants was carried out with Coot. Structural models of selected mutants were generated by the SWISS-MODEL server for comparison with the corresponding non-mutated structures. All structural figures were generated using PyMOL.
Results
We analyzed the conservation level of the single-point mutation sites and mapped them on different domains of MxA. Through individual structural analysis, we found that some mutations severely affect the stability and function of MxA either by disrupting the intra-/inter-molecular interactions supported by the original residues or by incurring unfavorable configuration alterations, whereas other mutations lead to gentle or no interference to the protein stability and function because of positions or polarity features. The potential clinical value of the mutations that lead to drastic influence on MxA protein is also assessed.
Conclusions
Among all of the reported tumor-associated single-point mutations, seven of them notably affect the structure and function of MxA and therefore deserve more attention with respect to potential clinical applications. Our research provides an example for systematic analysis and consequence evaluation of single-point mutations on a given cancer-related protein.
Keywords: Human cancer, MxA protein, Single-point mutation, Crystal structure, Domain distribution, Protein stability, Protein folding, Residue interaction, Stereochemistry, Polarity
Background
The myxovirus resistance 1 (Mx1) gene is one of the most prominent interferon (IFN)-stimulated genes in vertebrate that are highly activated when triggered by type I and III IFNs upon viral infection. Human Mx1 is located on chromosome 21 and encodes the myxovirus resistant protein A (MxA) protein, which is a major antiviral factor against a wide spectrum of RNA viruses, such as influenza virus and Thogotovirus [1, 2]. The 78-kDa MxA protein is a member of the dynamin superfamily of multi-domain GTPases whose function relies on oligomerization and GTP hydrolysis [3]. MxA has an N-terminal GTPase (G) domain, a stalk region responsible for oligomerization, a bundle-signaling-element domain (BSE) crucial for the communication between the G domain and the stalk region [4–6], and a 40-amino-acid-long loop (conventionally named L4) amid the Stalk on primary structure correlated with the pleckstrin homology (PH) domain in dynamin [3]. Besides, MxA possesses a disordered loop at the N-terminus that is not conserved within the dynamin superfamily. According to the current working model, MxA forms ring-like oligomers around viral ribonucleoparticles (RNPs) and disrupts the structure of RNPs via conformational changes induced by GTP hydrolysis [5].
Apart from its role as a prominent antiviral protein in innate immunity, MxA has been found to be associated with different types of human cancer. It has been reported that the deletion of Mx1 gene, as a consequence of certain gene fusion events, is closely related to prostate cancer with a high aggressive tendency [7]. Other studies of prostate cancer have suggested that the expression of MxA is suppressed in the highly metastatic human prostate carcinoma cell line PC-3M, and exogenous MxA can inhibit the mobility and invasiveness of PC-3M cells both in vivo and in vitro [8]. Prostate cancer cells in which the Mx1 gene is knocked out have been shown to be much less sensitive to docetaxel compared with MxA-positive cells [9]. The Mx1 gene has been found to be hypermethylated and suppressed in primary head and neck squamous cell carcinoma cell lines and tissue samples compared with normal lymphocytes [10]. These studies indicate that Mx1 is a potential tumor suppressor gene. On the other hand, as a traditional immunotherapeutic reagent [11], type I IFNs are widely used in clinical treatments against a number of human cancers, including renal cell carcinoma, follicular lymphoma, melanoma, and chronic myelogenous leukemia [12]. The effect of type III IFNs against different human cancers, including bladder carcinoma, Burkitt’s lymphoma, colorectal carcinoma, non-small cell lung cancer, and esophageal carcinoma, has also been investigated [13, 14]. As a key cytokine induced by types I and III IFNs, MxA is thought to be a useful biomarker for monitoring IFN activity and predicting clinical efficacy during IFN therapy in patients bearing certain types of cancer [15, 16]. A clinical study on early stage B cell chronic lymphocytic leukemia has suggested that patients with no MxA expression are likely to demonstrate a positive response to IFN therapy [16]. Moreover, the expression level of MxA is also employed to predict the efficacy of chemotherapy on several types of cancer. A worldwide multi-center study has indicated that robust MxA expression is a positive indicator for patients with breast carcinoma who might benefit from anthracycline-based chemotherapy [17].
In recent years, with the development of next-generation sequencing technique and its wide application in cancer studies [18], the landscape of somatic mutations has been revealed for many types of human cancer. According to these comprehensive data sets, Mx1 mutations have been discovered in a number of common cancer types, including colorectal cancer [19–22], head and neck squamous cell carcinoma [23], follicular lymphoma [24], cutaneous squamous cell carcinoma [25], mantle cell lymphoma [26], embryonal rhabdomyosarcoma [27], renal cell carcinoma [28], prostate cancer [29], lung adenocarcinoma [30], melanoma [31], medulloblastoma [32], and ovarian carcinoma [33]. Given the potential importance of MxA in tumorigenesis and metastasis as well as in the treatment and prognosis of different cancers, additional efforts are needed to investigate the biological associations between Mx1 mutations, especially the mutations that lead to the malfunction of its encoded protein MxA, and corresponding cancers. Mutations in coding regions can result in different consequences to the translated polypeptides, of which single amino acid alterations take up a considerable portion. These single-point mutations may give rise to various consequences to the function of MxA which, however, are difficult to be predicted and evaluated from the analysis of primary structure. In this study, we exploit the crystal structures of MxA and illustrate how these mutations specifically affect the structure and thus the function of MxA protein.
Methods
Data mining
Information on cancer-related mutations in human Mx1 gene was collected from the Catalogue of Somatic Mutations in Cancer (COSMIC) database [34]. All mutations were manually screened and only the mutations that cause single amino acid alterations in MxA were chosen for subsequent analysis.
Sequence alignment
Amino acid sequences of Homo sapiens (hs) MxA (UniProt code P20591) and MxB (P20592), Mus musculus (mm) Mx1 (P09922) and Mx2 (Q9WVP9), Gallus gallus (gg) Mx protein (Q90597), Danio rerio (dr) MxA protein (Q8JH68), Homo sapiens dynamin1 (Q05193), dynamin2 (P50570), and dynamin3 (Q9UQ16), Drosophila melanogaster (dm) dynamin (P27619), Caenorhabditis elegans (ce) dynamin (Q9U9I9), and Saccharomyces cerevisiae (sc) dynamin-related protein DNM1 (P54861) were aligned using Clustal W [35] and manually adjusted for non-conserved loop regions.
Structural analysis
Crystal structures of full-length MxA (PDB code 3SZR), MxA Stalk (3LJB), apo MxA G domain-BSE fusion construct (Stalkless MxA) (4P4U), and Stalkless MxA complexed with β,γ-methyleneguanosine 5′-triphosphate (GMPPCP, a non-hydrolysable GTP analogue) (4P4S) were used as reference structures. For optimal accuracy, the resolution was considered a prior factor during the selection of reference structures comprising corresponding mutated residues. For mutations within the G domain, the crystal structure of apo Stalkless MxA (1.9 Å resolution) was applied as reference with the exception of L95P and P96S on Switch I. For these two mutations, the GMPPCP-bound Stalkless MxA structure (3.3 Å) was taken as reference because only when a GTP analogue is bound to the G domain can Switch I be ordered and fully observable. For mutations in the stalk region, the MxA Stalk structure (2.4 Å) was used for structural analysis. For mutations in BSE and Hinge 1, the full-length MxA structure (3.5 Å) was the only choice because other published structural models do not contain these residues. Structural analysis for all mutations was performed using Coot [36]. Interactions among the residues were defined by the linear distance between the centers of the corresponding atoms in the reference models: 2.7–3.5 Å for hydrogen bonds and within 4 Å for salt bridges and hydrophobic interactions. Overall, all reference models provided sufficient information needed for reliable structural analysis.
Structural modeling of the mutants
Structural models of selected cancer-associated single-point mutants were calculated by the SWISS-MODEL server [37]. In the amino acid sequence input files for each MxA mutant, the original residue was substituted by the post-mutation residue. Mutated MxA sequences were then individually uploaded to the SWISS-MODEL server for model calculation. The templates used for structural model building were assigned according to the corresponding reference models for each mutant as previously mentioned. In the output models, the residues that were absent in the original templates were removed for better quality because these parts may contain severe errors as a result of lacking reference information. The final structural models of the mutants were individually superimposed on the corresponding original reference models using Coot, and the root mean square deviation (r. m. s. d.) values for steric positions of the corresponding atoms between each model pairs were subsequently calculated.
Structural figure preparation
All structural figures were generated using PyMOL [38]. Different types of atoms in stick or sphere representations were specified by the following colors: red for oxygen, blue for nitrogen, magenta for phosphorous, light green for magnesium, green for sulfur, yellow for carbon in residues that are mutated in human cancers, and gray for carbon in other amino acids that interact with residues of interest in this study. The spheres in the illustration represented Cα of the corresponding residues.
Results
Overview of single-point mutations in MxA in different types of cancer
In the COSMIC database, 122 tumor-related mutations were identified in the human Mx1 gene from whole-genome, whole-exome, and transcriptome sequencing data of human cancers. All 122 mutations reside in coding regions of the MxA protein. For these genetic aberrances, frameshift mutations and termination codon mutations were excluded, as they lead to obvious truncation of the polypeptide chain and therefore the obliteration of MxA structure and function. In addition, silent mutations were also excluded because the amino acid sequence of MxA remains intact in these cases. Consequently, 22 unique cancer-related single amino acid mutations in MxA protein were selected and subjected to subsequent analysis. These 22 MxA mutations, summarized in Table 1, were found across 12 types of human cancer, of which colorectal cancer accounted for half of the mutations, followed by cutaneous squamous cell carcinoma (3 mutations) and mantle cell lymphoma (2 mutations). The other 9 cancers each featured 1 mutation. Two mutations, namely T651M and R655C, both appeared twice. T651M was found in colorectal cancer and head and neck squamous cell carcinoma, whereas R655C was discovered in two separate sequencing projects for colorectal cancer. Overall, MxA single-point mutations are widely spread in different human cancers, and MxA mutates more frequently in colorectal cancer according to current data.
Table 1.
Summary of reported human myxovirus resistant protein A (MxA) single-point mutations in human cancers
| Cancer type | Mutation(s) |
|---|---|
| Colorectal cancer | S134L, N491K, R522C, T651M, R654Q, V263M, Y538C, S572Y, R655C (2)a |
| Head and neck squamous cell carcinoma | T651M |
| Follicular lymphoma | T27S |
| Cutaneous squamous cell carcinoma | L95P, P96S, P218S |
| Mantle cell lymphoma | G540D, L643V |
| Embryonal rhabdomyosarcoma | V449G |
| Renal cell carcinoma | G392V |
| Prostate cancer | K326N |
| Lung adenocarcinoma | R310S |
| Melanoma | E632K |
| Medulloblastoma | R649W |
| Ovarian carcinoma | L619I |
aR655C mutation was discovered in colorectal cancer in two individual sequencing projects
We then checked the conservation level of the amino acid residues mutated in cancers. Seven residues are highly conserved in Mx proteins and dynamins; 4 residues also show considerable overall conservation. In addition, 5 residues are conserved in Mx proteins but not in dynamins, whereas 6 residues display no conservations at all (Fig. 1). Intriguingly, T651 and R655, although their corresponding mutations appeared more frequently in sequencing data, belong to non-conserved group. This result suggests that it is obscure to infer the physiological importance of these single-point mutations only from their conservativeness in primary structure. To obtain more reliable information, individual analysis of these mutations by structural means is indispensable.
Fig. 1.
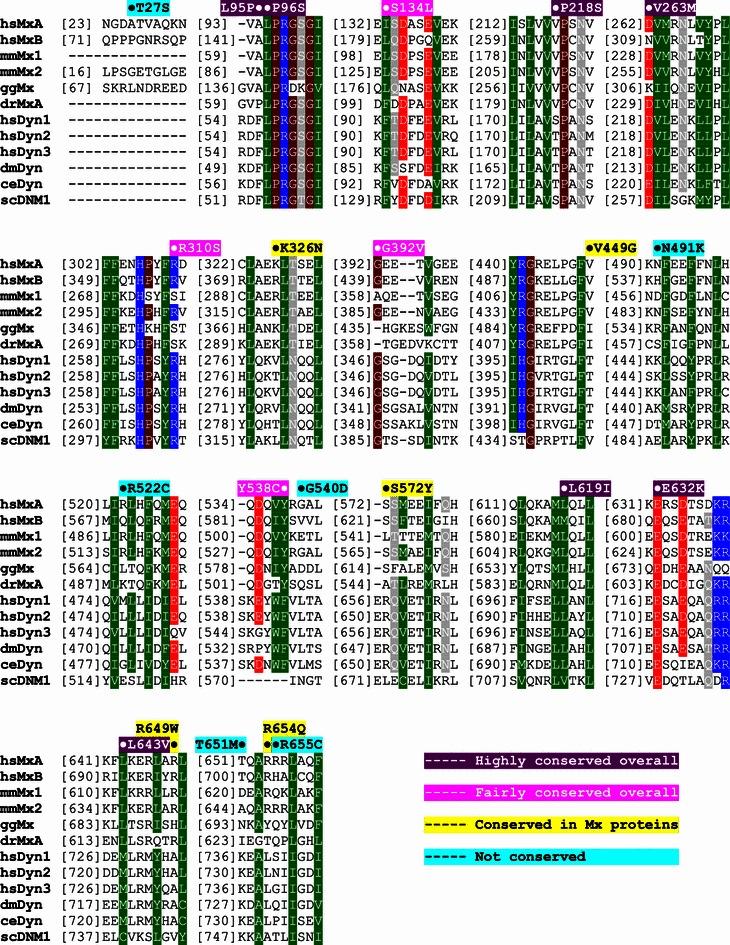
Amino acid sequence alignment of myxovirus resistance (Mx) proteins and dynamins. Amino acid sequences of Mx proteins and dynamins from human, mouse, chicken, zebra fish, fruit fly, C. elegans, and yeast are shown in the sequence alignment (see the “Methods” for details). Residues with a conservation of greater than 70% are color-coded (negatively charged amino acid residues D and E in red; positively charged R, K, and H in blue; polar N, Q, S, and T in grey; weak or nonpolar A, L, I, V, F, Y, W, M, and C in green; and special P and G in brown). Numbers in square brackets in front of each 10-residue sequence fragment indicate the ordinal position of the first residue of this fragment at the primary structure of the corresponding protein. Tumor-associated mutations are indicated at the corresponding positions. Residues highly conserved in Mx proteins and dynamins (L95, P96, P218, V263, L619, E632, and L643) are shown in violet, residues showing considerable overall conservation (S134, R310, G392, and Y538) in magenta, residues conserved in Mx proteins but not in dynamins (K326, V449, S572, R649, and R654) in yellow, and residues with no conservativeness (T27, N491, R522, G540, T651, and R655) in cyan
Distribution of cancer-related single-point mutations in MxA
Human MxA protein is a large dynamin-like GTPase composed of 5 structurally discriminated domains plus an N′-loop (Fig. 2a). Recently, several crystal structures of MxA have been reported, which collectively covered most of the molecule, except for the N′-loop and L4 [4–6]. We took advantage of these structures to perform an in-depth analysis of the 22 cancer-related mutations and their possible structural and functional outcomes on the protein. First, we mapped the mutated residues on MxA molecule and found that these 22 mutations are scattered throughout all regions (Fig. 2b). The G domain, which is largest in size, contains 7 mutation sites, whereas the smaller N′-loop and Hinge 1 each contains 1 mutation site. Considering the size, however, BSE and L4 are relatively more prone to mutation, where 5 and 3 mutation sites are spotted, respectively (Fig. 2c–f). The distribution of the mutations is summarized in Table 2. Next, all single-point mutations were individually investigated according to the domains.
Fig. 2.

Overview of the distribution of cancer-related mutations within the human myxovirus resistant protein A (MxA) domains. a schematic representation of the domain structure of human MxA. N′-loop N-terminal disordered loop; B bundle-signaling-element domain (BSE), G G domain; H Hinge 1, S Stalk. Borders of the domains are indicated by corresponding residue numbers. b overview of the position of all mutations in MxA. The G domain is colored in orange, BSE in red, Hinge 1 in sky-blue, and Stalk in green. The missing N-terminal 44 residues (shown in magenta) and L4 (shown in cyan) are indicated as dashed lines. Mutations that are included by the reference model are illustrated as yellow spheres. Residues that are missing in this reference model but are present in other reference models are illustrated as filled yellow circles. Residues missing in all reference structures are shown as yellow stars. c–f overview of the mutations in individual domains of MxA, as outlined by dashed rectangles in Fig. 2a at the corresponding areas: c G domain, d BSE, e Hinge 1, and f Stalk. Note that the representations of e Hinge 1 and f Stalk representations were rotated counter-clockwise 90° from those in Fig. 2b
Table 2.
Domain distribution of cancer-related MxA single-point mutations
| MxA segment | Mutation(s) in cancers |
|---|---|
| GTPase domain (G domain) | L95P, P96S, S134L, P218S, V263M, R310S, K326N |
| Bundle-signaling-element domain (BSE) | L643V, R649W, T651M, R654Q, R655C |
| Stalk | G392V, V449G, N491K, R522C, L619I |
| Hinge 1 | E632K |
| L4 | G540D, Y538C, S572Y |
| N′-loop | T27S |
Single-point mutations in G domain
The G domain of MxA is responsible for GTP hydrolysis, as well as for the inter-ring homo-dimerization via the nucleotide-binding pocket [39]. Both actions are crucial for the mechano-chemical coupling of the entire oligomer and thus for the function of the protein [5, 6].
Cutaneous squamous cell carcinoma-associated single-point mutations L95P and P96S sit within Switch I, a key component of the active site which harbors guanine nucleotides and exists in all known GTPases [40]. L95 is deeply buried in a comprehensive hydrophobic pocket composed of L87, V93, L107, L109, I143, and L164 (Fig. 3a). Its mutation to proline is less favored in this hydrophobic network and thereby affecting the stability of the whole domain. Besides, as proline and glycine are chemically different from other amino acids in ψ and φ dihedral angles about the peptide bond, L95P mutation may lead Switch I to a different bending direction, and this will hinder the binding of nucleotides (Fig. 3b). Similarly, P96S mutation tends to generate more freedom for Switch I to swing, which is also unfavorable for nucleotide binding and hydrolysis (Fig. 3b).
Fig. 3.
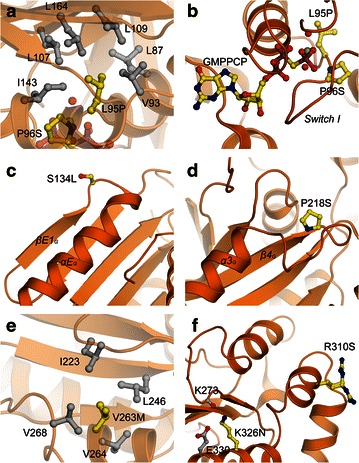
Cancer-related MxA mutations in G domain. a L95 is deeply buried in a hydrophobic cave. b P96 leads the direction of Switch I, which embraces guanine triphosphate nucleotide. c S134 is located on the surface of the G domain. d P218 sits at the end of β-strand 4 (β4 G). e V263 is loosely enwrapped by neighboring hydrophobic residues. f R310 is exposed to the solvent and takes two side chain conformations, whereas K326N interacts with vicinal residues. All cancer-related mutations are shown as the original (i.e., non-mutated) residues, and the post-mutation residues are included in the labels, as L(original)95P(post-mutation), and so on. This scheme is also applied to all of the following figures
S134L mutation occurs between the extra β-strand 1 (βE1G) and extra α-helix (αEG) of MxA (Fig. 3c). As the polar S134 is located on the surface of the molecule, and its side chain also protrudes outward, its mutation to hydrophobic leucine would not influence the global folding of the G domain, but just slightly change the surface entropy of the molecule.
P218S mutation emerges between β-strand 4 (β4G) and α-helix 3 of the G domain (α3G). P218 terminates β4G and turns the polypeptide chain to the opposite direction (Fig. 3d). Analogous to P96S, P218S may change the original trajectory of the following loop and therefore affect the folding of the protein.
V263 is surrounded by several other hydrophobic residues, including I223, L246, V264, and V268 (Fig. 3e). Compared with the hydrophobic pocket engulfing L95, the hydrophobic environment around V263 is much less extensive. As methionine is also a nonpolar residue, V263M mutation makes no alteration of the hydrophobic property of this area. Although methionine is physically a bulkier residue than valine, there is enough space at the top of V263 to accommodate a methionine side chain. Therefore, V263M mutation would cause very limited influence to the folding and stability of the protein.
R310 sits on the surface of the molecule. The side chain of R310 points outward and is quite flexible, as two conformations can be observed in the crystal structure, which suggests that this residue is not bound by any side-chain interaction from other parts of the protein (Fig. 3f). In this situation, its mutation to a polar serine residue will neither break any intra-molecular associations nor drastically change the surface polarity of MxA. Although R310S causes the loss of some positive charge, this mutation would not negatively affect the function of the protein.
K326 is located in α-helix 5 of the G domain (α5G) and also has an outward side-chain conformation. It forms a hydrogen bond with the oxygen of K273 and additionally a weak salt bridge with E330 which also sits in α5G (Fig. 3f). The K326 N mutation may not substantially affect these two interactions, as asparagine also has a polar side chain that can form hydrogen bonds with K273 and E330. Therefore, it is very likely that this mutation does not give rise to any major disruptions of MxA structure.
Single-point mutations in BSE
The BSE is composed of three α-helices which are widely dispersed at the very N-terminus, middle, and the very C-terminus of MxA protein, respectively [5]. BSE is the pivot for transmitting the mechanical force generated from GTP hydrolysis at the G domain to the stalk region, so as to regulate Stalk-dependent oligomerization of the molecule [41]. Five tumor-associated single-point mutations were found in BSE and, more precisely, the α-helix 3 of BSE (α3B), which is also close to the end of the protein (Fig. 4a). According to the crystal structure of full-length MxA, all these 5 residues exhibit an outward side chain conformation. L643 is hydrophobically linked to L357, but its mutation to nonpolar valine would not substantially disrupt this interaction, and thus does not adversely affect the structure of the protein. The side chains of R649, T651, and R655 are all located within a spacious environment and have no contact to other elements of the molecule. Therefore, their mutations, except for mutating to glycine or proline, will lead to negligible interference in the folding of the protein. However, since arginine and threonine are both strong polar residues, whereas methionine and tryptophan possess bulky hydrophobic side chains, and cysteine is a weak-polar residue prone to various post-translational modifications, the R649W, T651M, and R655C mutations bear the possibility to interrupt the association between the protein and other protein partners or incur unexpected modifications.
Fig. 4.

Cancer-related MxA mutations in BSE. a surface-located mutations and a surrounding residue. b the full-length MxA oligomer in the crystal lattice represented by a linear hexamer. All 6 monomers are distinguished by 6 different colors and indicted by corresponding numbers. c A magnified view from the dashed rectangle in Fig. 4b shows that the R654 is involved in inter-molecular Stalk-BSE interaction
In addition to its role as an intra-molecular messenger, BSE also contributes to the formation of functional homo-oligomers, which is a fundamental feature of dynamin superfamily members (Fig. 4b). R654 participates in BSE-Stalk interaction between parallel MxA monomers via a charged interaction with D478 on the other molecule (Fig. 4c). Disruption of this salt bridge by a D478A mutation results in abnormal GTPase activity and weakened oligomerization and antiviral abilities [4]. Not surprisingly, the colorectal cancer-associated R654Q mutation, which abolishes the D478-R654 salt bridge, should have a similar effect. On the other hand, glutamine still possesses the tendency to form a hydrogen bond with D478, and this can be deemed as a compensation for the loss of the salt bridge. As a result, this mutant would hardly cause negative consequence to MxA compared with the reported D478 mutant.
Single-point mutation in Hinge 1
Hinge 1 plays a crucial role in connecting BSE and the stalk region. Extensive interactions between the two loops of Hinge 1 (L1H and L2H) stabilize the ambient region so that the relative position of BSE and Stalk is confined from random movement [4]. The salt bridge formed by E632 on L1H and R640 on α3B is the determining factor in this stabilization effect (Fig. 5). Destruction of this salt bridge by mutating either E632 or R640 to alanine leads to tremendous change of GTPase activity, and almost completely abolishes the oligomerization ability and antiviral effect of MxA. Therefore, it can be well expected that the melanoma-related mutant E632K, which also disrupts the E632-R640 interaction, would be detrimental to the integrity of Hinge 1, and thus the global stability of MxA molecule.
Fig. 5.

Cancer-related MxA mutations in Hinge 1. E632 in Hinge 1 forms a salt bridge with R640 to stabilize the local conformation. Part of the Stalk was removed for clarity
Single-point mutations in the stalk region
The stalk region is essential for the functional assembly of dynamin superfamily members including MxA [5, 42, 43]. In crystal lattice, MxA stalk was found forming linear oligomers via several conserved interfaces, which were then proven to also mediate the architecture of ring-like oligomers in physiological conditions [4] (Fig. 6a). Therefore, it was necessary to first determine the mutations residing in these interfaces, namely G392V and V449G. G392 is located on interface 3 and highly conserved in dynamins and Mx proteins (Fig. 6b). The G392D mutation in MxA was demonstrated to disrupt the oligomerization of the protein [4]. Moreover, its counterpart mutation (G385) in yeast dynamin leads to the breakdown of tetramer into stable dimers [44]. It is therefore not astonishing that the G392V mutation found in renal cell carcinoma deprives the oligomerization capability of MxA and thus its biological activity. In addition, the V449 residue was found to be the interaction partner of G392 on the parallel monomer in the MxA oligomer (Fig. 6b). Its mutation to glycine tends to affect the integrity of interface 3, as well as the conformation of the subsequent Loop 2 on the stalk (L2S). Altogether, these two mutations (G392V and V449G) are likely to impair the physiological function of MxA.
Fig. 6.
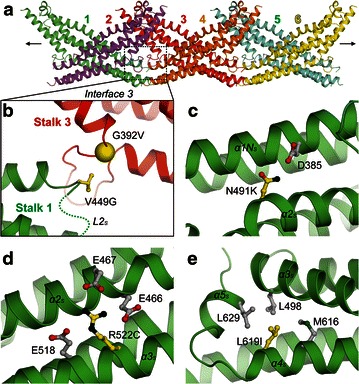
Cancer-related MxA mutations in Stalk. a the MxA Stalk oligomer in the crystal lattice, as represented by a linear hexamer. The monomers are color-coded and labeled in the same manner as in Fig. 4b. Note that compared with the relative direction of the full-length MxA hexamer in Fig. 4b, the Stalk hexamer has been rotated clockwise for 90° along the X axis, and then 180° along the Y axis. b A magnified view from the dashed rectangle in Fig. 6a corresponding to interface 3 shows the interaction between G392 and V449 from parallel monomers. The monomers 2 and 4 are removed for clarity. The invisible Loop 2 on the stalk (L2 S) in this model is indicated with a dashed line. c N491 forms a hydrogen bond with D385 from another α-helix. d R522 is enveloped by three nearby glutamates. e L619 is surrounded by several hydrophobic residues
On the other hand, three additional single-point mutations that are not involved in any oligomerization interfaces have been discovered in the stalk region. N491 is a surface-located residue situated on the α-helix 2 of Stalk (α2S), forming a hydrogen bond with D385 on the parallel α-helix 1N-terminal part (α1NS) (Fig. 6c). Its mutation to positively charged arginine may strengthen this interaction, as a salt bridge can be thus introduced. Therefore, this single-point mutation would play an insignificant role in tumorigenesis or in the development of colorectal cancer [22]. Another colorectal cancer-related mutation site, R522, is also a solvent-exposed residue on α3S. This residue, together with three neighboring negatively charged glutamates (E466 and E467 on α2S, and E518 on α3S), constitutes a vast network of charged interactions endorsed by the salt bridges, in which R522 is at the center, to provide positive charge (Fig. 6d). It is imaginable that its mutation to weak polar cysteine causes the absence of the pivotal positive charge and lead to interference of electrostatic balance on the protein surface. In this case, the folding and stability of the whole protein would become adversely influenced. Unlike N491 and R522, L619 located on α4S possesses an inward side chain conformation. It is part of a local hydrophobic core that includes L498 on α3S, M616 on α4S, and L629 on α5S. When mutated to isoleucine, a derivative comparable to leucine itself in both size and charge, the residue can still stably reside in and maintain this environment. As a result, this ovarian carcinoma-associated mutation L619I will lead to hardly detectable structural and functional aberrance to MxA.
Single-point mutations in the N′-loop and L4
The N′-loop and L4 are both intrinsically disordered loops that lack intra-molecular interactions with other residues, and therefore, it is impractical to observe them in crystal structures. The N′-loop is not conserved among Mx proteins, and its length varies among different species. This region was recently reported to be involved in the specification of viral targets [45]. Given the similar properties of threonine and serine, the T27S mutant would not chemically differ from wild-type MxA, but it is unpredictable whether this mutation would affect the protein’s interaction with viral structures. L4 is essential for membrane binding and viral resistance by direct interaction [46, 47]. However, currently only several residues in the middle of L4, but not Y538, G540, or S572, were proven to be functionally important for MxA [4, 47]. Therefore, it is difficult to predict the influence of these three mutations on MxA function, although they are not likely to disrupt the flexible conformation of L4.
Discussion
As next-generation sequencing data of different human cancers continue to accumulate, abounding tumor-associated mutations of various types are being discovered and further analyzed. Compared with frame shift and stop codon mutations in the coding regions of given genes, it is more tricky to judge the effect of single amino acid alterations on the physiological properties of the proteins. In principal, functional proteins entail orchestrated intra-molecular interactions of the component residues and proper folding of entire polypeptide chains, which can be substantiated as so-called protein structures. In regards to analyses of single-point mutations, if the structures of the corresponding proteins have been solved by X-ray crystallography or other techniques with a decent resolution, the prediction of the effects of these mutations becomes much more reliable.
When doing such predictions, one needs to take the following points into account: the types (or more precisely, the chemical properties) of the pre- and post-mutation residues, the position of the residue, and the known inter-molecular interactions this particular protein accounts for. In our structural analysis of 22 single amino acid mutations found in various human cancers, we scrutinized all target mutants by means of the above criteria and summarized the corresponding results in Table 3. According to our judgment of how severely the structure and function of MxA would be affected, the mutations were classified into 4 groups: drastic, moderate, very moderate, and unpredictable. For drastic mutations, most of them are relevant to glycine or proline. These two residues have distinct stereochemical features compared with the other 18 amino acids. Proline has a confined configuration and usually appears at sharp turns of a peptide chain but seldom in α-helices and β-strands. Therefore, mutations either from or to proline may mislead the peptide chain to inappropriate directions and/or give rise to unexpected kinks in secondary structure elements. In contrast, glycine is the most limber residue, and thus mutations that involve this amino acid may alter the flexibility of the peptide chain at particular positions. All these outcomes are fatal to the folding of the protein.
Table 3.
Summary of the structural analysis of cancer-related MxA single-point mutations
| Mutation influence | Position in protein | Polarity before mutation | Polarity after mutation |
|---|---|---|---|
| Drastic mutations | |||
| L95P | Buried | Nonpolar | Special |
| P96S | Exposed | Special | Polar |
| G392V | Exposed | Special | Nonpolar |
| V449G | Buried | Nonpolar | Special |
| P218S | Exposed | Special | Polar |
| R522C | Exposed | Positively charged | Weak polar |
| E632K | Buried | Negatively charged | Positively charged |
| Moderate mutations | |||
| V263M | Buried | Nonpolar | Nonpolar |
| K326N | Exposed | Positively charged | Polar |
| R649W | Exposed | Positively charged | Nonpolar |
| T651M | Exposed | Polar | Nonpolar |
| T654Q | Buried | Polar | Polar |
| R655C | Exposed | Positively charged | Weak polar |
| Very moderate mutations | |||
| S134L | Exposed | Polar | Nonpolar |
| R310S | Exposed | Positively charged | Polar |
| N491K | Exposed | Polar | Positively charged |
| L619I | Buried | Nonpolar | Nonpolar |
| L634V | Exposed | Nonpolar | Nonpolar |
| Unpredictable mutations | |||
| T27S | Exposed | Polar | Polar |
| Y538C | Exposed | Polar | Weak polar |
| G540D | Exposed | Special | Negatively charged |
| S572Y | Exposed | Polar | Polar |
According to our analysis, besides proline- and glycine-related mutations, other mutations cannot be associated with influencing levels only by single factor (position or chemical properties) (Table 3). To provide a more intuitionistic view of how these mutations may affect the structure of MxA, we performed modeling of several mutants selected from all four groups, and compared the output models to the original crystal structures. However, the calculated mutant models just bear tiny variations compared with the corresponding original structures (r. m. s. d. values less than 0.1 Å). The reason is that current structure prediction algorithms rely on the primary sequence alignment of reference structures, and differences for amino acids in stereochemistry and polarity are not taken into account during model calculation. Therefore, analyzing single-point mutations requires careful and comprehensive manual considerations in multiple aspects involving chemical properties of specific amino acid residues as well as those of the whole protein.
The seven drastic MxA mutations found in this study (L95P, P96S, G392V, V449G, P218S, R522C, and E632K) are likely to play important roles in tumorigenesis and development of corresponding human cancers, and therefore evolve to effective tumor-related biomarkers, although more biochemical and cellular assays are needed for their potential clinical applications. Moreover, it is noteworthy that all three cutaneous squamous cell carcinoma-related mutations (L95P, P96S, and P218S) lead to remarkable disruption of the protein structure, suggesting a strong association between this cancer type and MxA. Finally, our systematic approaches of structure-based analysis for single-point mutations in MxA protein are widely applicable to the evaluation of outcomes of mutations in different types of cancer for those proteins with available structural information.
Conclusions
In this study, by analyzing 22 unique tumor-associated mutations in the human MxA protein by structural methods, we found that 7 out of 22 mutations have a high propensity to affect tumorigenesis and the development of corresponding cancers. These seven mutations are therefore more prone to potential clinical application as useful biomarkers. In addition, our research provides a good example for thorough analysis and consequence evaluation of single-point mutations on a given cancer-related protein.
Authors’ contributions
J-LH, Y-JH, YC, and SG conceived the project, analyzed the data, and wrote the manuscript. J-LH, Y-JH, YC, and BY performed experiments. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by research grants from the National Natural Science Foundation of China (No. 31200553), the National Basic Research Program of China (No. 2013CB910500), the Program of New Century Excellent Talents in University (NCET-12-0567), and the Recruitment Program for Young Professionals.
We thank Prof. Chao-Nan Qian for the help during manuscript preparation.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Footnotes
Jia-Li Hu and Yi-Jun Hua contributed equally to this work
Contributor Information
Jia-Li Hu, Email: hujl@sysucc.org.cn.
Yi-Jun Hua, Email: huayj@sysucc.org.cn.
Yang Chen, Email: chenyang@sysucc.org.cn.
Bing Yu, Email: yubing@sysucc.org.cn.
Song Gao, Email: gaosong@sysucc.org.cn.
References
- 1.Haller O, Stertz S, Kochs G. The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect. 2007;9(14–15):1636–1643. doi: 10.1016/j.micinf.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Horisberger MA, Wathelet M, Szpirer J, Szpirer C, Islam Q, Levan G, et al. cDNA cloning and assignment to chromosome 21 of IFI-78K gene, the human equivalent of murine Mx gene. Somat Cell Mol Genet. 1988;14(2):123–131. doi: 10.1007/BF01534397. [DOI] [PubMed] [Google Scholar]
- 3.Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5(2):133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 4.Gao S, von der Malsburg A, Paeschke S, Behlke J, Haller O, Kochs G, et al. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature. 2010;465(7297):502–506. doi: 10.1038/nature08972. [DOI] [PubMed] [Google Scholar]
- 5.Gao S, von der Malsburg A, Dick A, Faelber K, Schroder GF, Haller O, et al. Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity. 2011;35(4):514–525. doi: 10.1016/j.immuni.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Rennie ML, McKelvie SA, Bulloch EM, Kingston RL. Transient dimerization of human MxA promotes GTP hydrolysis, resulting in a mechanical power stroke. Structure. 2014;22(10):1433–1445. doi: 10.1016/j.str.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Demichelis F, Fall K, Perner S, Andren O, Schmidt F, Setlur SR, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26(31):4596–4599. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 8.Mushinski JF, Nguyen P, Stevens LM, Khanna C, Lee S, Chung EJ, et al. Inhibition of tumor cell motility by the interferon-inducible GTPase MxA. J Biol Chem. 2009;284(22):15206–15214. doi: 10.1074/jbc.M806324200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown SG, Knowell AE, Hunt A, Patel D, Bhosle S, Chaudhary J. Interferon inducible antiviral MxA is inversely associated with prostate cancer and regulates cell cycle, invasion and Docetaxel induced apoptosis. Prostate. 2015;75(3):266–279. doi: 10.1002/pros.22912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calmon MF, Rodrigues RV, Kaneto CM, Moura RP, Silva SD, Mota LD, et al. Epigenetic silencing of CRABP2 and MX1 in head and neck tumors. Neoplasia. 2009;11(12):1329–1339. doi: 10.1593/neo.91110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goel G, Sun W. Cancer immunotherapy in clinical practice—the past, present, and future. Chin J Cancer. 2014;33(9):445–457. doi: 10.5732/cjc.014.10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonasch E, Haluska FG. Interferon in oncological practice: review of interferon biology, clinical applications, and toxicities. Oncologist. 2001;6(1):34–55. doi: 10.1634/theoncologist.6-1-34. [DOI] [PubMed] [Google Scholar]
- 13.Steen HC, Gamero AM. Interferon-lambda as a potential therapeutic agent in cancer treatment. J Interferon Cytokine Res. 2010;30(8):597–602. doi: 10.1089/jir.2010.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lasfar A, Abushahba W, Balan M, Cohen-Solal KA. Interferon lambda: a new sword in cancer immunotherapy. Clin Dev Immunol. 2011;2011:349575. doi: 10.1155/2011/349575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakschies D, Hochkeppel H, Horisberger M, Deicher H, von Wussow P. Emergence and decay of the human Mx homolog in cancer patients during and after interferon-alpha therapy. J Biol Response Mod. 1990;9(3):305–312. [PubMed] [Google Scholar]
- 16.Bezares F, Kohan S, Sacerdote de Lustig E, Farias E, Murro H, Horisberger MA. Treatment strategies for early-stage chronic lymphocytic leukemia: can interferon-inducible MxA protein and tumor necrosis factor play a role as predictive markers for response to interferon therapy? J Interferon Cytokine Res. 1996;16(7):501–505. doi: 10.1089/jir.1996.16.501. [DOI] [PubMed] [Google Scholar]
- 17.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–1309. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 18.Yli-Hietanen J, Ylipaa A, Yli-Harja O. Cancer research in the era of next-generation sequencing and big data calls for intelligent modeling. Chin J Cancer. 2015;34(1):12. doi: 10.1186/s40880-015-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giannakis M, Hodis E, Jasmine MuX, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46(12):1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014;74(12):3238–3247. doi: 10.1158/0008-5472.CAN-14-0013. [DOI] [PubMed] [Google Scholar]
- 21.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Network Cancer Genome Atlas. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6(1):130–140. doi: 10.1016/j.celrep.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20(24):6582–6592. doi: 10.1158/1078-0432.CCR-14-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013;24(6):710–724. doi: 10.1016/j.ccr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45(8):860–867. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 29.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488(7409):106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(Web Server issue):W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeLano WL. The PyMol Molecular Graphics System. 2002. http://pymol.org/. Accessed 3 June 2015.
- 39.Dick A, Graf L, Olal D, von der Malsburg A, Gao S, Kochs G, et al. Role of nucleotide binding and GTPase domain dimerization in dynamin-like myxovirus resistance protein A for GTPase activation and antiviral activity. J Biol Chem. 2015;290(20):12779–12792. doi: 10.1074/jbc.M115.650325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294(5545):1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 41.Chappie JS, Acharya S, Liu YW, Leonard M, Pucadyil TJ, Schmid SL. An intramolecular signaling element that modulates dynamin function in vitro and in vivo. Mol Biol Cell. 2009;20(15):3561–3571. doi: 10.1091/mbc.E09-04-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faelber K, Posor Y, Gao S, Held M, Roske Y, Schulze D, et al. Crystal structure of nucleotide-free dynamin. Nature. 2011;477(7366):556–560. doi: 10.1038/nature10369. [DOI] [PubMed] [Google Scholar]
- 43.Ford MG, Jenni S, Nunnari J. The crystal structure of dynamin. Nature. 2011;477(7366):561–566. doi: 10.1038/nature10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170(7):1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goujon C, Moncorge O, Bauby H, Doyle T, Barclay WS, Malim MH. Transfer of the amino-terminal nuclear envelope targeting domain of human MX2 converts MX1 into an HIV-1 resistance factor. J Virol. 2014;88(16):9017–9026. doi: 10.1128/JVI.01269-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von der Malsburg A, Abutbul-Ionita I, Haller O, Kochs G, Danino D. Stalk domain of the dynamin-like MxA GTPase protein mediates membrane binding and liposome tubulation via the unstructured L4 loop. J Biol Chem. 2011;286(43):37858–37865. doi: 10.1074/jbc.M111.249037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell PS, Patzina C, Emerman M, Haller O, Malik HS, Kochs G. Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe. 2012;12(4):598–604. doi: 10.1016/j.chom.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]