

Abstract
Understanding the mode of action (MOA) of many natural products can be puzzling with mechanistic clues that seem to lack a common thread. One such puzzle lies in the evaluation of the antitumor properties of the natural product withaferin A (WFA). A variety of seemingly unrelated pathways have been identified to explain its activity, suggesting a lack of selectivity. We now show that WFA acts as an inhibitor of the chaperone, p97, both in vitro and in cell models in addition to inhibiting the proteasome in vitro. Through medicinal chemistry, we have refined the activity of WFA toward p97 and away from the proteasome. Subsequent studies indicated that these WFA analogs retained p97 activity and cytostatic activity in cell models, suggesting that the modes of action reported for WFA could be connected by proteostasis modulation. Through this endeavor, we highlight how the parallel integration of medicinal chemistry with chemical biology offers a potent solution to one of natures’ intriguing molecular puzzles.
The withanolides are a large class of steroidal lactones that are found in a variety of plants in the Solanaceae or nightshade family (Figure 1). Withaferin A (WFA; 1), the first identified member of this family, was obtained from samples of the Winter Cherry (Withania somnifera),1 a plant whose extracts and preparations have been used for centuries as herbal remedies. Traditional uses are diverse and include treatment of affictions associated with immune modulation,2 protection from ischemia reperfusion injury,3 neurodegeneration,4 bacterial infections,5 and inflammation.6 Its use is perhaps best known in the Indian traditional medicine, Ayurveda, as a general chemopreventive agent, proclaimed to offer protection against many illnesses, enhance overall well-being, and extend lifespan.7
Figure 1.

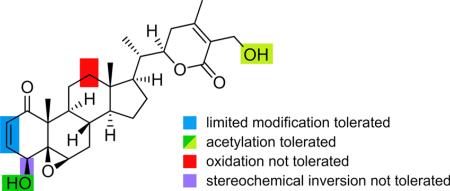
Structures of a panel of withanolides including withaferin A (1); viscosalactone B (14);8 congener 12;8 microbially transformed derivative 5;8 and semisynthetic derivatives 2–4, 6−8, 10, 11, 13, 15, and 16.8 WFA analogs 9, 17, 18, and 19 were prepared via semisynthesis (see Supporting Information). Color shading indicates regions that tolerate acetylation (green and chartreuse), tolerate limited functional modification (blue), do not tolerate oxidation (red), tolerate limited oxidation (orange and gray), or do not tolerate stereochemical inversion (violet).
Despite the long therapeutic history of preparations containing WA and other withanolides, deciphering their mode of antineoplastic activity, especially 1, has only become an area of intense interest over the past decade.9 As for many leads, identification of the precise mode of action (MOA) can be hard to accomplish. This is certainly the case for 1 and other members of this family of natural products. To date, multiple targets and targeted pathways have been assigned including inhibition of the core particle of the proteasome,10 inhibition of vimentin,11 inhibition of annexin II,12 as well as transcription factor regulation.13–17 However, many of these observations were made in a cellular context only and lack a detailed biochemical validation. That aside, the withanolides remain an exciting class of secondary metabolites with established commercial value and potential as chemotherapeutic leads.
RESULTS AND DISCUSSION
Our interest in this family of natural products arose through a program that used high-throughput and affinity techniques to identify natural products that target protein quality control machinery. We began by developing a screening platform to discover leads that modulate the activity of the ATPase associated with various cellular activities (AAA+), p97. To do so, we adapted a high-throughput ATPase assay from our work on another macromolecular machine, the chaperonin GroEL.18–21 This straightforward and well-characterized procedure uses malachite green to detect a phosphomolybdate complex that forms upon hydrolysis of the γ-phosphate of ATP. After screening for optimum conditions, we determined that 100 nM p97 and 100 μM ATP gave a robust signal after 1 h of incubation, while lower concentrations of either chaperone or ATP provided unreliable results even with prolonged incubation (data not shown). To further validate this assay for the present investigations, we assessed the Z factor in 384-well plates (Z factor = 0.75), which indicated that the assay to be used was robust.
We next employed this assay in 384-well plate format to screen a small but unique library of 880 plant and fungal derived extracts as well as 88 purified natural products from related sources. Initial screening was conducted at 10 μg/mL for extracts and 100 μM for pure compounds. As a positive control, we used EDTA, which chelates Mg2+ that is required for ATP hydrolysis. DMSO was used as a negative (vehicle) control. From this screen, we determined that 1 displayed moderate inhibition of p97, which piqued our interest due to its storied history and complex MOA. To validate 1 as a bona fide hit, a 12-point dose response was carried out yielding an IC50 value of 44.0 ± 0.2 μM.
Due to the modest activity of 1, we screened a panel of WFA analogs to see if the activity could be improved. In our possession (Figure 1), we had withanolides 1–88, 10–168, and synthesized 9 and 17–19 from 1 (see Supporting Information). Using a 12-point dose-response analysis, we determined the IC50 values for each of the withanolides 1–19 (Table 1). Prior studies have indicated the Michael acceptor in the A ring of 1 is essential for biological activity.8 This coupled with the presence of 12 cysteines in each p97 subunit argued that p97 might be covalently modified.
Table 1.
IC50 Values for wt-p97 and p97 Variants (CysO, C209A, and C522A)a
| compound | wt p97 | Cys0 | C209A | C522A |
|---|---|---|---|---|
| 1 | 44.0 ± 0.21 | >200 | 24.9 ± 1.91 | >200 |
| 2 | 21.6 ± 2.49 | >200 | 18.0 ± 0.74 | >200 |
| 3 | 78.1 ± 17.1 | >200 | 36.1 ± 6.46 | >200 |
| 4 | 19.0 ± 4.06 | >200 | 41.2 ± 24.5 | >200 |
| 5 | >200 | >200 | >200 | >200 |
| 6 | 21.9 ± 2.38 | >200 | 21.8 ± 1.87 | >200 |
| 7 | 48.3 ± 6.34 | >200 | >200 | >200 |
| 8 | 40.9 ± 14.8 | >200 | >200 | >200 |
| 9 | 17.7 ± 4.45 | >200 | 10.6 ± 1.51 | >200 |
| 10 | >200 | >200 | >200 | >200 |
| 11 | 32.2 ± 20.2 | >200 | >200 | >200 |
| 12 | >200 | >200 | >200 | >200 |
| 13 | >200 | >200 | >200 | >200 |
| 14 | >200 | >200 | >200 | >200 |
| 15 | >200 | >200 | >200 | >200 |
| 16 | >200 | >200 | >200 | >200 |
| 17 | 53.7 ± 4.22 | >200 | >200 | >200 |
| 18 | >200 | 19.4 ± 2.88 | 62.6 ± 12.7 | >200 |
| 19 | 21.9 ± 2.14 | >200 | 31.4 ± 10.2 | >200 |
IC50 values are given in μM inhibition of ATPase activity using the procedure provided in the Supporting Information.
As a preliminary test of covalent modification, we generated a cysteine-less p97 variant, p97–Cys0 (see Supporting Information) to determine if a nucleophilic cysteine was required for the observed activity. Using our ATPase assay, we confirmed that our panel of WFA analogs, with the exception of 18, showed no activity versus p97–Cys0 (Figure 2b and Table 1), indicating at least one cysteine was essential for activity. To test this more carefully, the most potent analogs 1, 4, 6, and 9 (Table 1) were incubated with wild-type p97 (wt-p97) followed by extensive dialysis to remove any noncovalently attached compounds, and finally the ATPase activity was measured. Incubation with the selected WFA analogs was completely reversible by extensive dialysis in the case of 4. Only slight inhibition remained in the presence of 1, and substantial residual inhibition remained for 6 and 9 (Figure S1). These data suggested a potential covalent modification in the case of 6 or 9, but that 1 and 4 are reversible inhibitors.
Figure 2.

Biochemical ATPase activity data. (a) IC50 values for the indicated withanolides versus p97 ATPase activity. (b) IC50 values versus the cysteine–less p97 variant, p97–Cys0. (c) IC50 values versus the p97 variant, p97–C209A. (d) IC50 values versus the bacterial, type I AAA+ protein, ClpX. (e) Inhibition of the bacterial chaperonin, GroEL. (f) Activity against the type II AAA+ chaperone, N-ethylmaleimide sensitive factor (NSF). Plots were generated from 0 to 100 μM; dots indicate IC50 values >200 μM.
To further explore this phenomenon, we generated single cysteine mutants of p97. Given that the activity of the WFA analogs was detected in an ATPase screen, we looked at the role of the two ATP-binding site cysteine residues, C209 (in the D1 pocket) and C522 (in the D2 pocket). The C522A mutants showed no inhibition of ATPase activity in the presence of all 19 WFA analogs (Table 1 and Figure S10), whereas the C209A mutants had IC50 values similar to those for wt-p97 (Figure 2c and Table 1). These data argued that 1–4, 6, 9, 18, and 19 targeted the D2 pocket either covalently or in a reversible manner.
We next measured the effects of ATP concentration on the IC50 values to get a further understanding of the mechanism of p97 inhibition (Figure S2). The parent withanolide, 1, seemed to interact in a competitive-like manner with p97, as there was a distinct shift in the IC50 value as the concentration of ATP increased. Intriguingly, the WFA analogs 2, 4, 9, and 19 did not show ATP-dependent shifts in IC50 values (Figure S2), indicating a more complex mechanism of inhibition, the elucidation of which is particularly daunting given the allosteric nature of p97.22–25
Since the activity of these WFA analogs was discovered using an ATPase assay, we were keen to test their biochemical specificity. To this end, we looked at a set of ATP-hydrolyzing molecular machines including the bacterial type I AAA+ chaperone ClpX (Figure 2d and Table 2), the chaperonin GroEL (Figure 2e and Table 2), and the type II AAA+ chaperone N-ethylmaleimide sensitive factor (NSF; Figure 2f and Table 2). In the case of GroEL (Figure 2e and Table 2), no inhibition was observed at concentrations up to 200 μM, which was not too surprising, as the GroEL ATP-binding pocket is quite dissimilar from that of p97. A more appropriate evaluation arose from the comparison between p97 and other AAA+ family members, in particular the AAA+ chaperones such as ClpX or NSF, the latter of these two being one of the most structurally and biochemically similar to p97. In these assays, WFA analogs 2, 15, and 18 showed inhibition of ClpX (Figure 2d and Table 2), but excitingly, none of the withanolides screened displayed activity against NSF with IC50 values >200 μM (Table 2).
Table 2.
IC50 Values for wt-p97 and Related ATPasesa
| compound | p97 | GroEL | ClpX | NSF |
|---|---|---|---|---|
| 1 | 44.0 ± 0.21 | >200 | >200 | >200 |
| 2 | 21.6 ± 2.49 | >200 | 9.47 ± 1.24 | >200 |
| 3 | 78.1 ± 17.1 | >200 | >200 | >200 |
| 4 | 19.0 ± 4.06 | >200 | >200 | >200 |
| 5 | >200 | >200 | >200 | >200 |
| 6 | 21.9 ± 2.38 | >200 | >200 | >200 |
| 7 | 48.3 ± 6.34 | >200 | >200 | >200 |
| 8 | 40.9 ± 14.8 | >200 | >200 | >200 |
| 9 | 17.7 ± 4.45 | >200 | >200 | >200 |
| 10 | >200 | >200 | >200 | >200 |
| 11 | 32.2 ± 20.2 | >200 | >200 | >200 |
| 12 | >200 | >200 | >200 | >200 |
| 13 | >200 | >200 | >200 | >200 |
| 14 | >200 | >200 | >200 | >200 |
| 15 | >200 | >200 | 35.5 ± 6.18 | >200 |
| 16 | >200 | >200 | >200 | >200 |
| 17 | 53.7 ± 4.22 | >200 | >200 | >200 |
| 18 | >200 | >200 | 21.8 ± 11.2 | >200 |
| 19 | 21.9 ± 2.14 | >200 | >200 | >200 |
IC50 values are given in μM inhibition of ATPase activity using the procedure provided in the Supporting Information.
Next, we explored the effects of 1, 2, 4, 9, and 19 on protein quality control, as p97 is a component of the proteostasis machinery. We first examined the level of total ubiquitylated proteins, which have been shown to increase with p97 inhibition.26–30 As shown in Figure 3a, total ubiquitylation increased for 1, 2, 4, and 19, indicating, at minimum, that these down-regulated some part of the ubiquitin proteasome system (UPS). We also examined the inhibition of the ubiquitin fusion degradation (UFD) reporter, UbG76VGFP (a ubiquitin (Ub) green fluorescent protein (GFP) fusion with a mutation to block deubiquitylase activity), in HEK293 cells. UbG76 VGFP cannot be processed by the proteasome independent of p97; thus an increase in fluorescence indicated inhibition of p97 (or the proteasome).26–30 As shown in Figure 3b, cells treated with the WFA analogs 1, 2, 4, and 19 returned dose-dependent, higher levels of reporter when surveyed by Western blot. Quantitation of the effects of each of these withanolides substantially increased the half-life of the UbG76VGFP reporter (Table 3 and Figure S3).
Figure 3.

Analysis of the ubiquitin proteasome system (UPS). (a) Total ubiquitylation was measured in HEK293 cells with an anti-ubiquitin (Ub) antibody. (b) UbG76VGFP (a Ub green fluorescent protein (GFP) fusion) was stably expressed and measured in HEK293 cells with an anti-GFP antibody. The negative controls used were DMSO and 12. The positive control (+) was generated with MG132 (a proteasome inhibitor).31 All experiments were conducted in triplicate, and exemplary blots are provided. Loading control is given by lamin A.
Table 3.
Protein Half-livesa
| UbG76VGFP |
TCRα-GFP |
HA-CD3δ |
||||
|---|---|---|---|---|---|---|
| compound | compound treated | DMSO control | compound treated | DMSO control | compound treated | DMSO control |
| 1 | 86.9 | 6.5 | 46.9 | 44.2 | 120.7 | 99.4 |
| 2 | 76.9 | 6.5 | 143.0 | 44.2 | 114.5 | 99.4 |
| 4 | 95.9 | 6.5 | 112.8 | 44.2 | 80.0 | 99.4 |
| 19 | 54.1 | 7.2 | 71.3 | 44.9 | 128.9 | 102.2 |
| MG132 | 102.2 | 70.7 | ||||
Half-lives are provided in minutes as obtained by Western blot analyses (Figures S3–S5).
As 1 has been shown to be an inhibitor of the proteasome,10 we wanted to differentiate between proteasome and p97 inhibition. To do so, we took advantage of two endoplasmic reticulum associated degradation (ERAD) reporters, CD3δ that has been shown to be degraded independently of p97 due to substantial cytosolic exposure (Figure 4a) and TCRα that has been shown to be degraded in a p97 dependent fashion due to limited cytosolic exposure (Figure 4b).23,26–30 In addition, TCRα has a deglycosylated form (dg-TCRα) that arises in the presence of proteasome inhibitors, but not p97 inhibitors (Figure 4a), therein providing a facile means of secondary validation.23,26–30 In the case of the TCRα assay, TCRα was fused to GFP (TCRα–GFP), allowing for multiple readouts, such as fluorescence microscopy (Figure 4c). In the absence of a p97 inhibitor, TCRα–GFP was rapidly degraded, leading to a lack of fluorescence in cells (left panel, Figure 4c), but when p97 is inhibited, TCRα–GFP was not extracted from the ER membrane, leading to the appearance of a fluorescent signal (center and right panels, Figure 4c).
Figure 4.

Differentiation of p97-dependent and p97-independent endoplasmic reticulum associated degradation (ERAD) pathways. (a) Poly-ubiquitylated CD3δ exposes a large portion of its sequence to the cytosol and can be extracted from the ER in a p97-independent manner. (b) Poly-ubiquitylated TCRα exposes only a small amount of sequence to the cytosol and requires the force of p97 to extract from the ER membrane. (c) TCRα–GFP can be visualized by fluorescence microscopy. No fluorescence obtains in the absence of a p97 inhibitor, but in the presence of a p97 inhibitor, 1 μM 19 or 1 μM 2, the protein is protected, leading to fluorescent cells. Bar denotes 10 μm.
To investigate both p97-dependent and p97-independent ERAD, we applied Western blotting to identify the levels of TCRα–GFP and hemagglutinin (HA) tagged CD3δ (HA– CD3δ) in HEK293 cells. Withanolide 12, a negative control, showed no increase of TCRα (Figure 5a) or CD3δ (Figure 5b), as expected. In contrast, interpretation of the WFA (1) data was complicated by toxicity for both TCRα (Figure 5a) and CD3δ (Figure 5b). WFA analogs 2, 4, and 19 each showed an increase in the amount of TCRα, but only 4 showed an increase in CD3δ, indicating 2 and 19 had cellular selectivity for p97 (Figure 5b). Interestingly, withanolide 9 was not active in these reporter assays despite potent biochemical activity (Figure 5a,b). Quantitation of the effects of 2, 4, and 19 substantially increased the half-life of the TCRα–GFP reporter, whereas 1 did not (Table 3, Figure S4), but 1 was highly toxic in this cell line, preventing detailed evaluation. In contrast, MG132 stabilized HA–CD3δ, whereas the other compounds showed little or no effect (Table 3, Figure S5).
Figure 5.

Western blot analyses of (a) TCRα–GFP and (b) hemagglutinin (HA)–CD3δ using anti-GFP and anti-HA antibodies, respectively. (c) Analysis of p97 function in HEK293 cells. The unfolded protein response (UPR) was evaluated using an anti-XBP1 antibody to analyze the appearance of spliced XBP1s, lower band. Autophagosome maturation was measured using an anti-LC3 antibody to determine the accumulation of LC3-II, lowest band. Loading controls are given by lamin A or GAPDH.
We then turned our focus to characterizing the effects on the unfolded protein response (UPR) and autophagosome maturation, which are controlled by p97. Using HEK293 cells, we looked at the Hrd1 arm of the UPR by analyzing XBP1 splicing (Figure 5c). The appearance of a lower band in a Western blot for XBP1 (XBP1s) indicated splicing and activation of this branch of the UPR.26–30 Next, we looked at failure of the autophagosomes to fuse with the lysosomes (Figure 5c). The appearance of a lower band in a blot for LC3 indicated the presence of LC3–II, which confirmed compromised maturation of the autophagosomes.26–30 As shown in Figure 5c, the negative controls, DMSO and 12, as well as 9, did not lead to an increase in XBP1s or LC3–II. WFA (1) did show an increase in XBP1s, but not LC3–II, at levels that are also toxic to the cells as indicated by the loss of loading controls (Figure 5c). WFA analogs 2, 4, and 19 all showed dose-dependent increases in both XBP1s and LC3–II, but the increase for 19 was modest relative to those of 2 and 4. These data confirm that the inhibition of p97 also translates in a cellular context.
As a final confirmation of p97 as a target of WFA (1) and its derivatives 2, 4, and 19, we used HEK293 cells expressing p97-C522A. As this p97 variant was shown to be resistant to the WFA molecules, a shift in the cytotoxicity (IC50) should be observed if p97 is the primary target. We used two cell lines, one expressing wt-p97 and one expressing p97-C522A, and determined the cytotoxicity using an eight-point dose– response and an MTT assay. Each experiment was repeated in triplicate. The cells expressing p97-C522A showed no statistically significant rescue in the case of WFA, but 2, 4, and 19 showed a statistically significant increase in the IC50's when p97-C522A was expressed, with compound 2 showing the largest relative shift (Figure 6). It should be noted that these experiments were done in a wt background, so the endogenous levels of p97 might explain the incomplete rescue.
Figure 6.

HEK 293 cells treated with WFA analogs are rescued by expression of p97-C522A. HEK 293 cells expressing wt-p97 (gray bars) or p97-C522A (black bars) were treated with the indicated WFA analogs, and cytotoxicity was measured using an eight-point dose response and an MTT assay. All experiments were done in triplicate, and error bars represent standard deviation from the mean.
Because p97 has been shown to be an essential protein,26–30 we examined the cytotoxicity of the WFA analogs 2, 4, 14, 18, and 19 in a selection of cell lines including mantle cell lymphoma and multiple myeloma cells (B95.8, Bjab, Ca46, NCEB1, Raji, Ramos, and SW-620 cells).31 We first quantified the level of p97 in each of the cell lines (Figure S6) and then measured LD50 values (Figure S7). As shown in Table 4, the effects of the WFA analogs 2 and 19 were cell type specific, with enhanced potency observed in the mantle cell lymphoma line, NCEB1, and reduced potency in the colorectal carcinoma cell line, SW-620, whereas 4 did not show specificity and was generally cytotoxic.
Table 4.
Cytotoxicity Analyses in Select Cell Linesa
| cell line | 2 | 4 | 14 | 18 | 19 |
|---|---|---|---|---|---|
| SW–620 | 29.7 | 5.40 | >100 | >100 | 95.0 |
| Ramos | 4.56 | 4.88 | >100 | >100 | 22.1 |
| Raji | 14.9 | 3.36 | >100 | >100 | 9.44 |
| NCEB1 | 1.86 | 6.80 | >100 | 94.7 | 3.47 |
| Ca46 | 16.2 | 21.4 | >100 | >100 | 26.9 |
| Bjab | 17.4 | 28.3 | >100 | >100 | 25.3 |
| B95.8 | 3.13 | 6.69 | >100 | >100 | 4.31 |
IC50 values are provided in μM using the MTS assay as described in the Supporting Information.
Next, because WFA (1) has been proposed to have several MOAs, we looked at some of these MOAs directly for the various WFA analogs. First, we examined the effects of 1, 2, 4, 9, and 19 on the core particle of the proteasome. Contrary to the cellular data, we found 1, 2, and 4 to inhibit the 20S proteasome but not 9 and 19 (Figure S8). A likely explanation for this discrepancy with the cellular data is that the core particle (20S) is not the operational proteasomal machine in cells and the effects are different for the 26S proteasome, which is the active cellular particle. To test the observation that 1 affects structural proteins, we used immunofluorescence to study the effects on actin (phalloidin), vimentin, and annexin II (Figure S9). As shown, at the concentrations used in the present cellular studies, we did not see effects on these structural proteins, possibly arguing against these as the primary targets of WFA.
From our preliminary panel, we were able to identify key structure–activity relationships (SAR) associated with p97 inhibitory activity, as colored in Figure 1. Oxidation on the C ring was not tolerated (red, Figure 1) as indicated in 5. Second, oxidation of the hydroxyl of the A ring (orange, Figure 1) was only tolerated when associated with modifications on the lactone ring (gray, Figure 1), as given by 9 and 11 retaining activity and the loss of activity in 10. Modification of the enone moiety in the A ring (blue, Figure 1) was only tolerated when substituted by a conjugate addition of an azide, as in 17 or 19. Interestingly, 16 did not bear activity when compared to 17 or 19, therein illustrating the remarkable selectivity within these molecules. The hydroxymethylene group on ring E (chartreuse, Figure 1), however, did accept modifications as in 2, 4, 6, 8, 9, 11, 17, and 19 (Table 1). In general, acylation of the hydroxymethylene group in ring E (chartreuse, Figure 1) and/or the A ring hydroxyl (green, Figure 1) leads to heightened activity and, importantly, increases p97 specificity in cells, as illustrated by 2 or 4.
Conclusion
We have studied 19 WFA analogs including 1 derived from members of the nightshade family (Figure 1). From this panel, we identified withanolides 2, 4, and 19 that effectively modulated the UPS in cells through inhibition of p97 function. Analysis against a panel of tumor cells indicated enhanced activity in Ramos, NCEB1, and B95.8, suggesting selectivity toward B-cell lymphomas. The level of this activity (i.e., 2–5 μM for 2) is remarkable given that p97 is one of the most abundant cytosolic proteins.32–37 p97 is a type II (having two ATP-binding cassettes) AAA+ (ATPase associated with various cellular activities) chaperone. Using the energy of ATP binding and hydrolysis, p97 acts to reshape protein complexes in a variety of biologically critical contexts. These functions include protein quality control through ERAD, cytosolic UFD, cell-cycle regulation, autophagy, and homotypic membrane fusion to name a few.38–41 These diverse actions also implicate p97 in a variety of pathological states including protein misfolding disorders and cancer.33 For over a decade, it has been known that increased p97 levels correlate with a poor clinical outcome in a broad array of malignancies, and recently, more direct links have been made between p97 and cancer.42–44 These factors have generated ever increasing enthusiasm for p97 as a potential therapeutic target.
In the present studies, we discovered that WFA (1) inhibits the ATPase activity of p97. We were intrigued by this, since protein quality control machinery can connect each of the reported modes of action of 1.38,40,42 However, further probing indicated 19 was not specific for p97 but also inhibited the proteasome. Using this information as a guiding principle, we tested WFA analogs 2–19 and were able to tune the activity away from the proteasome and toward p97. These “tuned” withanolides 2, 4, and 19 had good cellular activities and showed specificity for p97. Although the IC50's of these WFA analogs were below those of previously described molecules, the specificity for p97 in cells and modulation of p97-related pathways was found to be comparable to these previously reported molecules.28–30,45 In addition, these WFA analogs showed cytotoxicity against cancer cells with lymphoma specificity, as seen with other p97 inhibitors.28–30,45 Importantly, we have used these collective studies to help elucidate a possible primary mode of cytotoxic action for WFA (1), a natural product known to be responsible for the medicinal activities of a plant that has been used for thousands of years without a biochemical understanding. It is within such accolades that life's secondary metabolic puzzles unfold.
METHODS
Recombinant Protein Expression and Purification
Protein and mutant proteins used in this study were prepared by conventional recombinant protein expression followed by immobilized metal affinity chromatography (IMAC) or native purification in the case of GroEL.
p97.E. coli
BL21(DE3) cells containing pET14b-p97 were grown in Luria Broth (LB) medium containing 100 μg/mL ampicillin at 37 °C to an OD600 of 0.8, followed by induction with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h at 37 °C. Cells were collected by centrifugation (7000g for 10 min), resuspended in 40 mL of lysis buffer (50 mM HEPES, pH 7.4, 150 mM KCl, 5 mM MgCl2, 5% glycerol, 2 mM β-mercaptoethanol (BME), one complete EDTA-free protease inhibitor cocktail (Roche) per 50 mL of media), and lysed by single passage through an M110-T microfluidizer (Microfluidics). The lysate was clarified by centrifugation (118 834g, 1 h, 4 °C), and the resulting supernatant was incubated for 1 h at 4 °C with Talon metal affinity resin (Clontech) equilibrated in 50 mM HEPES, pH 7.4, 150 mM KCl, 5 mM MgCl2, and 5% glycerol. The resin and supernatant were then loaded into a 25 mL disposable column (BioRad), washed with 10 column volumes of wash buffer (50 mM HEPES, pH 7.4, 150 mM KCl, 5 mM MgCl2, 5% glycerol, 2 mM BME, 5 mM imidazole), followed by 10 column volumes of stringent wash buffer (50 mM HEPES, pH 7.4, 1 M KCl, 5 mM MgCl2, 5% glycerol, 2 mM BME, 20 mM imidazole) and eluted with elution buffer (50 mM HEPES, pH 7.4, 1 M KCl, 5 mM MgCl2, 5% glycerol, 2 mM BME, 250 mM imidazole). Fractions were analyzed by 12% SDS PAGE, and those containing p97 were pooled and dialyzed into a storage buffer (20 mM HEPES, 150 mM KCl, 5 mM MgCl2, 5% glycerol, 2 mM BME, pH 7.4), concentrated using a 30 kDa Ultra-15 centrifugal filter (Amicon) to yield a solution of p97 at 12 mg mL–1, which was aliquoted, frozen in liquid N2, and stored at −80 °C until needed.
p97–Cys0
Site directed mutagenesis was performed using wild type pET14b-p97 as a template. Ten of the 12 Cys residues in p97 (C69, C77, C104, C174, C184, C415, C535, C572, C691, and C695) were mutated to Val, and the remaining two Cys residues (C209 and C522) were mutated to alanine using a modified quick change procedure that employs offset oligonucleotides harboring the desired mutation.46 All mutations were confirmed by DNA sequencing (GeneWiz). Expression and purification of the p97–Cys0 construct were carried out as for wild-type p97.
p97–C522A and p97–C209A
Site directed mutagenesis was performed using wild type pET14b-p97 as a template. C209 or C522 residues were mutated to alanine, and mutagenesis was carried out as for the p97–Cys0 construct (see above paragraph). Mutations were confirmed by DNA sequencing. Expression and purification of p97– C522A and p97–C209A was performed as for wild-type p97 (see prior paragraph) then further purified through a Superose 6 10/300 GL preparative grade column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.4, 150 mM KCl, 1 mM MgCl2, 5% glycerol, and 1 mM dithiothreitol (DTT). Fractions containing p97–C522A were pooled, aliquoted, frozen in liquid N2, and stored at −80 °C until needed.
GroEL. E. coli
GroEL was cloned into a pTrc99a vector and expressed in E. coli DH5α cells. Transformed bacteria were grown in LB medium containing 100 μg/mL ampicillin at 37 °C until the OD600 was 0.8. The culture was then induced with 0.5 mM IPTG for 3 h at 37 °C. The cells were recovered by centrifugation (7000g for 10 min). Cell pellets from 4 L of culture were resuspended in 50 mL of buffer A (50 mM Tris, pH 7.4, 1 mM DTT). The cells were lysed by passage through an M110-T microfluidizer (Microfluidics), and the lysate was clarified by centrifugation (118 834g, 1 h, 4 °C) in a Ti45 rotor (Beckman). The cleared lysate was applied to a 75 mL FFQ column (Pharmacia) equilibrated in buffer A plus 20% buffer B (50 mM Tris, pH 7.4, 1 M NaCl, 1 mM DTT) and eluted with a gradient to 50% B over 10 column volumes. The fractions were analyzed by 12% SDS PAGE, and GroEL containing fractions were pooled. (NH4)2SO4 was added slowly to 1.2 M. This sample was then applied to a 25 mL Source ISO150 hydrophobic interaction column (Pharmacia) equilibrated in buffer C (50 mM Tris pH 7.4, 1 mM DTT, 1.2 M (NH4)2SO4). GroEL was eluted with a gradient to 100% buffer A over 10 column volumes. GroEL containing fractions were pooled and dialyzed into a storage buffer (50 mM Tris pH 7.4, 50 mM KCl, 1 mM DTT) and concentrated with a 30 kDa Ultra-15 centrifugal filter (Amicon) to 40 mg mL–1. Protein was aliquoted and stored at 4 °C until needed.
ClpX
ClpX was cloned into a pET14b vector, and the protein was expressed in E. coli BL21(DE3) cells. Transformed bacteria were grown in LB plus 100 μg/mL of ampicillin at 37 °C until the OD600 was 0.6 and cooled down to 21 °C for 1 h. Protein production was induced with 0.5 mM IPTG for 6 h at 21 °C. The cells were recovered by centrifugation (7000g for 10 min) and frozen at −80 °C. Cell pellets were resuspended in 20 mL of lysis buffer (25 mM HEPES–KOH, pH 7.4, 100 mM KCl, 400 mM NaCl, 10% glycerol, 2 mM BME, and complete EDTA-free protease inhibitor cocktail tablets (Roche)). The cells were lysed by passage through an M110-T microfluidizer (Microfluidics). The lysate was clarified by centrifugation (118 834g, 1 h, 4 °C) in a Beckman Ti45 rotor, and the resulting supernatant was loaded onto 3 mL of Talon resin, pre-equilibrated with 25 mM HEPES–KOH, pH 7.4, 100 mM KCl, 400 mM NaCl, and 10% glycerol. After incubation at 4 °C with stirring for 1 h, the column was washed with 60 mL of wash buffer (25 mM HEPES–KOH, pH 7.4, 100 mM KCl, 400 mM NaCl, 10% glycerol, 2 mM BME, 20 mM imidazole), and the ClpX protein was eluted with elution buffer (25 mM HEPES–KOH, pH 7.4, 100 mM KCl, 400 mM NaCl, 10% glycerol, 2 mM BME, and 250 mM imidazole). Fractions were analyzed by 12% SDS PAGE, and those containing ClpX were pooled, dialyzed into a storage buffer (50 mM Tris, pH 7.4, 50 mM KCl, 1 mM DTT), concentrated with a 10 kDa Ultra-15 centrifugal filter (Amicon), aliquoted, frozen in liquid N2, and stored at −80 °C until needed.
N-Ethylmaleimide Sensitive Factor (NSF)
A clone containing the gene for Chinese hamster (Cricetulus griseus) NSF was kindly provided by Dr. Zev Bryant (Stanford University). The NSF protein was expressed in E. coli Rosetta 2 (DE3) cells. Transformed bacteria were grown in LB medium plus 50 μg/mL kanamycin and 25 μg/mL chloramphenicol at 37 °C until the OD600 reached 0.8. Protein expression was induced with 0.5 mM IPTG for 4 h. The cells were recovered by centrifugation (7000g for 10 min) and stored at −80 °C. Cell pellets were thawed on ice and resuspended in 40 mL of lysis buffer (50 mM HEPES–KOH, pH 7.4, 400 mM KCl, 10% glycerol, 2 mM BME, 1 mM phenylmethylsulfonyl fluoride (PMSF) and complete EDTA-free protease inhibitor cocktail (Roche)). The cells were lysed by passage through an M110-T microfluidizer (Microfluidics). The lysate was clarified by centrifugation (118 834g, 1 h, 4 °C), and the resulting supernatant was loaded onto 2 mL of Talon resin pre-equilibrated with 50 mM HEPES, pH 7.4, 400 mM KCl, and 10% glycerol. After incubation at 4 °C while stirring for 1 h, the column was washed with 20 mL of wash buffer (lysis buffer with 20 mM imidazole). The NSF protein was eluted with elution buffer (lysis buffer with 250 mM imidazole). Fractions containing NSF were pooled and dialyzed into a dialysis buffer (20 mM HEPES, pH 7.4, 150 mM KCl, 1 mM MgCl2, 5% glycerol, 1 mM DTT) and concentrated with a 30 kDa Ultra-15 centrifugal filter (Amicon) to reduce the volume to 2 mL. The protein was then run through a HiLoad 26/600 Superdex 200 preparative grade column (GE Healthcare) with 20 mM HEPES, pH 7.4, 150 mM KCl, 1 mM MgCl2, 5% glycerol, and 1 mM DTT. Fractions containing NSF were pooled, aliquoted, frozen in liquid N2, and stored at −80 °C until needed.
ATPase Assays
ATPase activity was evaluated using Malachite green as an indicator. An identical procedure was used for each protein. Briefly, assay buffer (50 mM Tris pH 7.4, 150 mM KCl, 10 mM MgCl2, 1 mM DTT) containing either 100 nM p97–Cys0, 100 nM p97–C522A, 100 nM p97–C209A, 50 nM GroEL, 50 nM ClpX, or 200 nM NSF was dispensed into each well of a 96 well plate, and the assay was run. Test compounds at the desired concentrations (0.0004 μM, 0.002 μM, 0.004 μM, 0.02 μM, 0.04 μM, 0.2 μM, 2 μM, 10 μM, 20 μM, 40 μM, 60 μM, and 100 μM) in DMSO (2 μL) were added to each well. DMSO (2 μL) and 50 mM EDTA (final concentration) were used as negative and positive controls, respectively. Following incubation at 21 °C for 10 min, ATP hydrolysis was initiated by adding 100 μM ATP to each well followed by incubation at 21 °C. Aliquots (20 μL) were taken at 30, 60, 90 and 120 min and added immediately into 50 μL of malachite green solution (9.3 μM malachite green, 53 mM (NH4)2MoO4, 1 M HCl, 10% Tween 20). After 5 min, 10 μL of 34% sodium citrate was added, and the OD670 was read on a GEN5 plate reader (BioTek Synergy 2). IC50 values were calculated by fitting the percentage inhibition at a given compound concentration plotted on semi-log scale using KaleidaGraph (Synergy Software).
Analysis of Covalent Reactivity
Aliquots of compounds 1, 4, 6, or 9 were added at 1 μM concentration to a 1 μM solution of p97 in assay buffer (50 mM Tris, 150 mM KCl, 10 mM MgCl2, 1 mM DTT, pH 7.4) and incubated at 4 °C for 12 h, 21 °C for 6 h, or 37 °C for 2 h. DMSO (1%) was used as a control. The reaction was dialyzed into assay buffer for 2 h using Slide-A-Lyzer Dialysis Cassette (Thermo Scientific), and the buffer was exchanged for fresh buffer three times every 2 h. The ATPase activity was then measured. To initiate the reaction, 2 mM ATP was added. A 10 μL aliquot was taken every 10 min, up to 1 h, and added to 800 μL of malachite green solution. After 5 min, 100 μL of 34% sodium citrate was added. OD660 values were collected on a Genesis 10S VIS Spectrophotometer (Thermo Scientific), converted to nanomoles of inorganic phosphate using a standard curve, and plotted as a function of time using KaleidaGraph (Synergy Software).
Confocal Microscopy on UbG76V-GFP and TCRα-GFP
HEK293 cells stably expressing UbG76VGFP or TCRα-GFP were kindly provided by Dr. Ron Kopito (Stanford University). For live cell imaging, cells were seeded at 2 × 106 cells in phenol-red-free Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) on 35 mm glass-bottom dishes (Bio Express). After incubation at 37 °C under a 5% CO2 atmosphere for 18 h, each dish was treated with either 5 μM 1, 5 μM 2, or 5 μM 3 from DMSO stock solutions (0.1% final DMSO concentration). DMSO (0.1%) was used as a negative control. The cells were imaged in phenol-red-free DMEM supplemented with 10% FBS. Images were captured with an Observer Z1 microscope (Zeiss) by using the Slidebook 5.0 software (Intelligent Imaging Innovations).
Western Blot Analyses
Cells were cultured with the ascribed compound in DMEM supplemented with 10% FBS for 24 h (select markers) or 48 h (apoptotic markers) in 1.9 cm2 culture dishes (Greiner Bio–One). The medium was removed, and the cells were harvested in sample buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 100 mM DTT, 0.1% bromophenol blue) and lysed via sonication followed by clearance of the cell debris by centrifugation. The resulting supernatants were then applied to 4–20% gradient SDS PAGE gels (Invitrogen) and transferred to a Nitrobind nitrocellulose transfer membrane (Maine Manufacturing) using a Bolt Miniblot Module (Life Technologies) gel box. The blots were blocked in 5% milk for 1 h. Primary antibodies were applied in 5% milk at 1:1000 rabbit anti-GFP (GeneTex), 1:1000 anti-caspase 3 (Cell Signaling), 1:1000 anti-eIF2α (pS52) (Invitrogen), 1:1000 mouse anti-VCP (Santa Cruz), 1:1000 mouse anti-β-Actin (Santa Cruz), 1:1000 mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz), 1:1000 anti-ubiquitin (Sigma), 1:1000 anti-XBP1s (Biolegend), 1:1000 anti-HA epitope (Covance), or 1:1000 mouse anti-LC3 I/II (Sigma). The blots were washed three times at 10 min intervals with wash buffer (1 × PBS, 0.1% Tween 20). Secondary antibodies were applied in 5% milk at 1:3000 goat anti-mouse (Santa Cruz) or 1:3000 goat anti-rabbit (Santa Cruz). The blots were washed four times at 10 min intervals with wash buffer, incubated in Supersignal West Pico, Dura, or Femto Substrate (Thermo Scientific), and imaged using ChemiDoc XRS (Bio-Rad). The blots were analyzed using Quantity One 1-D Analysis software (Bio–Rad).
Cytotoxicity Analyses
Cells were seeded at a density of 10 000 cells in 90 μL of DMEM containing 10% FBS in a 96-well plate format. After incubation for 12 h at 37 °C in a 5% CO2 environment, the cells were treated with compounds 2, 4, 14, 18, and 19 in DMEM to provide a concentration gradient at a 5–6 log range in 0.5 log steps. Negative controls were conducted using cells without any additional materials. Analysis was conducted using a CellTiter 96 Aqueous Assay (Promega). Briefly, plates were analyzed after incubation for 48 h by the addition of 0.5 mg mL–1 of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and phenazine methosulfate (PMS). After 2 h of further incubation, the plate was analyzed using a Synergy HT microtiter plate (Biotech) reader at 490 nm. Negative “no-cell” control absorbance wells were prepared with culture media ± compound only to account for background interference. The data were corrected with the absorbance of compounds in the negative “no-cell” control and normalized to the “not-treated” control to obtain the relative absorbance intensity. Absorbance values were averaged from technical triplicate samples to generate individual biological replicates. LD50 values were determined from the plot of relative absorbance intensity versus compound concentration with Prism software (GraphPad) using non-linear regression modeling.
Half-life Measurements
HEK293 cells stably expressing UbG76VGFP, stably expressing TCRα–GFP, or transiently transfected with HA–CD3δ were cultured for 24 h before treatment. At this point, the cells were either left untreated (DMSO) or treated with compounds 1. 2, 4, 19, or MG132 followed by 50 μM cycloheximide until protein synthesis was fully blocked. Total cell lysates were collected at indicated time points and subjected to Western blot analysis with anti-GFP, anti-HA, or anti-tubulin (loading control) or anti-lamin A (loading control) antibodies (see Western Blot Analyses section for protocols). The relative intensities of the bands were quantified for each time point using the CRS gel documentation system (ChemiDoc) and Quantity one software (BioRad) and plotted to determine the half-lives of UbG76VGFP, TCRα–GFP, or HA–CD3δ.
Proteasome Inhibition
Chymotrypsin-like activity of 20S proteasome activity was determined using a 20S Proteasome Assay Kit K-900 (Boston Biochem) using a 96 well plate format. The provided 20S proteasome (1.2 nM final concentration) was incubated with compounds 1, 2, 4, 9, or 19 (eight-point dose response using 3-fold serial dilutions) at 37 °C for 10 min in a reaction buffer (Boston Biochem). A 50 μM aliquot of substrate Suc-LLVY-AMC (Boston Biochem) was then added into each well followed by incubation at 37 °C for 1 h. Hydrolysis of Suc-LLVY-AMC was measured using a Spectramax Gemini XS (Molecular Devices) microplate reader with excitation at 380 nm and emission at 440 nm. IC50 values were determined from a semi-log scale of the plot of relative fluorescence intensity versus compound concentration using KaleidaGraph (Synergy Software).
Confocal Microscopy
HEK293 cells were grown on round glass coverslips (Fisher Scientific) in 35 mm cell culture dishes. After treatment with 1, 2, 4, or 19 for 4 h, the cells were fixed with pre-chilled methanol for 20 min. The coverslips were washed with phosphate-buffered saline (PBS), and blocked with 2% bovine serum albumin (BSA) in PBS for 30 min and then incubated with primary antibody (Texas Red-antiphalloidin, Life Tecnologies; antivimentin, Sigma; antiannexin II – Santa Cruz) for 1 h (see Western Blot Analyses section for discussion of antibodies). After removal, the coverslips were washed three times for 5 min washes with PBS and then incubated with a fluorescently labeled secondary antibody (Alexa Fluor 647 antimouse and Alexa Fluor 555 antirabbit, Invitrogen) for another 50 min. After washing with PBS, the coverslips were mounted onto glass slides with anti-fade mounting medium (Invitrogen). Images were captured with a Zeiss Observer Z1 microscope by using Slidebook 4.2.0.11 (Intelligent Imaging Innovations).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by start-up funds provided by the University of Arizona (E.C.), U.S. National Institute of Environmental Health Sciences Grant R01 ES023758 and ES006694 (E.C. and D.D.Z.), U.S. National Cancer Institute Grant R01 CA90265 (A.A.L.G.), and U.S. National Cancer Institute R01 CA154377 (D.D.Z.).
Footnotes
Author Contributions
E.C., D.D.Z., S.T., and J.T. designed experiments. S.T., J.T., M.K., T.W., E.C.L., C.M., D.J.M., and R.V.B. conducted biochemical assays. S.T. and J.T. conducted the confocal microscopic studies. S.T. and J.T. conducted the Western blot analyses; E.M.K.W. and Y.-M.X prepared the compounds and assigned the NMR data. A.A.L.G. guided the compound isolation and medicinal chemistry efforts. J.J.L. assisted with the data interpretation and presentation. D.D.Z. and E.C. guided the program and led efforts for the biological studies. All authors contributed to the writing.
The authors declare no competing financial interest.
Supporting Information
Supporting Information includes synthetic methods, additional figures, and copies of select NMR spectra. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00367.
REFERENCES
- 1.Lavie D, Glotter E, Shvo Y. Constituents of Withania somnifera Dun. III. The Side Chain of Withaferin A. J. Org. Chem. 1965;30:1774–1778. [Google Scholar]
- 2.Agarwal R, Diwanay S, Patki P, Patwardhan B. Studies on immunomodulatory activity of Withania somnifera (Ashwagandha) extracts in experimental immune inflammation. J. Ethnopharmacol. 1999;67:27–35. doi: 10.1016/s0378-8741(99)00065-3. [DOI] [PubMed] [Google Scholar]
- 3.Gupta SK, Talwar KK, Dinda A, Joshi S, Bansal P, Saxena A, Arya DS. Cardioprotection from ischemia and reperfusion injury by Withania somnifera: a hemodynamic, biochemical and histopathological assessment. Mol. Cell. Biochem. 2004;260:39–47. doi: 10.1023/b:mcbi.0000026051.16803.03. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad M, Saleem S, Ahmed AS, Ansari MA, Yousuf S, Hod N. Neuroprotective effects of Withania somnifera on 6-hydroxydopamine induced Parkinsonism in rats. Hum. Exp. Toxicol. 2005;24:131–147. doi: 10.1191/0960327105ht509oa. [DOI] [PubMed] [Google Scholar]
- 5.Arora S, Dhillon S, Rani G, Nagpal A. The in vitro antibacterial/synergistic activities of Withania somnifera extracts. Fitoterapia. 2004;75:385–388. doi: 10.1016/j.fitote.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Kaileh M, Vanden Berghe W, Heyerick A, Horion J, Piette J, Libert C, De Keukeleire D, Essawi T, Haegeman G. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J. Biol. Chem. 2006;282:4253–4264. doi: 10.1074/jbc.M606728200. [DOI] [PubMed] [Google Scholar]
- 7.Singh N, Bhalla M, de Jager P, Gilca M. An overview on ashwagandha: a Rasayana (rejuvenator) of Ayurveda. Afr. J. Tradit. Complement. Altern. Med. 2011;8:208–213. doi: 10.4314/ajtcam.v8i5S.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wijeratne EMK, Xu YM, Scherz-Shouval R, Marron MT, Rocha DD, Liu MX, Costa-Lotufo LV, Santagata S, Lindquist S, Whitesell L, Gunatilaka AAL. Structure-activity relationships for withanolides as inducers of the cellular heat-shock response. J. Med. Chem. 2014;57:2851–2863. doi: 10.1021/jm401279n. [DOI] [PubMed] [Google Scholar]
- 9.Vanden Berghe W, Sabbe L, Kaileh M, Haegeman G, Heyninck K. Molecular insight in the multifunctional activities of Withaferin A. Biochem. Pharmacol. 2012;84:1282–1291. doi: 10.1016/j.bcp.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 10.Mohan R, Hammers HJ, Baragna-Mohan P, Zhan XH, Herbstritt CJ, Ruiz A, Zhang L, Hanson AD, Conner BP, Rougas J, Pribluda VS. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis. 2004;7:115–122. doi: 10.1007/s10456-004-1026-3. [DOI] [PubMed] [Google Scholar]
- 11.Bargagna-Mohan P, Hamza A, Kim YE, Khuan Abby Ho Y, Mor-Vaknin N, Wendschlag N, Liu J, Evans RM, Markovitz DM, Zhan CG, Kim KB, Mohan R. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 2007;14:623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falsey RR, Marron MT, Gunaherath GM, Shirahatti N, Mahadevan D, Gunatilaka AAL, Whitesell L. Actin microfilament aggregation induced by withaferin A is mediated by annexin II. Nat. Chem. Biol. 2006;2:33–38. doi: 10.1038/nchembio755. [DOI] [PubMed] [Google Scholar]
- 13.Choi MJ, Park EJ, Min KJ, Park JW, Kwon TK. Endoplasmic reticulum stress mediates withaferin A-induced apoptosis in human renal carcinoma cells. Toxicol. In Vitro. 2011;25:692–698. doi: 10.1016/j.tiv.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Grogan PT, Sleder KD, Samadi AK, Zhang H, Timmermann BN, Cohen MS. Cytotoxicity of withaferin A in glioblastomas involves induction of an oxidative stress-mediated heat shock response while altering Akt/mTOR and MAPK signaling pathways. Invest. New Drugs. 2013;31:545–557. doi: 10.1007/s10637-012-9888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santagata S, Xu YM, Wijeratne EMK, Kontnik R, Rooney C, Perley CC, Kwon H, Clardy J, Kesari S, Whitesell L, Lindquist S, Gunatilaka AAL. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem. Biol. 2012;7:340–349. doi: 10.1021/cb200353m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh D, Aggarwal A, Maurya R, Naik S. Withania somnifera inhibits NF-kappaB and AP-1 transcription factors in human peripheral blood and synovial fluid mononuclear cells. Phytother. Res. 2007;21:905–913. doi: 10.1002/ptr.2180. [DOI] [PubMed] [Google Scholar]
- 17.Stan SD, Hahm ER, Warin R, Singh SV. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008;68:7661–7669. doi: 10.1158/0008-5472.CAN-08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson SM, Sharif O, Mak PA, Wang HT, Engels IH, Brinker A, Schultz PG, Horwich AL, Chapman E. A biochemical screen for GroEL/GroES inhibitors. Bioorg. Med. Chem. Lett. 2014;24:786–789. doi: 10.1016/j.bmcl.2013.12.100. [DOI] [PubMed] [Google Scholar]
- 19.Chapman E, Farr GW, Fenton WA, Johnson SM, Horwich AL. Requirement for binding multiple ATPs to convert a GroEL ring to the folding-active state. Proc. Natl. Acad. Sci. U.S.A. 2008;105:19205–19210. doi: 10.1073/pnas.0810657105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang L, Bertelsen EB, Wisen S, Larsen EM, Zuiderweg ER, Gestwicki JE. High-throughput screen for small molecules that modulate the ATPase activity of the molecular chaperone DnaK. Anal. Biochem. 2007;372:167–176. doi: 10.1016/j.ab.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Rowlands MG, Newbatt YM, Prodromou C, Pearl LH, Workman P, Aherne W. High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal. Biochem. 2004;327:176–183. doi: 10.1016/j.ab.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 22.Song C, Wang Q, Li CC. nATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J. Biol. Chem. 2003;278:3648–3655. doi: 10.1074/jbc.M208422200. [DOI] [PubMed] [Google Scholar]
- 23.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. Central pore residues mediate the p97/VCP activity required for ERAD. Mol. Cell. 2006;22:451–462. doi: 10.1016/j.molcel.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 24.Huyton T, Pye VE, Briggs LC, Sands C, Beuron F, Zhang X, Freemont PS. The crystal structure of murine p97/VCP at 3.6A. J. Struct. Biol. 2003;144:337–348. doi: 10.1016/j.jsb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Pye VE, Dreveny I, Briggs LC, Sands C, Beuron F, Zhang X, Freemont PS. Going through the motions: the ATPase cycle of p97. J. Struct. Biol. 2006;156:12–28. doi: 10.1016/j.jsb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Wójcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell. 2006;17:4606–4618. doi: 10.1091/mbc.E06-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Li L, Ye Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 2008;283:7445–7454. doi: 10.1074/jbc.M708347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou T, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4834–4839. doi: 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou T, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem. 2013;8:297–312. doi: 10.1002/cmdc.201200520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, Polucci P, Ballinari D, Perrera C, Leone A, Cervi G, Casale E, Xiao Y, Wong C, Anderson DJ, Galvani A, Donati D, O'Brien T, Jackson PK, Isacchi A. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013;9:548–556. doi: 10.1038/nchembio.1313. [DOI] [PubMed] [Google Scholar]
- 31.Goldberg AL. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 2012;199:583–588. doi: 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunger AT, DeLaBarre B. NSF and p97/VCP: similar at first, different at last. FEBS Lett. 2013;555:126–133. doi: 10.1016/s0014-5793(03)01107-4. [DOI] [PubMed] [Google Scholar]
- 33.Chapman E, Fry AN, Kang M. The complexities of p97 function in health and disease. Mol. Biosys. 2011;7:700–710. doi: 10.1039/c0mb00176g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D. Halawani D, Latterich M. p97: The cell's molecular purgatory? Mol. Cell. 2006;22:713–717. doi: 10.1016/j.molcel.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Wang Q, Song C, Li CC. Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J. Struct. Biol. 2004;146:44–57. doi: 10.1016/j.jsb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 36.Ye Y. Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J. Struct. Biol. 2006;156:29–40. doi: 10.1016/j.jsb.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Rumpf S, Jentsch SS. Cdc48 (p97): a “molecular gearbox” in the ubiquitin pathway? Trends Biochem. Sci. 2007;21:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Baek GH, Cheng H, Choe V, Bao X, Shao J, Luo S, Rao H. Cdc48: a swiss army knife of cell biology. J. Amino Acids. 2013;2013:183421. doi: 10.1155/2013/183421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bug M, Meyer H. Expanding into new markets–VCP/p97 in endocytosis and autophagy. J. Struct. Biol. 2012;179:78–82. doi: 10.1016/j.jsb.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Franz A, Ackermann L, Hoppe T. Create and preserve: proteostasis in development and aging is governed by Cdc48/p97/VCP. Biochem. Biophys. Acta. 2014;1843:205–215. doi: 10.1016/j.bbamcr.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 41.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 42.Fessart D, Marza E, Taouji S, Delom F, Chevet E. P97/CDC-48: proteostasis control in tumor cell biology. Cancer Lett. 2013;337:26–34. doi: 10.1016/j.canlet.2013.05.030. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Hei Y, Shu Q, Dong J, Gao Y, Fu H, Zheng X, Yang G. VCP/p97, down-regulated by microRNA-129–5p, could regulate the progression of hepatocellular carcinoma. PLoS One. 2012;7:e35800. doi: 10.1371/journal.pone.0035800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valle CW, Min T, Bodas M, Mazur S, Begum S, Tang D, Vij N. Critical role of VCP/p97 in the pathogenesis and progression of non-small cell lung carcinoma. PLoS One. 2011;6:e29073. doi: 10.1371/journal.pone.0029073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapman E, Maksim N, de la Cruz F, La Clair JJ. Inhibitors of the AAA+ chaperone p97. Molecules. 2015;20:3027–3049. doi: 10.3390/molecules20023027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.