

1. Introduction
One of the most complex and dispersed organs in the human body is the immune system which functions to identify and destroy invading infectious agents such as bacteria and viruses. It includes cells of discrete organs such as the spleen and thymus, but also components of other organs including bones (bone marrow) and the intestine (Peyer’s patch). Additionally, it uses a network of blood and lymphatic vessels that circulate molecules and cells through much of the body. When an infection threatens the body, various cells and molecules of the immune system work together to destroy the infectious particles. This represents a formidable defensive wall in healthy individuals against foreign invaders and is rarely breached. Of interest here are a series of programmed DNA alterations initiated by an enzyme, activation-induced deaminase (AID), that are essential for an effective immune response. The molecular mechanism of AID action and the response of the cell in the form of DNA repair will be discussed in detail below.
A useful way to look at the immune system is to divide the immune response to an infection into two parts- the cellular response and the humoral response. The first of these refers to the action of cells like the killer T cells and involves direct interactions of these cells with other cells of the immune system and infected cells in the body. The other part, the humoral response, acts instead through antibodies. These proteins can have such a variety of structures that they bind an apparently limitless number of different small molecules such as fragments of proteins and lipopolysaccharides (collectively called antigens) derived from infectious agents. Antibodies are made by B lymphocytes (B cells) and form tight specific complexes with the antigens. This recognition of foreign antigens by antibodies helps other molecules and cells of the immune system to kill and destroy the infectious organism. The two types of immune responses are not completely separate and in fact work together. In particular, T cells play a crucial role in activating B cells to undergo genetic rearrangements described below. We will focus only on the humoral response in this review and cover the progress made in the field since 1999. However, we shall first describe some aspects of the immune response relevant to these alterations in an outline form and the reader is referred to a standard immunology textbook (See Ref. 1; for example) for additional details.
2. Background
2.1 General Structure of Antibody Genes and Proteins
An antibody is a homodimer of a heterodimer consisting of a longer polypeptide chain (called the heavy chain) and a shorter (light) chain (Fig. 1A). The homodimer as well as the heterodimer is partly held together by disulfide bridges and the complete protein can bind two identical antigen molecules. The amino terminal parts of the heavy and light chains, which form the binding pocket, accomplish antigen binding. These protein segments are called variable domains because antibodies that bind different antigens have different primary sequences within these segments. Although the remaining part of each chain is referred to as the “constant” domain, there are five different types of constant domains-α, γ, δ, ε, and μ. The antibodies with these domains are respectively said to be of IgA, IgG, IgD, IgE and IgM isotypes 1.
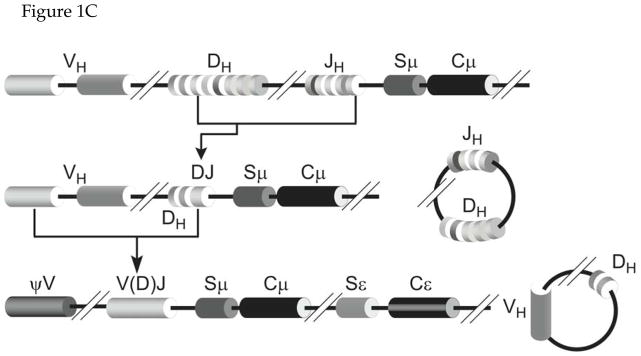
Figure 1. Antibody Structure and V(D)J Recombination.

A. Schematic representation of an antibody molecule. The longer and shorter chains are respectively called heavy (IgH) and light (IgL) chains. Disulfide links between the chains (-S-S-) are also shown. Each chain is divided into variable (lightly shaded) and constant (dark) domains. For convenience, only IgH genes are shown in figures below.
B. Schematic representation of an IgH gene. PVH and Pμ are promoters and V(D)J and Cμ are exons that code the variable and constant for an IgM isotype antibody. Eμ and Sμ are respectively an enhancer for the promoter PVH and the switch sequence for Cμ.
C. V(D)J Recombination. The human chromosome contains multiple tandem segments for V (variable), D (diversity) and J (junction) sequences. Recombination occurs in two steps; first involving a D and a J segment followed by recombination between a V segment and the already rearranged DJ segment. The recombined VDJ segment is the exon that codes for the variable domain. This is typically shown as V(D)J in recognition of the fact that the segment that codes light chain variable domain does not contain a D segment.
The variable and the constant domains of the antibodies are coded by separate exons in the immunoglobulin (Ig) gene (Fig. 1B). The multiple constant domains are encoded by separate exons and the choice of which constant domain is combined with a particular variable domain is made through genetic recombination (see section 2.4 below). The transcription from promoters for the Ig genes occurs at high levels due to the presence of enhancers which for the heay chain lie downstream of the exon for the variable segment (Fig. 1B). The level of transcription of the Ig genes is regulated in part by genetic rearrangements within B cells that bring the downstream enhancers closer to the promoter 2.
2.2 Generation of Antibody Diversity
A remarkable feature of the immune response is its ability to produce secreted antibodies and cell surface receptors that recognize a limitless number of foreign molecules, antigens, using only a limited number of genes. The antigen-binding pockets of antibody proteins are very malleable in their three-dimensional structure and this diversity arises because the variable domain can acquire an almost limitless diversity of amino acid sequences. Consequently, the immune system is thought to be capable of producing antibodies that can collectively bind over 1011 different antigens. One of the early paradoxes regarding immune system was that the antibody proteins can bind so many different antigens although the total number genes in the human genome is thought not to exceed 50,000. Some of the molecular mechanisms that create this amazing diversity within the antibodies are the subject of this review.
This molecular diversity is due, in part, to a series of recombination events that create the variable segment called V(D)J in Figure 1B. This is a combinatorial process that combines three types of protein coding DNA units called V, D and J segments. There are scores of different V segments and a few copies each of the D (only for the heavy chains) and the J segments in the genome (Fig. 1C). During early development, each B cell creates a variable segment from a unique combination of V, D and J (for heavy chains) or V and J (light chains) segments (Fig. 1C). This genetic rearrangement (V(D)J recombination) occurs prior to the exposure of B cells to any antigen and creates millions of clones, each capable of making a distinct antibody. These antibodies are of IgM isotype and are displayed on the cell surface such that they can bind antigens. The molecular mechanisms underlying V(D)J recombination are widely covered in advanced biology textbooks and reviews (e.g. 1,3,4) and will not be discussed here.
2.3 Clonal Selection Theory
In higher vertebrates, B lymphocytes undergo additional genetic changes when the cells are exposed to an antigen. This helps many of these cells produce antibodies that bind antigens with higher affinity. This evolutionary process of making better antibodies is explained by the “clonal selection theory” of Burnet and Talmage 5,6 and a modern version of this proposal is presented in Figure 2.
Figure 2. Clonal Selection Theory.
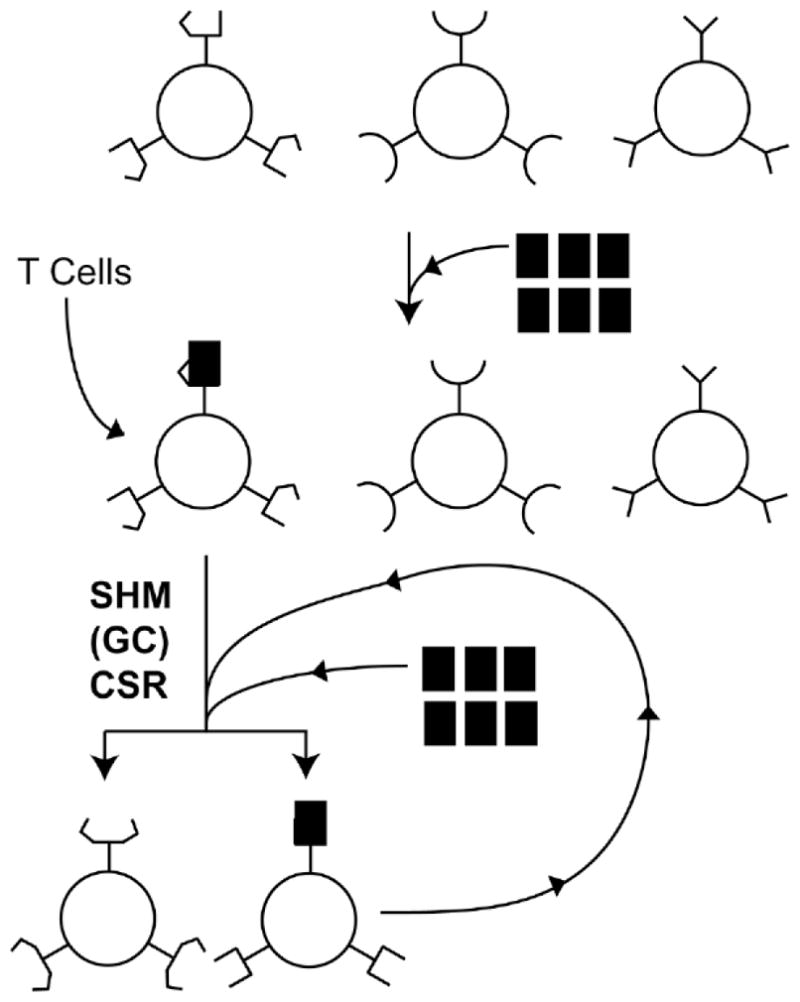
V(D)J recombination creates clones of B cells each coding for a different antibody. Three such B cells with different cell surface antibodies are schematically shown. When a specific antigen (filled rectangle) appears, it has significant affinity towards only one of the clonal antibodies. T cells recognize this antigen-antibody complex and stimulate the B cell to divide. The dividing cells also undergo genetic rearrangements abbreviated as SHM, GC and CSR (see text for details). This changes the structure of the antibodies made creating antibodies with worse (bottom left) or better affinity (bottom right) towards the antigen. The cell producing the antibody that binds the antigen can undergo the same selection and amplification to further increase antibody affinity (semi-circular arrow).
The current version of this model for antibody maturation starts with V(D)J recombination creating millions of clones of B cells, with each clone expressing a unique antibody on its cell surface. When the organism is exposed to a foreign agent such as a virus, only a small fraction of these clones are capable of binding foreign antigens using the antibodies displayed on their surface (Fig. 2). These antigen presenting B cells interact with T cells; which then stimulate the B cells to undergo division and further differentiation. This results in the amplification only those B cell clones that are capable of producing antibodies specific for the foreign antigen 7. At the same time, the cells undergo additional genetic alterations that create antibodies of even higher affinity towards the antigen. These latter alterations in the Ig genes are a critical part of the “affinity maturation” of antibodies.
2.4 Genetic Alterations during Affinity Maturation
The vertebrate Ig genes in maturing B lymphocytes are known to undergo three genetic changes-somatic hypermutations (SHM), class switch recombination (CSR) and gene conversion (GC; Fig. 3; Ref. 8). Of these, SHM and GC are principally mutational processes that introduce (mostly) base substitutions within the V(D)J rearranged Ig genes at a rate of ~10−3 per base pair per generation. This mutation frequency is ~106-fold higher than normal 9 and is restricted to the V(D)J segment of Ig genes. GC involves recombination between a rearranged V(D)J segment and a pseudo-V gene and is presumed to require homologous recombination events (Fig. 3). It is found in some animals (rabbits and chickens), but not in humans and will not be discussed here.
Figure 3. Genetic rearrangements during affinity maturation of antibody genes.

An IgH gene resulting from V(D)J recombination is shown at the top. Depending on the organism the gene undergoes somatic hypermutations (SHM; part a) or gene conversion (GC; part b). Point mutations introduced in the V(D)J segment during SHM are shown by darker lines. The part of V(D)J converted to the sequence of a pseudo V segment (ψV) during GC is shown as a dark patch. IgH genes also undergo class switch recombination (CSR; part c). In this case double-strand breaks within two different switch regions (Sμ and Sε) and the rejoining of open ends create two products. One contains an IgH gene that codes for IgE isotype antibody and a circular DNA product with DNA between the two double-strand breaks.
SHM introduces point mutations in the Ig gene starting at the promoter for the Ig gene and ending around the 5′ end of the intron between V(D)J and the Cμ segments. They do not extend into the constant domain segments 10,11. These mutations are scattered over the variable segment and include transitions as well as transversions. The hypermutations occur about equally at C:G and A:T base pairs creating approximately one amino acid change per cell per generation. Among the many interesting features of SHM is its ability to target a ~1500 bp segment out of a genome of ~3×109 bp and the presence of hypermutational “hotspots” within the V(D)J segment. Another curious feature of SHM is its strict requirement for transcription of the Ig gene 12–14. These and other aspects of SHM are described below in some detail.
Some of these mutated B cell clones produce antibodies that have higher affinity towards the foreign antigen and are further selected for cell division and amplification (Fig. 2). This is an iterative process involving mutations in the Ig variable segment and selection of antigen-binding antibody-producing cells. This means that if the infection that triggered affinity maturation persists in the body then the humoral response creates antibodies with higher and higher affinities for the antigens with the passage of time. For the same reason, repeated immunization of an animal with the same vaccine helps it better able to combat an infection. In contrast to cells that produce antibodies that can bind the antigen, cells that express mutated antibodies that do not bind the antigen are no longer stimulated for cell division and are eliminated from the B cell population. The final stage of the development of B cells producing antibodies against circulating antigens is their conversion to plasma cells that secrete the antibody molecules, which then diffuse into blood and lymphatic vessels 1.
The introns separating the exons for the different constant segments contain two features that are relevant to the third genetic rearrangement, CSR. One feature is a sequence referred to as the “switch” (S) region and the second is a promoter within the intron that transcribes each switch region prior to the genetic rearrangements within the constant domains. The S regions contain short repetitive sequences (GGGGT and GAGCT, for example) and typically have different base composition in the two DNA strands. CSR is a region-specific recombination process that requires double-strand breaks in two different S regions and the joining of the open DNA ends eliminating intervening constant segments as a circle (Fig. 3). In maturing B cells this exchanges the μ constant segments of the immature Ig genes with one of the other constant segments (say ε) causing a switch from IgM type antibodies to a different isotype (IgE; Fig. 3; Ref. 2,8). The strand breaks essential for CSR occur within the S-regions and require transcription (but not translation) of these sequences. The molecular mechanism of CSR is poorly understood and will be discussed below mostly in the context of SHM.
2.5 Antibody Maturation and Immunodeficiency Syndrome
Defects in affinity maturation of antibodies lead to immune deficiency referred to as hyper-IgM syndrome (HIGM). HIGM is a rare immunodeficiency characterized by normal or elevated serum IgM levels with absence of IgG, IgA, and IgE, resulting in a profound susceptibility to bacterial infections and an increased susceptibility to opportunistic infections. While the lack of antibody types other than IgM in these patients is due to defective CSR, many of these patients are also defective in SHM. It is the latter defect that reduces the ability of these patients to fight infections. HIGM is divided into five subgroups, HIGM1 through 5. While two of these subgroups (HIGM1 and HIGM3) have genetic defects that prevent activation of B lymphocytes for maturation by an antigen, two others (HIGM2 and HIGM5) have defects in the DNA processing that creates more diverse antibodies. The latter two types of genetic defects will be discussed in sections 3, 4 and 6. The remaining subgroup (HIGM4) may also be defective in a DNA processing step required for CSR, but its molecular cause is unknown (see OMIM database for additional details- http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM).
3. Discovery and Biology of AID
In recent years, there have been two conceptual breakthroughs in our understanding of the molecular processes by which immunoglobulin genes are altered in response to the exposure of naive B cell clones to antigen. The first of these was the discovery of a gene whose protein product, activation-induced deaminase (AID), is required for both SHM and CSR in murine lymphoid cells 15. The second breakthrough in our understanding of the mechanics of antibody maturation came with the observation that AID is a mutator in E. coli 16 and the suggestion that AID may be a DNA-cytosine deaminase.
We discuss below the molecular mechanisms underlying antibody maturation with a greater emphasis on trying to understand the role of the mutator protein, AID, and of a number of DNA repair processes involved in SHM. The mechanisms of CSR and GC will be discussed only in the context of SHM.
3.1 AID is Required for Antibody Maturation
T. Honjo and colleagues discovered AID while studying a cell line that required stimulation by cytokines to undergo CSR. They found that expression of AID from a tet-contolled promoter alleviated the requirement for cytokines suggesting that cytokines may stimulate the expression of AID to promote CSR. Furthermore, an AID−/− mouse was defective in both CSR and SHM 15 demonstrating the absolute requirement for AID in these processes. In other experiments, the expression of AID was studied in various murine and human tissues and the protein was detected in germinal center B cells 17, and various lymphoid organs 18. Another study investigated the importance of this protein in a clinical setting by sequencing AID gene from 18 patients with one form of the hyper-IgM syndrome (HIGM2). All the patients had mutations in their AID gene and ten different mutant alleles, which included missense mutations as well as frame-shift mutations that cause premature chain termination, were discovered (Ref. 19 and Table 1).
Table 1.
Phenotypes of AID and UDG Mutations
| Amino acid change | Nucleotide change | Phenotype | Additional Comments | References | ||||
|---|---|---|---|---|---|---|---|---|
| WT (aa) | AA position | Mutation (aa) | SHM | CSR | ||||
| AID, point mutations | ||||||||
| S, N | 3, 168 | G, S | NR | Low | Low | Artificial | 48 | |
| K | 10 | R | NR | moderate | moderate | Artificial | 48 | |
| F, C | 11, 117 | V, X | T 31 G, 226 ins 1bp | No | No | HIGM2 | 145 | |
| Y | 13 | H | NR | Low | Normal | Murine, nuclear transport defect | 57 | |
| V | 18 | R | NR | Low | Normal | Murine, nuclear transport defect | 57 | |
| V, R | 18, 19 | S, V | NR | Low | Normal | Murine, nuclear transport defect | 57 | |
| W | 20 | K | NR | Low | Normal | Murine, nuclear transport defect | 57 | |
| G | 23 | S | NR | Low | Normal | Murine | 57 | |
| R | 24 | W | C 70 T | No | No | HIGM2 | 19, 48, 145 | |
| S | 43 | P | T 127 C | Low | No | HIGM2 | 145 | |
| H | 56 | R | NR | NR | NR | Artificial, no deamination | 44 | |
| H | 56 | Y | NR | No | No | HIGM2 | 48 | |
| H | 56 | X | C 166 T | NR | No | HIGM2 | 146 | |
| E | 58 | Q | NR | NR | NR | Artificial, no deamination | 44, 33, 35 | |
| L, W | 59, 68 | F, X | 175 del 9bp, G 203 A | No | No | HIGM2 | 19 | |
| W | 68 | X | G 203 A | No | No | HIGM2 | 48 | |
| W | 80 | R | T 238 C | No | No | HIGM2 | 19, 48 | |
| W | 84 | X | G 251 A | NR | No | HIGM2 | 147 | |
| C | 87 | A | NR | NR | NR | Artificial, no deamination | 33 | |
| C | 87 | R | T 259 C | NR | No | HIGM2 | 81,146 | |
| C | 90 | A | NR | NR | NR | Artificial, no deamination | 33 | |
| L | 98 | R | T 292 G | NR | No | HIGM2 | 81 | |
| L | 106 | P | T 317 C | No | No | HIGM2 | 19, 48,146 | |
| R | 112, 208 | C, X | C 334 T, 544 del 1bp | moderate | No | HIGM2 | 145, 48 | |
| R | 112 | C | C 334 T | No | j No/Normal | HIGM2 | 147, 48, 81 | |
| R | 112 | H | G 335 A | j No/Low | j No/Low | HIGM2 | 147, 48, 145 | |
| I | 136 | K | T 407 A | NR | No | HIGM2 | 145 | |
| M | 139 | V | A 415 G | Low | No | HIGM2 | 19, 48 | |
| C | 147 | X | C 441 A | Low | No | HIGM2 | 19, 48 | |
| F | 151 | S | T 452 C | NR | No | HIGM2 | 19, 146 | |
| R | 174 | S | A 522 C | NR | No | HIGM2 | 146 | |
| AID, addition/deletions | ||||||||
| R | a 190 | X | C 568 T | Normal | No | HIGM2, dominant negative | 148, 48 | |
| b N | 7 | X | 21 del 19bp | NR | No | HIGM2 | 146 | |
| b F | 15 | X | 21 del 19bp | No | No | HIGM2 | 19,48 | |
| W | 68 | X | 175 del 9bp, G 203 A | No | No | HIGM2 | 19 | |
| 3 aa del | 235 del 9bp | No | NR | HIGM2 | 145 | |||
| R | 112, 208 | C, X | C 334 T, 544 del 1bp | moderate | No | HIGM2 | 145, 48 | |
| 183–208 | c 544 del 1bp | Normal | No | Artificial | 48 | |||
| 182–215 | d 182 ins 102bp | Normal | No | HIGM2 | 48 | |||
| 189–198 | e 565 del 30bp | Normal | No | Artificial | 56 | |||
| f G +1 T (exon 2 ret) | NR | Low | HIGM2 | 149 | ||||
| g G +1 C (exon 4 del) | NR | No | HIGM2 | 145 | ||||
| UNG | ||||||||
| 159 | X | 497 del TA | h Normal | Low | HIGM5 | 80, 81 | ||
| F | 151 | S | ||||||
| 141, 224 | X, X | 462 del C, 639 del TA | h Normal | Low | HIGM5 | 80 | ||
| F | 251 | S | T 822 C | i NR | No | HIGM5 | 80, 79 | |
Seven patients with AIDR190X/+ genotype have been described
deletion of 19bp starting from position 21 led to premature stop codon at position 26. This is reported differently in references 146 and 19.
frameshift replacement of c-terminus with 26 amino acids (CMRLMTYETHFVLWDFDSNFQECHTR) at position 183
Insertion of 34 amino acids (VTKPSTQFRRLSGPTDPQPRFEAIHSICFSLSLR) at position 182
C-terminal deletion of 10 amino acids starting at position 189
Splice donor site mutation (G > T) at position of +1 of intron 2 leading to retention of part of intron 2
Splice donor site mutation (G > C) at position of +1 of intron 4 leading to deletion of exon 4
normal mutation frequency but biased toward transitions at G/C residues
mutant protein was fully active when purified from E. coli
Different patients show different phenotypes
HIGM patients with different genetic defects are classified as follows: CD40 ligand (CD40L)-HIGM1; AID-HIGM2; CD40-HIGM3; Unknown defect-HIGM4; UDG-HIGM5
Abreviation: aa, amino acid; X, stop codon; ins, insertion; del, deletion; ret, retention; NR, not reported
AID is required for two additional mutational processes associated with antibody maturation. As noted earlier, some animals use GC instead of SHM as the principal mechanism for generating sequence diversity in maturing B lymphocytes. Using targeted gene disruption in chicken cells, Arakawa et al 20 and Harris et al 21 showed that AID was required for gene conversion in Ig genes. The other mutational process occurs within the switch regions upstream of the constant domain segments in Ig genes. When B lymphocytes are stimulated to undergo CSR, the switch regions acquire point mutations and small addition/deletions regardless of whether or not they have undergone recombination 22. Nagaoka et al 23 found that in a murine AID−/− cell line the Sμ region upstream of non-switched Cμ segments did not acquire mutations when B cells were stimulated. Transfection of these cells with an AID expressing retrovirus restored the hypermutation phenotype in the switch region. Thus AID is required for all known mutational and recombinational processes involved in antibody maturation and plays a critical role in producing a robust immune response against infections.
3.2 AID as an RNA-editing Enzyme
A comparison of AID sequence with available sequences suggested a function for the protein. The protein shares sequence similarity with bacterial cytidine (rC) and cytidylate (rCMP) deaminases 17. This suggested that AID may also be a cytidine deaminase and was accordingly named activation-induced cytidine deaminase. This activity was apparently confirmed biochemically for a GST-AID hybrid protein purified from Escherichia coli and indirect results also suggested that the protein may contain catalytically important zinc ion(s) 17. More intriguingly, AID was most similar in sequence to a RNA-cytosine deaminase, APOBEC1 17. This enzyme converts the cytosine at position 6666 in the mRNA for apolipoprotein B100 to uracil changing a glutamine codon to a termination codon. The resulting shortened protein (apolipoprotein B48) has different physical properties and is processed differently by liver cells. The sequence conservation between AID and APOBEC1 led Muramatsu et al 15 to suggest that AID may act on an mRNA encoding as yet unknown protein changing its product into a CSR recombinase and hypermutator. In the latter case, double-strand breaks (DSBs) caused by this protein within the variable segments of Ig genes would be repaired and the errors in rejoining the broken DNA ends would result in hypermutations. They further suggested that, like APOBEC1 24, AID may also require an accessory protein factor(s) to provide its substrate specificity 15.
DSBs are a clear prerequisite for CSR and hence a model that invokes the synthesis of a new DNA endonuclease in response to AID induction is attractive. However, several key pieces in this hypothesis are missing and there are serious questions about its validity. First, if the repair of DSBs in the variable region lead to SHM, then the predominant signature of this event should be addition/deletion mutations and not base substitutions. Typically, less than ~10% of mutations in hypermutating cells are addition/deletion type 25. Second, there may not be a strict requirement for DSBs for SHM although some reports do suggest a correlation between the two events 26–28. Instead, single-strand breaks in Ig genes may be converted to DSBs during replication 29. Third, SHM does not require DNA-dependent protein kinase catalytic subunit 30, or Rad54 and Rad54B 31 suggesting that neither non-homologous-end joining (NHEJ) nor homologous recombination machinary is required for SHM. Thus the process by which the proposed DSBs in the variable segments would be repaired remains unclear. Fourth, although AID does bind non-specific RNA pools 32,33, as yet no specific mRNA has been identified as its target for cytosine deamination. Finally, if AID does require another protein to target it to a specific mRNA, the identity of this accessory protein is also currently unknown. It seems clear that much work remains to be done to validate the RNA editing model for AID action.
3.3 AID as a Mutator
M. Neuberger and colleagues used four different forward mutation assays to show that expression of AID in E. coli was moderately mutagenic. In wild-type (WT) cells, AID increased the frequency of mutations ~3 to 6-fold and shifted the spectrum of mutations in favor of transition mutation at C:G base pairs. In particular, in the absence of AID, only 31% of mutations in the rpoB gene creating a rifampicin-resistant phenotype (RifR) had a C:G to T:A mutation (hereafter referred to as C to T mutation), but these mutations were 80% of the total in AID expressing cells. The mutations in the gyrA gene (phenotype-nalidixic acid-resistance) showed a similar picture. In this case, C:G to T:A transitions were 34% of all the mutations without AID and 70% with the enzyme 16. Subsequently, the mutator effect of AID was confirmed in other genetic selection systems in E. coli 34–36 and yeast 37,38.
Petersen-Mahrt et al 16 suggested that AID acts directly on DNA converting cytosines to uracils. As uracil in DNA pairs with adenine causing C:G to T:A transitions (Fig. 4) this explains the increase in this class of mutations when AID is expressed in E. coli. Recently, Martomo et al 39 used biochemical techniques to confirm that uracil accumulates in E. coli DNA upon expression of AID. Uracil is excised from DNA by the uracil-DNA glycosylase (UDG), which is present in all organisms (Ref.s 40–43 and Fig. 4). It hydrolyzes the N-glycosidic linkage between the uracil and deoxyribose sugar to create an abasic site, which is processed further by the base excision repair (BER) pathway to restore the cytosine base (Fig. 4). The role of this enzyme in SHM and CSR is discussed more fully in section 5.1.
Figure 4. Cytosine deamination and C to T mutations.

The possible consequences of the deamination of cytosine to uracil are shown. Repair of the lesion through the action of UDG and base excision repair (BER) restores the original C:G pair. Instead, if replication occurs prior to repair, half the daughter molecules contain C to T mutations.
3.4 AID is a DNA-cytosine Deaminase, not a Cytidine Deaminase
Direct evidence for the ability of AID to catalyze deamination of cytosines in DNA was obtained by four different research groups. Bransteitter et al 32 used a GST-AID fusion protein purified from insect cells to show that it converts cytosines in single-stranded (SS) DNA to uracil, but not in double-stranded (DS) DNA, SS RNA or in a DNA-RNA hybrid. Furthermore, they found that different pools of non-specific RNAs from E. coli or mammalian cells were inhibitory towards the SS DNA deamination activity of AID 32. Chaudhuri et al 44 showed that partially purified B-cell extracts were capable of converting 3H-cytosines in DNA to 3H-uracils which could then be released from DNA using UDG. This activity was inhibited by 20 μM tetrahydrouridine and was confirmed further by converting the abasic site created by UDG to strand breaks using alkali 44. Sohail et al and Dickerson et al 33,36 used respectively GST- and Strep-tagged AID purified from E. coli and demonstrated its activity on DS DNA with a bubble and SS DNA. The GST-AID purified partially from E. coli specifically deaminated cytosines in a 5 nt SS bubble to uracils without affecting the cytosines in DS portion of the same molecule and the reaction was inhibited by 1,10-phenathroline, but not by EDTA 36. It is known that Zn2+ ion within APOBEC1 can be extracted with 1,10-phenathroline, but not EDTA 45 and hence these data suggest that AID also contains Zn2+ in its active site. Other investigators have also shown that AID can act on the SS portion of a DNA bubble substrate and molecules with larger bubbles are better for it 32. Dickerson et al 33 found that Strep-AID bound tightly to SS RNA and DNA, but deaminated cytosines in only the latter nucleic acid. These and other studies have established firmly that AID is a SS DNA-specific DNA-cytosine deaminase that has little effect on RNAs that have been tested.
Despite its original naming, AID is not a cytidine deaminase. It does not complement E. coli cdd defective in cytidine deaminase activity (M. C. and A.S.B., unpublished results). It is also not a cytidylate or deoxycytidylate deaminase. However, as mentioned above, one study has reported that GST-AID can deaminate cytidine 17. In contrast, Dickerson et al 33 reported that rC, rCTP or dCTP were not detectably converted to their deamination products by Strep-AID. A possible difference between these two studies is the level of purity of the protein used for the biochemical assays. While the former group purified the protein hybrid on an affinity column for GST, the latter group used two ion exchange columns to purify the protein. The Strep-AID protein was shown to be near homogenous by silverstaining, while the purity of GST-AID was not reported. It is possible that the GST-AID used by Muramatsu et al 17 was contaminated with E. coli Cdd protein. Beale et al 34 raised this very possibility in their study of AID, APOBEC1 and APOBEC3G (another enzyme in the AID-Apobec family). They found that the level of deoxycytidine (dC) deaminase activity in their proteins purified from E. coli varied from preparation to preparation and could be completely eliminated by the addition of tertrahydrouridine (THU), a known inhibitor of cytidine deaminase. Furthermore, a preparation of the catalytically inactive mutant of APOBEC1 (C93A) also had high level dC deaminase activity, which could also be inhibited with THU 34. In contrast, DNA-cytosine deaminase activity of APOBEC1 was unaffected by THU 34,46. These data suggest that purified AID (as well as APOBEC1 and APOBEC3G) is not a nucleoside or mononucleotide deaminase and should be considered a DNA-cytosine deaminase. For this reason, we prefer to call it an activation-induced deaminase, rather than cytidine deaminase.
4. Structure of AID
4.1 Gene for AID
AID gene is located on chromosome 12 in Homo sapiens in a region of microsynteny (12p13) from mammals to pufferfish 47 and is close to the APOBEC1 gene. The human gene contains 5 exons over 10,677 bp and is transcribed into a 2791 nt mRNA. This message is translated into a small 198 amino acid protein (MW 23,954). Mutations that lie in the AID gene exons and in intron-exon boundaries have been discovered in the human population and these individuals suffer from the hyper-IgM type 2 (HIGM2) (Ref. 19 and Table 1).
4.2 Subunit Composition
Several lines of evidence suggest that AID dimerizes or forms higher order multimers, but the number of subunits within active AID remains unclear. One of the HIGM2 mutations (8 aa deletion from C-terminus, Table 1) has a dominant negative phenotype 48 suggesting multimer formation. Additionally, when AID with two different tags were expressed in murine cells, they immunoprecipitated together when antibody against either tag was used 48. The structure of yeast CDD1, which is an ortholog of APOBEC1, has been used to argue that AID may be a dimer 49. However, the biochemical evidence regarding AID composition is conflicting. Chaudhuri et al 44 purified partially AID from mammalian cells and found that it sediments on a glycerol gradient as a 30,000–60,000 MW size range and they have suggested that AID may exist as a dimer 50. However, Dickerson et al 33 reported that Strep-AID purified from E. coli was strongly resistant to dissociation and migrated on the gel as a tetramer. Consequently, the subunit composition of AID remains a matter of debate.
4.3 Subcellular Localization Signals
When AID is tagged at its N-terminus with GFP and expressed in Ramos cells, the protein is predominantly found in the cytoplasm 51. This observation initially suggested that AID does not directly act on DNA. However, Ito et al 52 constructed AID tagged at its C-terminus with GFP and found that the protein shuttles between the cytoplasm and nucleus. Specifically, the fusion protein accumulated in the nucleus following the treatment of cells with an inhibitor of nuclear export. They also found that C-terminal 16 amino acids in AID were essential for the export 52. Similar results were also reported by two other groups 53,54. Furthermore, Ito et al reported the existence of a nuclear localization signal (NLS) in the N-terminus and a similar motif has been found in the N-terminus of APOBEC1 52. However, this sequence may not constitute a true NLS as its removal does not eliminate AID from the nuclei 53. It is possible that AID is kept in the cytoplasm by specific chaperones until the stimulation of B-cells for maturation actively translocates AID to the nucleus 55. Additional work is needed to fully illuminate the mechanisms that regulate AID transport in and out of the nucleus.
4.4 Functional Domains
The carboxy terminus of AID has a second biochemical function; it is required for CSR, but not for SHM. One HIGM2 patient had an AID allele with the terminal 8 amino acids deleted (Table 1) and this protein was shown to be defective in CSR 48. However, this mutant has normal SHM activity in the RifR assay in E. coli. Additionally, a mutant with a 34 aa insertion after codon 181 and another frameshift mutant with changes starting after codon 182 also had a similar split phenotype 48. Similarly, Baretto et al 56 found that a deletion of 10 aa from the carboxy terminus of AID eliminates CSR, but not SHM. Furthermore, these investigators found that hypermutations in the Sμ region were normal showing that CSR was not required for switch region mutations 56.
Shinkura et al 57 reported several mutations near the N-terminus of AID that had reduced SHM activity, but had near normal CSR (Table 1). Based on these mutations these investigators argue that SHM-specific factors bind near the N-terminus of AID that are not required for CSR. However, there are some concerns regarding such a conclusion. First, none of the mutants is completely defective in SHM. Depending on the assay used, some mutants have up to 50% of the WT SHM activity. Second, all the mutations lie within the putative NLS mentioned above. Consistent with their location, five out of the six mutants described are defective in transport into the nucleus. Thus a possible simple explanation for their reduced SHM phenotype is a reduced accumulation in the nucleus 57. Despite these reservations, there appear to be functionally distinct domains at the two ends of the protein. The N-terminal domain (1–23 aa) has a role in nuclear localization and may bind SHM-specific factors, while the C-terminal domain (~180–198 aa) is required for export from the nucleus and is required only for CSR. All the known mutations in the central region affect both SHM and CSR and have little effect on protein localization (Table 1).
4.5 Catalytic Mechanism
Figure 5 shows a model for the active site of AID and a possible reaction mechanism. It is based on the structure and mechanism of E. coli cytidine deaminase 58 and properties of some of the AID mutants listed in Table 1. Briefly, a water molecule is activated and split by the combined action of glutamate 58 and the zinc (II) cation within the active site (step 1). A cytosine within SS DNA is inserted into the active site and stabilized by π interactions with Trp-80 (Fig. 5A). The positioning of W80 within the active site is based on a suggestion regarding APOBEC1 structure by Harris et al 59. This allows the coordinated hydroxide, acting as a nucleophile, to attack at C4 of the cytosine. The π bond between C4 and N3 is lost and the N3 deprotonates glutamate 58 (step 2; Fig. 5B). The result is interrupted ring resonance as C4 is now tetrahedral (step 3). Some rearrangement follows as glutamate 58 deprotonates the hydroxyl and protonates the amine, making it a good leaving group (steps 3 & 4). The reaction cycle completes as the negative charge on O4 forms a π bond with C4, kicking off the positively charged ammonium as ammonia and restoring the ring resonance (step 5; Fig. 5B). No mechanism-based inhibitors of AID or other DNA-cytosine deaminases have been reported and the ability of the product-mimic tetrahydrouridine (THU) to inhibit AID is controversial (Ref.s 17,34,44,50 and see section 3.4). Consequently, much work remains to be done to validate the proposed mechanism.
Figure 5. Active site structure and proposed reaction mechanism for AID.

A. Structure of the active site. The enzyme is presumed to contain a zinc atom, which is coordinated by two cysteines, a histidine and a water molecule. These residues have been identified based on sequence alignments and mutational studies. It is expected that a tryptophan or some other aromatic residue in the protein will stabilize the cytosine. Harris et al {Harris, 2002 #132} have suggested that Trp-80 serves this function. The amino acid residues are numbered to correspond to the human AID sequence.
B. Reaction mechanism of AID. The proposed mechanism based on a mechanism of E. coli cytidine deaminase {Betts, 1994 #80}. E-58 alternately acts as a general base and a general acid, activating the water molecule bound to the zinc atom for an attack at C4 of cytosine and protonating N3. The same residue undergoes one more round of acid-base catalysis to protonate N4 and making it a better leaving group.
4.6 Structural Model for AID
An X-ray crystal structure is currently unavailable for AID, APOBEC1 or other members of this family. A structure is available for the yeast RNA editing cytosine deaminase yCDD1 49. Ta et al have suggested dividing AID into four domains-helix, active site, linker and pseudo active site 48. This division was based on similar proposed division for APOBEC1 60, but is not found in the yCDD1 or modeling efforts based on yCDD1 done by Xie et al 49 and by us.
An alignment of AID sequence with its sequence homologs with known structures is shown in Figure 6A. The model of human AID protein was constructed using the fold-recognition approach 61, followed by recombination of fragments and the optimization of the sequence-structure fit of the “FRankenstein monster” approach 62, combined with remodeling of uncertain regions with ROSETTA 63. All fold-recognition servers generated reliable matches between the AID sequence and the structure of several different deaminases, with the yeast cytosine deaminase (yCD; Ref. 64) singled out as the unequivocally best template for modeling of hAID, in agreement with the earlier suggestion 65. Importantly, no server produced a match that would agree with another prediction, that AID comprises two domains similar to the yeast yCDD1 enzyme 49. Thus, we modeled AID based on the structure of the yCD dimer. The substrate SS DNA was docked manually based on the superposition of the target base with the ligand in the yCD structure.
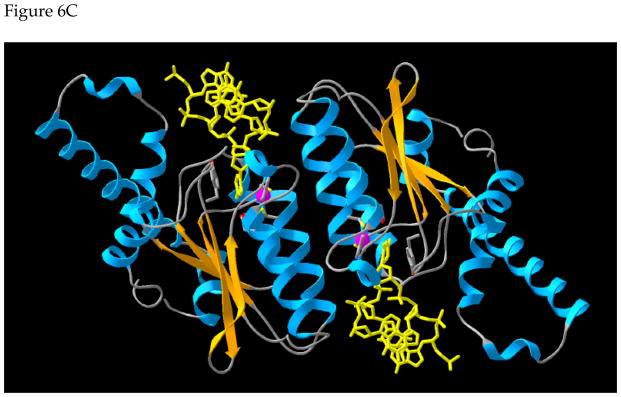
Figure 6. A model for the structure of AID.

A. Structure-based alignment of AID with other deaminases. The deaminases other than AID are identified by their protein data bank, PDB, identification number (1p60, 1wkq etc). Consensus alignment between the human AID and deaminases with known structure was constructed using the protein fold-recognition methods. The mutual alignment of deaminase structures was guided by their structural superposition. Residues identified as being important for the reaction mechanism figure are indicated with asterisks. Amino acids omitted for clarity are indicated in brackets.
B. Structure of the AID monomer. The residues thought to be involved in SHM, but not CSR, are shown in red. The residues in green, when mutated, are known to abolish both SHM and CSR. The C-terminal residues involved in CSR, but not, SHM are shown as light blue trace. The zinc is shown as a magenta sphere. The substrate deoxycytidine is shown in yellow.
C. Structure of the AID dimer. The protein backbone is shown in the ribbon representation, with helices in cyan and strands in orange. SS DNA is shown in yellow and was docked into the dimer so as to insert a cytosine in the active site. The Zn-binding residues C87, C90, and H56, as well as the putative catalytic residue E58 and Y28, which stacks with the target base are shown in the wire-frame representation (oxygen atoms are in red, sulfur atoms are in yellow).
The monomer structure contains a five stranded β sheet which is sandwiched between multiple α helices (Figure 6B). Significantly, the C-terminal residues of the protein required for CSR, but not SHM, fold partly into a helix (aa 190–198; light blue in Figure 6B) and are well separated from the residues thought to be required for SHM, but not CSR (shown in red). While some of the mutations that affect both SHM and CSR (shown in green) are within the proposed active site, some, such as M139 are quite far away. Presumably, these latter class of mutations disrupt overall protein stability.
We modeled the protein as a dimer and docked two SS DNAs into it (Figure 6C). The protein dimerizes as a result of interactions between two central α helices which are also involved in catalysis. The two active sites may interact through the dimer interface and may be sensitive to each other’s structural changes during catalysis. Consequently, it is possible to visualize a model for the enzyme in which binding of the substrate (or catalysis) by one active site affects the structure of the second active site. These structural models serve only as a basis for designing experiments and will have to be modified when additional biochemical or physical data become available.
5. Enzymatic Activity of AID
5.1 Sequence-specificity of AID
One of the key features of SHM is that a significant fraction of them appear within the consensus sequence WRCY (W is A or T, R is purine and Y is pyrimidine; 66) or TW 67. These will be referred respectively as C:G and T:A hotspots. These hotspots could, in principle, have several different origins. They could represent susceptible DNA structures present in Ig genes undergoing SHM, Ig protein domains that are in contact with the antigen or sequences in DNA that reflect the DNA sequence specificity of one or more enzymes involved in SHM. Although it is likely that all these factors contribute to the observed sequence preferences in SHM, the last of these potential causes may make the largest contribution.
When SS M13 DNA was used as the substrate, AID converted multiple cytosines in each substrate molecule in a 230 nt lacZα segment to uracil 68,69. Many of the same cytosines were found mutated in multiple independent clones while some cytosines were rarely targeted by AID. A sequence analysis of the mutants revealed that while the hotspots had the consensus WRC, the coldspot consensus was SYC (S is G or C; Ref. 69 and Table 2). These data show that targeting of DNA by AID is based largely on two bases 5′ to the substrate cytosine and that its selectivity (or avoidance) of the target is a synergistic effect of selectivity at each of the two sites.
Table 2.
*. Sequence preference of AID
| Hotspots | Coldspots | |||
|---|---|---|---|---|
| Sequence | No. of C to U | Sequence | No. of C to U | |
| ATACGC | 34 | TGCCCG | 0 | |
| ATGCTT | 34 | TCCCGA | 0 | |
| CAGCTA | 33 | CGCCTT | 0 | |
| CAACTT | 32 | CGCCAG | 0 | |
| ATGCAG | 31 | GGCCGA | 0 | |
| AAACCC | 31 | TtTCAC | 1 | |
| TTACCC | 31 | CGCCTC | 1 | |
| AAACCA | 29 | TCaCAC | 1 | |
| TAGCTG | 28 | ACaCAG | 1 | |
| AcGCAA | 28 | GGCCGT | 1 | |
| AAACCG | 28 | CGTCGT | 2 | |
| TTGCAG | 28 | TaaCAA | 2 | |
| CAGCAC | 28 | TGgCCG | 3 | |
| AAACAG | 26 | CGCCCA | 3 | |
| TTACGA | 26 | TCTCAC | 3 | |
| CGaCAG | 3 | |||
| CGTCGT | 4 | |||
| Consensus | WRC | SYC | ||
Based on Table 1 in Ref. 69. W is A or T; R is purine, Y is pyrimidine and S is G or C. © Nature Publishing. Reproduced with permission.
The consensus target for AID is very similar to the consensus sequence of the C:G hotspots in SHM 66,70 and hence it is likely that the former causes the latter. This correlation between targeting by AID and SHM hotspots is yet another piece of evidence that supports the idea that AID acts directly on Ig gene DNA rather than on an RNA. As AID is required for all hypermutations, it is also required for mutations at T:A pairs. However, the role played by AID in promoting mutations at T:A pairs is less clear and this is discussed in section 6.
5.2 Processivity of AID
When an SS DNA substrate was used for AID in vitro and the DNA was subsequently introduced into ung E. coli, a large number of clustered mutations were observed (10 to 70 per clone in a 230 nt segment; Ref. 69). This preponderence of multiple closely spaced mutations is due either to multiple interactions of the substrate and AID, or processivity of the protein on DNA. If AID is processive, it may explain a subclass of SHM that are clustered 71. However, most SHM are not clustered and hence the biological significance of this observation remains unclear. Furthermore, some additional considerations cast doubt on the idea that AID acts processively in SHM. One of those considerations is that the likely target for AID in vivo is Ig genes undergoing transcription and not SS DNA (see below).
When lacZ gene fragment undergoing transcription from a T7 RNA polymerase promoter was used as the target for AID, the average number of mutations per clone was only ~3 and about 50% of the LacZ− mutants had single mutations 68. This is in stark contrast with the high degree of multiple clustered mutations reported when SS DNA form of the same substrate was used 9 see above). Additionally, we have never observed multiple mutations in a genetic reversion assay where a transcribing kan gene was the target for AID (Ref. 72; and M. S. and A.S.B., unpublished results). This genetic system is capable of detecting revertants with multiple mutations including those mutants in which adjacent cytosines have been converted to thymines. Furthermore, when the same DS DNA is transcribed using T7 RNA polymerase, the fraction of revertants with multiple mutations appear to be restricted to a minority subpopulation of DNAs that are arrested during transcription (C. Canugovi and A.S.B., unpublished results). This contrasts with an average of 10 to 70 mutations per clone observed by Pham et al 69 who used a SS DNA substrate. Thus the use of a non-physiological substrate, SS DNA, may be responsible for the observation of apparent processivity by AID. It may not act in a processive manner on actively transcribing Ig genes. However, the reported processive action of AID 69 has been incorporated into certain models of SHM and is discussed further in section 9.
6. Role of DNA Repair in SHM
6.1 Uracil Excision Repair
Early evidence for the involvement of uracil excision in modulating the mutagenicity of AID was obtained by comparing wild-type E. coli with ung cells (phenotype-UDG−) The RifR frequency was nine-fold higher in ung cells compared to ung+ cells suggesting that AID causes the conversion of cytosines in the chromosome to uracils 16. In a related study using mice lacking UDG, SHM was affected by a UDG defect. Although the study did not determine overall mutation frequencies, the percent of mutations among hypermutated Ig genes that were C to T was 31% in UDG+/+ mice and 52% in UDG−/− mice 73. Interestingly, the distribution of mutations within the intronic region that was sequenced, was similar in the two genetic backgrounds suggesting that UDG was involved in determining the type of base substitutions found in SHM, but not their local distribution 73. Similar results were also obtained in a chicken cell line where UDG was inhibited by expressing a specific inhibitor of the enzyme, UGI 74. In this case, the frequency of C to T mutations increased from 38% of the total to 86% when UGI was expressed in the cell line 75. Both the studies point to an important role for UDG in SHM and suggest that an intermediate in the SHM pathway is DNA containing uracils.
A couple of additional points should be made here. When UDG−/− mice were first described 76, no phenotype could be attributed to the mutation. In fact, unlike E. coli, murine UDG−/− cells did not have a significant increase in mutation frequency. This was explained by the investigators as a consequence of the activity of a backup uracil-DNA glycosylase, SMUG1 76,77. However, subsequent studies not only revealed altered SHM spectra and reduced CSR in these mice 73, but also the presence of B cell lymphomas and a slightly shortened life span 78. These mice consistently showed lymphoproliferation and developed macroscopic hyperplasia of spleen and lymph nodes at 22 times the rate of the WT mice 78. Although this study did not assay for mutations directly it is reasonable to conclude that UDG must be the principal uracil removal enzyme in lymphatic cells and in its absence mutations and/or other genetic rearrangements occur in the cells at a much higher frequency. Thus the load of uracils in DNA must be particularly high in these cells, supporting the hypothesis that AID (which is expressed only in lymphatic tissue) converts cytosines in DNA to uracils. These conclusions were recently confirmed in a separate study that investigated the relative importance of UDG and SMUG1 in the removal of uracil from DNA in lymphoid tissue 79.
Some HIGM patients (HIGM5) have been found to have mutations in the UDG gene (Ref. 80,81 and Table 1). Three of the four UDG mutations found in these patients contain deletions that result in premature termination and a substantial shortening of the protein. It is reasonable to assume that the truncated proteins expressed in these cells are completely defective in uracil excision. The remaining patient was homozygous for the mutation F251S and somewhat surprisingly the mutant protein, when purified from E. coli, was fully active 79. However, it was defective in transport to the nucleus and as a result uracil excision activity was substantially reduced in nuclear extracts from B lymphocyte cell lines derived from this patient. The extracts had 0.4% residual activity compared to extracts from UDG+/+ individuals 79. These results support an important role for UDG in CSR and have generally been interpreted to mean that UDG is required for the formation of DSBs in the switch regions that precede CSR 73.
Begum et al 82 have questioned such a role for UDG in CSR, indirectly questioning the importance of the catalytic ability of AID to convert cytosines in DNA to uracil. They found that the formation of γH2AX (i.e. phosphorylation of the minor histone H2AX) required AID, but not UDG. H2AX is phosphorylated in response DNA strand breaks and is a used as readout for DSBs that occur during CSR. These investigators expressed UGI, a specific inhibitor of UDG, and found that γH2AX foci could still be observed in response to AID expression 82. When any of the UDG single mutants, D145N, N204V, H268L or F242S (equivalent to the human UDG mutant F251S mentioned above), were expressed in UDG−/− B cells, CSR was normal as evidenced by the titer of IgG. However, neither of the double mutants tested, D145N-N204V and H268L-D145N, could complement this defect. This apparent requirement for UDG in CSR was interpreted as “structural” rather than a catalytic requirement 82.
It has been pointed out (Ref. 83 and G. Baldwin, personal communication) that the three single mutants, D145N, N204V and H268L, of UDG are very powerful catalysts and can excise uracil from duplex DNA with a half-life of about 1 min. Thus the CSR observed in the presence of these mutant proteins may simply be due to residual catalytic activities of these mutants. As mentioned above, the human equivalent of F242S mutation is catalytically active and the overexpression of this mutant from a retroviral vector is likely to result in nuclear accumulation of the active enzyme restoring CSR 79. Furthermore, the double mutants of UDG should be substantially more defective in catalytic activity than the single mutants and may have inadequate activity to promote CSR. These and other considerations suggest 79,83 that results of Begum et al 82 are, in fact, consistent with a role for both AID and UDG in the formation of DSBs that trigger CSR.
6.2 Translesion Synthesis DNA Polymerases
A number of DNA polymerases capable of synthesis across a variety of DNA lesions (translesion synthesis DNA polymerases or TLS Pols) have been implicated in SHM 84. In particular the role of the so called Y-family DNA polymerases (Pol η, Pol ι, Pol κ and Rev1) have been investigated most thoroughly because of their propensity to perform synthesis across bulky lesions and abasic sites. None is absolutely required for SHM, and eliminating some of the TLS Pols only modulates the hypermutation spectrum. It is likely that multiple polymerases participate in the steps that lead to SHM and may be able to compensate for each other.
The DNA polymerase whose involvement in SHM is best understood is Pol η. This polymerase is capable of synthesis across cyclobutane pyrimidine dimers and is missing in Xeroderma pigmentosum variant (XP-V) patients 85,86. This synthesis is relatively “error-free”. However, pol η can also insert nucleotides across from an abasic site causing mutations 87 and this may be its role in SHM (see below). The XP-V patients do not show HIGM syndrome and in XP-V cells the frequency of hypermutations is normal 88. Interestingly, however, the targeting of hypermutations changes in these cells. In the absence of Pol η, the mutations T:A pairs were reduced from 54% to only 18% 88. A similar bias towards mutations at G:C pairs was also observed during SHM in Pol η knockout mice 89,90. In these animals, 79% 90 or 85% 89 of the total mutations occurred at G:C pairs. These studies also confirmed that a lack of Pol η does not strongly affect overall hypermutation frequencies. These studies identify Pol η as a major player in the targeting of T:A hotspots during SHM.
Additional support for this role comes from work in which in vitro copying of the kappa light chain gene using Pol η gave rise to mutations that were consistent with the T:A hotspot mutations 67,91. The same data also suggest that Pol η may be preferentially copying the transcriptional template strand of the Ig genes to cause these hypermutations 91. Finally, there are data that suggest a linkage between the roles of DNA mismatch repair and Pol η, and this will be discussed in section 6.3.
TLS Pol κ, μ and λ have been shown not to play an essential role in SHM 92–94. The other TLS Pols with a connection with SHM are Pol ι, ζ and θ. However, there are contradictory data regarding the roles of these polymerases in the literature, and their precise role in SHM remains unclear. It was reported that when Pol ι gene was knocked out in a Burkitt’s lymphoma cell line in which SHM can be induced, SHM was also eliminated 95. This suggested that Pol ι must be required for SHM. However, this conclusion was contradicted by the observation that in mice with a nonsense mutation in the Pol ι gene, the frequency and distribution of SHM was normal 96. Furthermore, a mouse lacking both Pol η and Pol ι underwent hypermutations and the mutation spectrum was similar to that in a Pol η−/− mice 89. These data cast further doubt about a role for Pol ι in SHM. In an earlier study, Pol ζ transcripts in human B cells were reduced by the use of Pol ζ-specific antisense oligonucleotides and this resulted in a reduction in SHM by a factor of up to 3 97. Similar decrease in hypermutation frequency was also observed in mice expressing antiPol ζ anti-sense RNA 98. Curiously, both the studies found that the hypermutation spectrum remained unchanged in Pol ζ-deficient cells 97,98. This would suggest that Pol ζ plays a major role in causing hypermutations, but a mechanism with a mutational specificity similar to Pol ζ acts as a backup in SHM. However, this conclusion is inconsistent with data that suggest that Pol θ may play a major role in determining SHM frequency and/or spectrum.
Zan et al 99 reported that in Pol θ knockout mice, the frequency of SHM decreased 2.6 to 5.0-fold without changing the ratio of mutations at C:G and A:T pairs. Thus the SHM phenotype of cells deficient in Pol θ and Pol ζ are quite similar to each other. It should be noted that Pol θ and Pol ζ belong to different DNA polymerase families, Class B and Class A, respectively. Further complicating our understanding of the role of TLS Pols in SHM is a recent study by Masuda et al 100. This study found that a different mouse knockout of Pol θ exhibited only a slight reduction in overall SHM frequency (0.8% compared to 1.0% in WT mice). They also found a 41% reduction in mutation frequencies at C:G pairs (0.28% vs 0.48% in WT mice). It is also of interest to note that PolQ, the gene that encodes Pol θ, is specifically expressed in lymphoid tissues and abundant polQ transcripts are detected in germinal center B cells, the target cells for both SHM and CSR 101.
It should be clear from the discussion above that the role of TLS Pols in SHM is poorly understood at this time. This reflects partly our lack of good understanding of the physiology of these enzymes, and as to when and how they participate in DNA synthesis. This is particularly true when it comes to the role of these enzymes in DNA synthesis during BERor mismatch repair (see the next section) as opposed to replicative DNA synthesis. For example, it is possible that two or more of these enzymes form a complex and the absence of one enzyme disrupts the whole complex. This would explain the reports of similar mutational phenotypes in cell lines missing different TLS Pols. Additionally, different Pols may partially compensate for each other preventing a “clean” knockout phenotype. Finally, different TLS Pols may be involved in causing mutations at C:G and A:T sites making the analysis of mutation spectra difficult.
6.3 DNA Mismatch Repair
All organisms possess the ability to correct replication errors that have been overlooked by the proof-reading ability of DNA polymerases. Because it works on two normal DNA bases that are incorrectly paired together, it is referred to as mismatch repair (MMR). The molecular steps of MMR will be described below in a cartoon fashion, and the reader is referred to a specialized review (see Ref. 102 for further details).
In human cells, MMR is initiated by the binding of heterodimer of MSH2 and MSH6 at the mismatch (Fig. 7). A second heterodimer containing MSH2 and MSH3 can initiate the repair of short extrahelical loops and is probably not relevant to antibody maturation. The mismatch-bound MSH2/MSH6 heterodimer undergoes an ATP-dependent conformational change, which converts it to a sliding clamp capable of translocating along the DNA. The MSH2/MSH6•ATP•DNA complex is bound by a second heterodimer, composed of MLHl and PMS2 in a second ATP-dependent step. This complex can translocate in either direction, in search of a strand discontinuity (Fig. 7). A key requirement of MMR is that it must replace the base from the newly synthesized strand and not the “old” strand. The only known mechanism for this discrimination in eukaryotes are the gaps between Okazaki fragments on the lagging strand, or the 3′-termini on the leading strand. In vitro, MMR can be reconstituted using DNA substrates that are not actively undergoing replication, but contain nicks or gaps and hence it is plausible that this could also occur in antibody maturation. EXO1, a 5′ to 3′ exonuclease, is stimulated by the traveling MSH2/MSH6•MLH1/PMS2 complex and can start from a nick situated 5′ from the mispair and travel towards the mispair creating a gap. The region of single-stranded DNA is stabilized by replication protein A (RPA) (Fig. 7). A 3′ to 5′ exonuclease activity (probably within EXO1 itself) can similarly travel from a nick 3′ to the mismatch creating a gap in the other direction and allowing bidirectional MMR. Other proteins known to play a role in MMR are RFC and PCNA (Fig. 7).
Figure 7. Principal Steps during Mismatch Repair.

The repair of a T•G mispair generated as a result of replication error is shown. The proteins involved in this repair are shown in a cartoon fashion and identified in the figure. The DNA substrate contains a strand discontinuity (bottom strand) presumably due to a gap between two Okazaki fragments. For details see the text. Adapted from a figure in Ref. 102.
A series of papers a few years ago showed that mouse knockouts of MSH2, MSH6, MLH1 or PMS2 genes continue to undergo SHM, but at a reduced frequency 103–108. Although there are some differences in the reported decreases in SHM levels, the more interesting observation is that in the MMR deficient animals, SHM showed a stronger bias towards mutating C:G pairs than T:A pairs. In one report 107, hypermutations at C:G pairs increased from 42% in WT to 91% in MSH2−/− mice. Similarly, in MSH6−/− mice the mutations at C:G were 87% of the total compared to 46% in WT animals 103. A smaller increase in mutations at C:G pairs was reported for MLH1−/− and PMS2−/− mice in one study 105, while another report did not find any significant increases in PMS2−/− or MLH1−/− animals 106,107. Although there are some inconsistencies in the data, it is clear that the mismatch recognizing proteins MSH2 and MSH6 have a stronger effect on the mutation spectra than MLH1 and PMS2.
Another interesting observation regarding mutations in mice deficient in MSH2 or MSH6 is that they occurred at the same WRCY hotspots found in WT animals, and the hotspots tended to get hotter 103,107,108. This observation led Rada et al 108 to suggest that there are two phases in targeting of mutations in Ig genes. In the first phase, mutations were targeted at C:G pairs within WRCY sequence motifs by an unknown factor and in the second phase, the mutations at C:G pairs were suppressed by MMR while increasing mutations at T:A pairs at the same time 108. It is likely that the first phase of mutational targeting is performed by AID by its ability to deaminate cytosines within WRCY sequences in DNA 69. The mechanism by which MMR increases mutations at T:A sites in phase II is less well understood.
The studies of SHM and CSR in EXO1−/− mice also confirm a role for MMR in antibody maturation. These mice are defective in CSR compared to WT and their heterozygote siblings 109. SHM was also affected in these animals. While the frequency of hypermutations remained unchanged, the mutations shifted to C:G pairs. Furthermore, mutations within WRCY hotspots also increased. Thus the effects of EXO1 defect and MSH2 defects are very similar suggesting that they both affect the same mutational subpathways that contribute to SHM and CSR.
As noted above, cells deficient in Pol η also show a bias towards C:G targeted mutations. This suggests that MMR may use DNA synthesis by Pol η in extending the mutational process that begins at a C:G pair (presumably within a WRCY context) to T:A base pairs. Evidence has also been presented that both the MSH2-MSH6 and MSH2-MSH3 complexes bind Pol η, but not Pol ι 110. Furthermore, MSH2-MSH6, but not MSH2-MSH3, stimulated primer extension by Pol η. The rate enhancement by MSH2-MSH6 was ~6 fold, while KM also increased by 2.6 fold 110. Thus MSH2-MSH6 increased the catalytic efficiency of Pol η by a factor of only 2.3 fold. MSH2-MSH6 also had little effect on processivity of the polymerase or its fidelity. Thus the overall effect of MSH2-MSH6 complex on Pol η activity is small raising concerns about whether such an interaction has a significant effect on antibody maturation.
When an MSH2 defect in mice is combined with a UDG defect, SHM alters in an interesting way. The overall hypermutation frequency remains unchanged but now essentally all the mutations are targeted at C:G pairs and are C to T 111. This contrasts the MSH2−/− mice where 26% of the mutations were still at T:A pairs. This dramatic shift in the mutation spectrum in the double mutant suggests that MSH2-MSH6 is not the only complex that directs mutations to T:A base pairs. It is likely that in the absence of MMR, processing of U•G mismatches by BER somehow results in shifting some mutations to T:A pairs. The near complete absence of non-C-to-T hypermutations in MSH2−/− UDG−/− mice also suggests that no DNA glycosylase is available to process U•G mismatches in activated B lymphocytes and the uracil is accurately copied by replication polymerases. Also, the fact that not all the mutations are C to T in UDG−/− MMR+ mice 51, says that MMR process finds a way of using the U•G mismatches created by AID for error-prone repair that shifts mutations away from the U•G mispair.
Despite a number of interesting observations and some tantalizing clues, the role of MMR in SHM remains far from clear. The principal difficulty in understanding this role is that MMR acts during SHM in ways that are contrary to its perceived function-avoidance of replication errors. It appears to actively promote misincorporations in DNA by recruiting error-prone DNA polymerases such as Pol η. This would create a paradoxical and potentially explosive situation where MMR promotes creation of mismatches which it then must try to repair! This futile cycle cannot be sustained and in other situations, such as repair of O6-methyl-G:C pairs, is known to cause cell death 112. Clearly, MMR cannot work this way during SHM.
There are other gaps in our understanding of how MMR is involved in SHM. One concerns the possibility that the MSH2-MSH6 complex may bind U•G pairs generated by AID or an abasic site arising from it. This is the simplest explanation for the mutational spectrum in MSH2−/− UDG−/− mice 111, but is puzzling. About 80 U•G mismatches are created in the human genome per generation from non-enzymatic hydrolytic deamination of cytosines 113. These occur throughout the cell cycle and are unrelated to DNA replication. It is clear that although MSH2-MSH6 can bind a U•G mispair in vitro (it is really not that different from a T•G mispair it must frequently repair) MMR is thought not to interact with these non-enzymatically generated mismatches and they are handled exclusively by UDG and other BER enzymes. In fact, interference by MMR in BER of these U•G pairs would be disastrous as MMR does not have any intrinsic discrimination between a U and a G. The repair of these non-enzymatically generated mismatches by MMR would create about 40 C to T mutations (one-half of 80) per generation. It is believed that the rate of mutations in human cells is about 50 times lower (less than one mutation per cell per generation; Ref. 114). Thus the possibility that MMR routinely “repairs” most U•G mispairs is inconsistent with the observed mutation rate in human cells. If MMR does not repair U•G pairs obtained from non-enzymatic deaminations, how can it act on AID generated U•G pairs? In other words, what factor(s) target MMR proteins to Ig gene undergoing SHM?
Another problem is that in contrast to prokaryotes, MMR in eukaryotes is incapable of generating the free 3′-OH needed for EXO1 action and must rely on preexisting nicks or gaps in DNA. What is the source of these nicks in the Ig genes for MMR to function? Processing of uracils by BER does generate transient nicks and gaps, but this also eliminates the U•G mismatches that MSH2-MSH6 may need to bind to participate in SHM. Additionally, a UDG−/− mouse has a different hypermutational spectrum than a UDG−/− MSH2−/− mouse 73,111 suggesting that MMR does affect the SHM spectrum in mice defective in UDG. In principle, it is possible that there is a yet undiscovered U•G mismatch-specific endonuclease that nicks either DNA strand and helps initiate MMR. However, it would have to be B cell-specific as it would otherwise interfere with BER of U•G pairs elsewhere. Another solution to this problem may lie with the reported processivity of AID 69. If AID generates a large number of uracils in Ig genes, some may be partially repaired by BER, while others may remain unrepaired. In such a situation, MMR may step in and initiate repair of U•G mispairs that have not been repaired by BER and use the nearby nicks generated by the partial repair of other U•G mispairs by BER to initiate DNA synthesis. A similar model for the role of MMR in CSR has recently been proposed by Schrader, Stavnezer and colleagues 115,116.
Finally, MMR is thought to be a “long patch” repair process and this is not compatible with the low processivity of the translesion synthesis DNA polymerases. In other words, most scenarios for the involvement of TLS Pols and MMR in SHM force the latter to either become a “short patch” repair process or require a switch to a high fidelity polymerase such as Pol δ after one or two nucleotide incorporation. One observation that lends support to “short patch” repair by MMR during SHM is the relatively modest effects of MLH1 and PMS2 mutations on SHM (see above). As the binding of MLH1/PMS2 dimer to MSH2/MSH6 complex is believed to precede the translocation of of the latter molecule along DNA (Fig. 7 and Ref. 102), the absence of the former dimer may keep the latter complex near the mismatch it binds to. However, many biochemical details including the logistics of a polymerase switch during DNA synthesis are poorly understood at this time 117,118. In summary, we know that the MSH2•MSH6 complex plays a key role in shaping SHM spectrum, especially at A:T pairs, that it may act without the aid of the MLH1/PMS2 dimer and probably acts through a direct interaction with Pol η (and/or some other TLS Pols). However, a conceptual (or experimental) breakthrough is needed before a detailed molecular model for this process can be constructed.
7. Mutagenesis by AID
7.1 AID and C to T Hypermutations
The simplest explanation for the C to T mutations within SHM and switch region mutations is that they result from unrepaired uracils generated by AID. In this model, a certain fraction of uracils created by AID through deamination of cytosines escape repair by UDG and these are eventually replicated to create C to T mutations (Fig. 4). In E. coli an ung mutant has a ~10-fold higher frequency of C to T mutations than its WT parent suggesting that 9 out of 10 uracils in chromosomal DNA resulting from cytosine deamination are excised by UDG. If UDG has a similar efficiency in B lymphocytes, AID must deaminate ~10 times as many cytosines as there are C to T hypermutations. Typically, C to T mutations are ~25% of all SHM and hence AID may generate ~two and one half times as many uracils in DNA as there are SHM. Alternately, uracil repair in B lymphocytes could be much less efficient than in E. coli and most uracil generated by AID may ultimately result in SHM. Which one of these two models is correct can be determined if the amount of uracil generated by AID in the variable segment of Ig genes could be quantified. This is a technically challenging goal where the presence of uracil must be determined in a specific 0.00003% (~1,000 bp out of 3×109 bp) of the genome at a sensitivity of ~1 in 300 nt or better. Although, this has never been done before, a complete understanding of SHM cannot be achieved without it.
7.2 AID and non-C-to-T Hypermutations
While it is easier to understand how uracils generated by AID in DNA may cause C to T mutations, the origin of all other base substitutions and frame-shift mutations (hereafter referred to as non-C-to-T mutations) is much less clear. One possibility is that incomplete repair of uracils in DNA may generate non-C-to-T mutations. As mentioned above, even when MMR is absent, a significant fraction of the mutations occur at T:A pairs. The likely mechanism for these mutations is incomplete BER that leaves a nick which is converted to a gap by exonucleases and the filling-in of these gaps by Pol η or other TLS Pols creates mutations at T:A pairs (Fig. 8). Thus repair of U•G can have three consequences- (1) complete, accurate BER resulting in no mutations; (2) incomplete BER resulting in mutations at C:G as well as T:A; and (3) no repair resulting in C to T mutations (Fig. 8).
Figure 8. Processing of U•G mismatches generated by AID.

The three possible pathways by which U•G mispairs created by AID may be processed are shown. The mutational consequences, if any, are also indicated in each case.
However, many more non-C-to-T mutations are created because of the involvement of MMR. It is also clear that TLS Pols, especially Pol η, play key roles in this process. Unfortunately, no plausible detailed molecular model for the involvement of MMR in SHM exists currently and hence the non-C-to-T mutations in SHM cannot be satisfactorily explained.
8. Role of Transcription in SHM
It has been recognized for some time that transcription of the rearranged Ig gene is essential for both SHM and CSR 14,119,120. Recently, several lines of evidence have converged to highlight the connection between transcription and SHM and CSR. Immunoprecipitation experiments have found that AID associates with a complex containing RNA polymerase II (RNAP II) 121. Other experiments have found that there is a quantitative correlation between the level of expression of the target gene for mutations and the frequency of SHM within it. In B cells this requirement for high transcription is met by the presence of two enhancers upstream of the V segments, but many experiments have shown that the effect is not specific for the V(D)J promoter or the enhancers 122. For example, a defective GFP gene expressed from a tetracycline-controlled promoter in a hypermutation-active pre-B cell line accumulated mutations at a rate that was proportional to the level of transcription of the GFP gene 123. Similar results were also obtained in a fibroblast cell line transfected with AID gene 124. Similarly, CSR is also stimulated by transcription of the switch region 122 and the directionality of transcription may be important for this effect 125.
Ramiro et al 35 and Sohail et al 36 showed that when AID is expressed in E. coli from a native promoter, its mutagenicity is enhanced 20 to 50-fold by the transcription of the target gene. Further, Chaudhuri et al 44 and Sohail et al 36 showed that the same was true in vitro. When AID partially purified from human cells 44 or from E. coli 36 was used in an in vitro transcription reaction involving T7 RNA polymerase (T7 RNAP), the cytosine deaminations caused by AID increased 10 to ~1000-fold. The fact that AID acts in a transcription-dependent manner when the target gene is transcribed by either the E. coli or T7 RNAP suggests that AID recognizes some feature of the transcription bubble rather than a specific RNAP.
8.1 Strand-bias in AID Action
A remarkable property of the transcription-dependence of AID action is its strand bias. Both in E. coli and in vitro AID preferentially deaminates cytosines in the non-transcribed strand (non-template strand; NTS) compared to the transcribed strand (template strand; TS). Consequently, when an ung (i.e. UDG-deficient) host is used, AID promotes C to T mutations in E. coli preferentially in the NTS of the target gene. In vivo the cytosines are 20 to 50 times more frequent targets for deamination when they are in the NTS compared to TS 35,36. Recently, Martomo et al 39 confirmed this observation biochemically and showed that uracils accumulate preferentially in the NTS of a gene in E. coli expressing AID. Their results differed somewhat from the results of genetic assays in that the biochemical assays found only a two-fold difference in the accumulation of uracils in the NTS compared to the TS 39. The reasons for this discrepancy between the genetic and biochemical assays for the magnitude of the strand bias in AID action are unclear. However, when DNA being transcribed in vitro is treated with AID, the bias in favor of converting cytosines in the NTS is at least 10-fold and may be as high as 100-fold 36,44 suggesting that the bias is likely to be much greater than two-fold.
We have previously shown that the NTS in transcribed genes of E. coli is much more accessible to reactive chemicals and acquires more DNA damage 126,127. Specifically, non-enzymatic conversion of cytosines to uracil by water and guanine to 8-oxoguanine by reactive oxygen species occurs at 6 to 40 times higher frequency in NTS than in TS 126–131. Thus AID shows the same transcriptional strand bias as seen with simple reactive chemicals in E. coli. The preference of AID for SS DNA may partially explain this strand bias. However, this cannot be the whole story, because not all chemicals can access NTS as well as water and reactive oxygen species (M. Sanath Kumar and A.S.B., unpublished results). As mentioned earlier, it is likely that AID somehow recognizes the transcription bubble itself and not just the NTS.
This observed strand bias of AID was not anticipated because a strand bias in C to T (G to A) mutations is not found in SHM. In other words, WRCY sequences in either DNA strand can be hotspots for hypermutations. Thus the observed strand bias of AID is either an aberration of the experimental system or the original bias in AID action is somehow “lost” during subsequent DNA processing. This contradiction between the action of AID in E. coli and in vitro and the absence of bias in SHM has led to the several alternate models for the involvement of transcription in AID action.
8.2 Roles of Phosphorylation and RPA in AID Action
F. Alt and colleagues have struck a somewhat different theme 50,132 regarding the propensity of AID to act on genes undergoing transcription. They report that this activity is regulated by the phosphorylation of AID on Ser-38 132. Another residue, Tyr-184, is also phosphorylated in B cells, but the significance of this phosphorylation to AID activity is unclear 132. Both the phosphorylations are performed by protein kinase A (PKA) and this creates the physiologically active form of the protein. Thus PKA and a phosphatase modify AID to respectively turn it on and off 132.
They also make a distinction between the activity of AID on SS DNA and the presumed physiological substrate, DS DNA undergoing transcription (DS-T DNA). They find that partially purified phosphorylated form of AID (AID-P) deaminates cytosines from both SS and DS-T DNAs, the unphosphorylated form (AID-UP) acts only on SS DNA 50. Furthermore, when the AID-P is purified to apparent homogeneity from B cells, it loses its ability to act upon DS-T DNA. This activity is restored the single-strand DNA-binding protein, RPA, is added to the reaction 50. Co-immunoprecipitation and other biochemical assays have been used to show that the 32 kDa subunit of RPA interacts with AID-P, but not AID-UP. Thus in this view of how AID finds transcriptionally active Ig genes, RPA plays a critical role.
These results are not consistent with data presented by other research groups 36,68,69. In the latter studies AID purified from insect cells 68,69 or E. coli 36, which is likely to be unphosphorylated, acts robustly on genes actively transcribed in vitro. Additionally, the ability of AID to act on E. coli genes in vivo depends strongly on the transcription of the genes 35,36. It has been suggested 44,50 that some genes form R-loops when transcribed and the SS DNA within the R-loop may be targeted by AID-UP explaining the difference between the two sets of results. However, we find no correlation between the presence of R-loops and AID activity on DS-T DNA (C. Canugovi and A.S.B., unpublished results). Furthermore, there is little support in the transcription factor literature that RPA is part of the transcription elongation complex. It is still possible that the differences between the two sets of results reflect some subtle differences in the biochemical assays employed and that they can be reconciled.
9. Models for how AID may Target Transcribing Genes
Some earlier models regarding the role of transcription in SHM and CSR included the involvement of specific transcription factors or the RNAP II itself in recruiting AID to specific promoters. While such interactions can not be ruled out, the work with AID in E. coli and in vitro strongly suggests that they are not a requirement for the transcription dependence of SHM. Subsequently, several other models have been proposed to either explain the dependence of SHM and CSR on transcription or other specific properties of SHM. These properties include strand bias in mutations (or the lack thereof), clustering of mutations and acquisition of non-C-to-T mutations. These models for the involvement of transcription in SHM are outlined below (Fig. 9). (It should be noted that these models are not mutually exclusive; two or more mechanisms may be active in causing AID promoted mutations in SHM).
Figure 9. Models for the role of transcription in SHM.

Various models regarding the role of transcription in SHM are presented (A through E). The position of the melted segment of DNA in part D and of the cruciform structure in part E is arbitrary. These structures can form anywhere between the point of attachment of the DNA upstream of the promoter and the transcription bubble. See the text for additional details.
9.1 Transcriptional Pause Model
RNAP often pauses at certain sequences and arrests at others. Several years ago, it was proposed 14,133 that a “mutator factor” (now believed to be AID) would act at pause sites. In the original formulation the mutations were attributed to faulty transcription-coupled repair, but must now be ascribed to the action of AID itself. The NTS in the bubble at a pause site is more accessible than the TS and hence this model would predict that more mutations arise due to damage to cytosines in the NTS than in the TS (Fig. 9A). It may also provide an extended time during which AID can repeatedly act within the bubble. This could create clustered mutations observed in some studies of SHM 71,134 and create multiple U•G mismatches needed for some models of SHM.
9.2 Bubble-access Model
We have argued for some time 126 that the NTS in an elongation complex is accessible to chemicals and that this is true even without any pausing or arrest of the RNAP (Fig. 9B). The action of AID in E. coli 35,36 and in vitro 36 is consistent with this idea. This model differs from the transcriptional pause model above in that it does not predict clustering of hypermutations. This is because AID catalysis is unlikely to keep pace with the speed of the transcription bubble, ~30 nt/sec. We have pointed out that 72 most sequence-specific DNA-binding enzymes such as DNA methyltransferases and restriction endonucleases turn over at a rate of ~1 per min. If AID is similarly slow in its catalytic turnover, it would fall off DNA as soon as the transcription bubble passes it by. No studies of the interaction of AID with a stable elongation complex have been reported.
9.3 R-loop Model
It has been suggested that the pre-mRNA resulting from transcription of Ig gene may not be removed, thus creating R-loops. These have been specifically observed in Ig genes undergoing CSR 135, but have also been suggested in case of SHM 44. In this case, a large section of the NTS would be accessible to AID for a prolonged period of time (Fig. 9C). The TS in the R-loop should largely be protected by the RNA, but may also be available to AID at the edges of the bubble. Recent studies of the interaction of AID with an artificial R-loop shows that the DNA in NTS is indeed accessible to AID 136. This model would also predict multiple U•G mismatches in the Ig gene.
9.4 Superhelical Domain Model
Transcription of a gene creates a wave of positive supercoiling ahead of the RNAP and negative supercoiling behind it. This “twin-domain” model of transcription-induced topological changes in DNA 137 suggests that the DNA near the 5′ end of a gene (or upstream of a gene) tends to be underwound and can “breathe” more easily. Shen and Storb suggest 138 that heavy transcription of rearranged Ig genes underwinds the 5′ end of the gene and transient opening of DNA here makes it accessible to AID (Fig. 9D). It is possible that AID, once bound to SS DNA in underwound regions, can travel some distance before falling off and creating multiple cytosine deaminations.
An attractive feature of this model is that negative superhelicity exposes both the DNA strands to AID and hence there is no strand-bias in the resulting cytosine deaminations. While this is consistent with the mutational spectra in SHM, it is inconsistent with the data obtained using E. coli and in vitro transcription. As noted earlier, uracils accumulate in a strand-biased fashion in E. coli and in vitro when the target gene is transcribed 35,36,39,44.
Another point to note is that transcription-driven superhelical domains should not be restricted to the gene being transcribed. They can extend both upstream and downstream of the gene. This would predict that the 5′ edge of SHM could be upstream of its promoter. However, the SHM data clearly show that hypermutations rarely occur 5′ of the promoter 10,139. Some modifications to this model may be necessary to accommodate this fact.
9.5 Stem-loop Structure Model
This is a variation on the superhelical domain model. Wright has pointed out that 140,141 if certain sequences in Ig genes contain inversely repeated sequences, they would tend to form stem-loop structures (SLS) when the DNA becomes underwound. The stability of these structures would be different from structure to structure based on the length of the stem, length of the loop, G+C content etc. If such structures are moderately stable, the cytosines in their loops would be accessible to AID (Fig. 9E). She has found some correlation between the stability of the potential SLS and the occurrence of hypermutation hotspots 141. Most of the predictions of this model are similar to the superhelical domain model discussed above and has many of the same strengths and weaknesses.
10. Antibody Maturation and Cancer
Malignant transformation is frequently associated with genomic instability and chromosome translocations. In particular, lymphomas often contain translocations involving the immunoglobulin (Ig) genes and oncogenes such as c-myc and bcl2 142. One such translocation between IgH and c-myc is induced by IL6 and was studied in Balb/c mice expressing an IL6 transgene 143. Two types of mice were used in this study, one type was defective in AID (genotype AID−/−), these mice did not undergo antibody maturation. When lymphatic hyperplasia were studied, the control group (genotype AID+/−), but not their AID−/− siblings, contained translocations between IgH and c-myc genes 143. Thus the DNA rearrangements initiated during SHM and CSR may sometimes lead to chromosomal translocations that activate protooncogenes and contribute to tumorigenesis.
Another link between antibody maturation and cancer was demonstrated by Okazaki et al 144 by studying a mouse with an AID transgene. They found that mice expressing an AID transgene constitutively had substantially shorter lifespans and enlarged lymphoid organs. These mice developed T cell lymphomas and micro-adenomas or dysgenetic lesions of respiratory bronchiole 144. Although the lymphomas did not contain higher than normal frequency of translocations, the c-myc gene did accumulated high levels of mutations in the region encompassing the exon 1 and intron 1, the hotspot of mutations and breakpoints of translocations in B cell. These and other studies show that if the DNA processing steps required for antibody maturation occur ectopically, they can lead to mutagenesis and cancer.
11. Antibody Maturation and Molecular Evolution
A bedrock principle of biology for the past 100 years has been that organisms try to maintain a low rate of mutations. This is because most mutations are harmful to the organism. Consequently, cells make extensive efforts to prevent DNA damage and to repair any damage that escapes the preventive mechanisms. Affinity maturation of antibodies in higher vertebrates is an exception to this rule. Here a class of cells, B lymphocytes, in the body introduce damage to DNA in a programmed fashion leading to mutations in one part of one (or a few) chromosomal gene. This creates a population of cells with different levels of Darwinian “fitness” for combating an infection. Furthermore, the process that acts upon these mutant cell populations and selectively expands the clones to make better antibodies uses polypeptides and other molecules derived from the agent that caused antibody maturation in the first place. Thus the infectious agent itself causes the evolution of B cells making them more adept at the destruction of the agent. This is Lamarckian evolution at work-something that does not occur during the evolution of whole organisms. These cellular events have no clear parallels elsewhere in biology and are of intrinsic interest for understanding the interplay between mutations and selection that is inherent to biological evolution that has been ongoing for millions of years.
12. Concluding Remarks
In the past five years, considerable progress has been made in understanding how higher eukaryotes alter the antibody proteins so that the circulating antigens fit well within their binding pockets. Many key proteins required for this unique mutational pathway have been identified and the role of some of these proteins in SHM and CSR is fairly well understood. In particular, the discovery of AID, an enzyme essential for antibody maturation, and the demonstration that it actively damages DNA have been exciting developments. However, there are some areas of chemistry and enzymology where important challenges lie ahead. These include- (1) Structure and reaction mechanism of AID. Very little is known in this regard directly from studies of AID. There is no detailed kinetics of this enzyme published, nor have inhibitors of the enzyme been validated. Considering the potential role of this family of enzymes in promoting mutations in oncogenes, designing and testing inhibitors for them should be an important goal. (2) What directs AID, UDG, MSH2 and other proteins to the rearranged transcribing Ig genes? Clearly, high transcription of the Ig genes play a role in this selectivity, but additional factors should be involved in preventing genome wide mutations during antibody maturation. These factors are likely to be associated with chromatin-modifying enzymes to allow greater access to the Ig locus. (3) Major hurdles remain in understanding the role of TLS Pols and MMR in antibody maturation. The richness of the TLS Pol families in mammalian cells in itself makes it difficult to present testable models for their role in SHM. This role will become clearer only when we have a better understanding of the biochemical interactions of these enzymes with other components of DNA replication apparatus and MMR.
Finally, is antibody maturation the only process in biology that performs programmed genetic rearrangements that depend on enzymatically damaging DNA? Given its success in mammals and other eukaryotes, it would be reasonable to assume that it has evolved in other contexts where high degree of structural variability is needed. Possible biological systems where such a program of DNA damage and mutations may be useful are receptors that recognize a large number of structurally similar chemicals, and pathogens that must evade host attack based on proteins such as antibodies. It would be exciting if we were to find that other biological systems have also used this inherently risky path of controlled mutagenesis to their advantage.
Acknowledgments
This work was supported by funds from the NIH grants GM57200 (to A.S.B.) and CA97899 (to A.S.B. and J.B.). We would like to thank Tami Zellner for help with some of the figures. We would like to thank all the reviewers for providing thoughtful comments on the manuscript and would like to particularly single out reviewer #1 for his/her detailed insightful critique.
References
- 1.Janeway CA, Travers P, Walport M, Shlomchik M. Imunobiology. 5. Garland Publishing; London: 2001. [Google Scholar]
- 2.Stavnezer J, Kinoshita K, Muramatsu M, Honjo T. In: Molecular Biology of B Cells. Honjo T, Alt FW, Neuberger MS, editors. Elsevier Academic Press; London: 2004. [Google Scholar]
- 3.Gellert M. Annu Rev Genet. 1992;26:425. doi: 10.1146/annurev.ge.26.120192.002233. [DOI] [PubMed] [Google Scholar]
- 4.Sekiguchi J, Alt FW, Oettinger M. In: Molecular Biology of B Cells. Honjo T, Alt FW, Neuberger MS, editors. Elsevier Academic Press; London: 2004. [Google Scholar]
- 5.Burnet FM. The Clonal Selection Theory of Acquired Immunity. Cambridge University Press; Cambridge, England: 1959. [Google Scholar]
- 6.Talmage DW. Science. 1959;129:1643. doi: 10.1126/science.129.3364.1643. [DOI] [PubMed] [Google Scholar]
- 7.Maclennan ICM, Hardie DL. In: Molecular Biology of B Cells. Honjo T, Alt FW, Neuberger MS, editors. Elsevier Academic Press; London: 2004. [Google Scholar]
- 8.Papavasiliou FN, Schatz DG. Cell. 2002;109:S35. doi: 10.1016/s0092-8674(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 9.Berek C, Milstein C. Immunol Rev. 1988;105:5. doi: 10.1111/j.1600-065x.1988.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 10.Lebecque SG, Gearhart PJ. J Exp Med. 1990;172:1717. doi: 10.1084/jem.172.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rada C, Yelamos J, Dean W, Milstein C. Eur J Immunol. 1997;27:3115. doi: 10.1002/eji.1830271206. [DOI] [PubMed] [Google Scholar]
- 12.Maizels N, Scharff MD. In: Molecular Biology of B Cells. Honjo T, Alt FW, Neuberger MS, editors. Elsevier Academic Press; London: 2004. [Google Scholar]
- 13.Neuberger MS, Milstein C. Curr Opin Immunol. 1995;7:248. doi: 10.1016/0952-7915(95)80010-7. [DOI] [PubMed] [Google Scholar]
- 14.Storb U. Curr Opin Immunol. 1996;8:206. doi: 10.1016/s0952-7915(96)80059-8. [DOI] [PubMed] [Google Scholar]
- 15.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Cell. 2000;102:553. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 16.Petersen-Mahrt SK, Harris RS, Neuberger MS. Nature. 2002;418:99. [PubMed] [Google Scholar]
- 17.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. J Biol Chem. 1999;274:18470. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 18.Muto T, Muramatsu M, Taniwaki M, Kinoshita K, Honjo T. Genomics. 2000;68:85. doi: 10.1006/geno.2000.6268. [DOI] [PubMed] [Google Scholar]
- 19.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Cell. 2000;102:565. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 20.Arakawa H, Hauschild J, Buerstedde JM. Science. 2002;295:1301. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 21.Harris RS, Sale JE, Petersen-Mahrt SK, Neuberger MS. Curr Biol. 2002;12:435. doi: 10.1016/s0960-9822(02)00717-0. [DOI] [PubMed] [Google Scholar]
- 22.Dunnick W, Wilson M, Stavnezer J. Mol Cell Biol. 1989;9:1850. doi: 10.1128/mcb.9.5.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, Honjo T. J Exp Med. 2002;195:529. doi: 10.1084/jem.20012144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehta A, Kinter MT, Sherman NE, Driscoll DM. Mol Cell Biol. 2000;20:1846. doi: 10.1128/mcb.20.5.1846-1854.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goossens T, Klein U, Kuppers R. Proc Natl Acad Sci U S A. 1998;95:2463. doi: 10.1073/pnas.95.5.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bross L, Fukita Y, McBlane F, Demolliere C, Rajewsky K, Jacobs H. Immunity. 2000;13:589. doi: 10.1016/s1074-7613(00)00059-5. [DOI] [PubMed] [Google Scholar]
- 27.Papavasiliou FN, Schatz DG. Nature. 2000;408:216. doi: 10.1038/35041599. [DOI] [PubMed] [Google Scholar]
- 28.Sale JE, Neuberger MS. Immunity. 1998;9:859. doi: 10.1016/s1074-7613(00)80651-2. [DOI] [PubMed] [Google Scholar]
- 29.Kong Q, Maizels N. Genetics. 2001;158:369. doi: 10.1093/genetics/158.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bemark M, Sale JE, Kim HJ, Berek C, Cosgrove RA, Neuberger MS. J Exp Med. 2000;192:1509. doi: 10.1084/jem.192.10.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bross L, Wesoly J, Buerstedde JM, Kanaar R, Jacobs H. Eur J Immunol. 2003;33:352. doi: 10.1002/immu.200310009. [DOI] [PubMed] [Google Scholar]
- 32.Bransteitter R, Pham P, Scharff MD, Goodman MF. Proc Natl Acad Sci U S A. 2003;100:4102. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dickerson SK, Market E, Besmer E, Papavasiliou FN. J Exp Med. 2003;197:1291. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beale RC, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. J Mol Biol. 2004;337:585. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 35.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Nat Immunol. 2003;4:452. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 36.Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS. Nucleic Acids Res. 2003;31:2990. doi: 10.1093/nar/gkg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayorov VI, Rogozin IB, Adkison LR, Frahm CR, Kunkel TA, Pavlov YI. BMC Immunol. 2005;6:10. doi: 10.1186/1471-2172-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poltoratsky VP, Wilson SH, Kunkel TA, Pavlov YI. J Immunol. 2004;172:4308. doi: 10.4049/jimmunol.172.7.4308. [DOI] [PubMed] [Google Scholar]
- 39.Martomo SA, Fu D, Yang WW, Joshi NS, Gearhart PJ. J Immunol. 2005;174:7787. doi: 10.4049/jimmunol.174.12.7787. [DOI] [PubMed] [Google Scholar]
- 40.Duncan BK, Weiss B. J Bacteriol. 1982;151:750. doi: 10.1128/jb.151.2.750-755.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krokan H, Wittwer CU. Nucleic Acids Res. 1981;9:2599. doi: 10.1093/nar/9.11.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krokan HE, Drablos F, Slupphaug G. Oncogene. 2002;21:8935. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 43.Lindahl T. Proc Natl Acad Sci U S A. 1974;71:3649. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Nature. 2003;422:726. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 45.Navaratnam N, Morrison JR, Bhattacharya S, Patel D, Funahashi T, Giannoni F, Teng BB, Davidson NO, Scott J. J Biol Chem. 1993;268:20709. [PubMed] [Google Scholar]
- 46.Petersen-Mahrt SK, Neuberger MS. J Biol Chem. 2003;278:19583. doi: 10.1074/jbc.C300114200. [DOI] [PubMed] [Google Scholar]
- 47.Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Mol Biol Evol. 2005;22:367. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- 48.Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, Nonoyama S, Tashiro J, Ikegawa M, Ito S, Kinoshita K, Muramatsu M, Honjo T. Nat Immunol. 2003;4:843. doi: 10.1038/ni964. [DOI] [PubMed] [Google Scholar]
- 49.Xie K, Sowden MP, Dance GS, Torelli AT, Smith HC, Wedekind JE. Proc Natl Acad Sci U S A. 2004;101:8114. doi: 10.1073/pnas.0400493101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaudhuri J, Khuong C, Alt FW. Nature. 2004;430:992. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 51.Rada C, Jarvis JM, Milstein C. Proc Natl Acad Sci U S A. 2002;99:7003. doi: 10.1073/pnas.092160999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, Honjo T. Proc Natl Acad Sci U S A. 2004;101:1975. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brar SS, Watson M, Diaz M. J Biol Chem. 2004;279:26395. doi: 10.1074/jbc.M403503200. [DOI] [PubMed] [Google Scholar]
- 54.McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. J Exp Med. 2004;199:1235. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reynaud CA, Aoufouchi S, Faili A, Weill JC. Nat Immunol. 2003;4:631. doi: 10.1038/ni0703-631. [DOI] [PubMed] [Google Scholar]
- 56.Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. Mol Cell. 2003;12:501. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 57.Shinkura R, Ito S, Begum NA, Nagaoka H, Muramatsu M, Kinoshita K, Sakakibara Y, Hijikata H, Honjo T. Nat Immunol. 2004;5:707. doi: 10.1038/ni1086. [DOI] [PubMed] [Google Scholar]
- 58.Betts L, Xiang S, Short SA, Wolfenden R, Carter CW., Jr J Mol Biol. 1994;235:635. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- 59.Harris RS, Petersen-Mahrt SK, Neuberger MS. Mol Cell. 2002;10:1247. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 60.Navaratnam N, Fujino T, Bayliss J, Jarmuz A, How A, Richardson N, Somasekaram A, Bhattacharya S, Carter C, Scott J. J Mol Biol. 1998;275:695. doi: 10.1006/jmbi.1997.1506. [DOI] [PubMed] [Google Scholar]
- 61.Kurowski MA, Bujnicki JM. Nucleic Acids Res. 2003;31:3305. doi: 10.1093/nar/gkg557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kosinski J, Cymerman IA, Feder M, Kurowski MA, Sasin JM, Bujnicki JM. Proteins. 2003;53(Suppl 6):369. doi: 10.1002/prot.10545. [DOI] [PubMed] [Google Scholar]
- 63.Simons KT, Kooperberg C, Huang E, Baker D. J Mol Biol. 1997;268:209. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 64.Ko TP, Lin JJ, Hu CY, Hsu YH, Wang AH, Liaw SH. J Biol Chem. 2003;278:19111. doi: 10.1074/jbc.M300874200. [DOI] [PubMed] [Google Scholar]
- 65.Zaim J, Kierzek AM. Nat Immunol. 2003;4:1153. doi: 10.1038/ni1203-1153. author reply 1154. [DOI] [PubMed] [Google Scholar]
- 66.Rogozin IB, Kolchanov NA. Biochim Biophys Acta. 1992;1171:11. doi: 10.1016/0167-4781(92)90134-l. [DOI] [PubMed] [Google Scholar]
- 67.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Nat Immunol. 2001;2:530. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 68.Bransteitter R, Pham P, Calabrese P, Goodman MF. J Biol Chem. 2004;279:51612. doi: 10.1074/jbc.M408135200. [DOI] [PubMed] [Google Scholar]
- 69.Pham P, Bransteitter R, Petruska J, Goodman MF. Nature. 2003;424:103. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 70.Rogozin IB, Diaz M. J Immunol. 2004;172:3382. doi: 10.4049/jimmunol.172.6.3382. [DOI] [PubMed] [Google Scholar]
- 71.Gearhart PJ, Bogenhagen DF. Proc Natl Acad Sci U S A. 1983;80:3439. doi: 10.1073/pnas.80.11.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bhagwat AS. DNA Repair (Amst) 2004;3:85. doi: 10.1016/j.dnarep.2003.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Curr Biol. 2002;12:1748. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 74.Wang Z, Mosbaugh DW. J Biol Chem. 1989;264:1163. [PubMed] [Google Scholar]
- 75.Di Noia J, Neuberger MS. Nature. 2002;419:43. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 76.Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. Mol Cell. 2000;5:1059. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- 77.Nilsen H, Haushalter KA, Robins P, Barnes DE, Verdine GL, Lindahl T. Embo J. 2001;20:4278. doi: 10.1093/emboj/20.15.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nilsen H, Stamp G, Andersen S, Hrivnak G, Krokan HE, Lindahl T, Barnes DE. Oncogene. 2003;22:5381. doi: 10.1038/sj.onc.1206860. [DOI] [PubMed] [Google Scholar]
- 79.Kavli B, Andersen S, Otterlei M, Liabakk NB, Imai K, Fischer A, Durandy A, Krokan HE, Slupphaug G. J Exp Med. 2005;201:2011. doi: 10.1084/jem.20050042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, Durandy A. Nat Immunol. 2003;4:1023. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- 81.Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Blood. 2005;105:1881. doi: 10.1182/blood-2003-12-4420. [DOI] [PubMed] [Google Scholar]
- 82.Begum NA, Kinoshita K, Kakazu N, Muramatsu M, Nagaoka H, Shinkura R, Biniszkiewicz D, Boyer LA, Jaenisch R, Honjo T. Science. 2004;305:1160. doi: 10.1126/science.1098444. [DOI] [PubMed] [Google Scholar]
- 83.Stivers JT. Science. 2004;306:2042. doi: 10.1126/science.1105225. author reply 2042. [DOI] [PubMed] [Google Scholar]
- 84.Diaz M, Lawrence C. Trends Immunol. 2005;26:215. doi: 10.1016/j.it.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 85.Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F. Embo J. 1999;18:3491. doi: 10.1093/emboj/18.12.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. Nature. 1999;399:700. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 87.Zhang Y, Yuan F, Wu X, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Nucleic Acids Res. 2000;28:4717. doi: 10.1093/nar/28.23.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. Nat Immunol. 2001;2:537. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 89.Delbos F, De Smet A, Faili A, Aoufouchi S, Weill JC, Reynaud CA. J Exp Med. 2005;201:1191. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martomo SA, Yang WW, Wersto RP, Ohkumo T, Kondo Y, Yokoi M, Masutani C, Hanaoka F, Gearhart PJ. Proc Natl Acad Sci U S A. 2005;102:8656. doi: 10.1073/pnas.0501852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pavlov YI, Rogozin IB, Galkin AP, Aksenova AY, Hanaoka F, Rada C, Kunkel TA. Proc Natl Acad Sci U S A. 2002;99:9954. doi: 10.1073/pnas.152126799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bertocci B, De Smet A, Flatter E, Dahan A, Bories JC, Landreau C, Weill JC, Reynaud CA. J Immunol. 2002;168:3702. doi: 10.4049/jimmunol.168.8.3702. [DOI] [PubMed] [Google Scholar]
- 93.Schenten D, Gerlach VL, Guo C, Velasco-Miguel S, Hladik CL, White CL, Friedberg EC, Rajewsky K, Esposito G. Eur J Immunol. 2002;32:3152. doi: 10.1002/1521-4141(200211)32:11<3152::AID-IMMU3152>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 94.Shimizu T, Shinkai Y, Ogi T, Ohmori H, Azuma T. Immunol Lett. 2003;86:265. doi: 10.1016/s0165-2478(03)00046-4. [DOI] [PubMed] [Google Scholar]
- 95.Faili A, Aoufouchi S, Flatter E, Gueranger Q, Reynaud CA, Weill JC. Nature. 2002;419:944. doi: 10.1038/nature01117. [DOI] [PubMed] [Google Scholar]
- 96.McDonald JP, Frank EG, Plosky BS, Rogozin IB, Masutani C, Hanaoka F, Woodgate R, Gearhart PJ. J Exp Med. 2003;198:635. doi: 10.1084/jem.20030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zan H, Komori A, Li Z, Cerutti A, Schaffer A, Flajnik MF, Diaz M, Casali P. Immunity. 2001;14:643. doi: 10.1016/s1074-7613(01)00142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Diaz M, Verkoczy LK, Flajnik MF, Klinman NR. J Immunol. 2001;167:327. doi: 10.4049/jimmunol.167.1.327. [DOI] [PubMed] [Google Scholar]
- 99.Zan H, Shima N, Xu Z, Al-Qahtani A, Evinger AJ, III, Zhong Y, Schimenti JC, Casali P. Embo J. 2005;24:3757. doi: 10.1038/sj.emboj.7600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Masuda K, Ouchida R, Takeuchi A, Saito T, Koseki H, Kawamura K, Tagawa M, Tokuhisa T, Azuma T, JOW Proc Natl Acad Sci U S A. 2005;102:13986. doi: 10.1073/pnas.0505636102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kawamura K, Bahar R, Seimiya M, Chiyo M, Wada A, Okada S, Hatano M, Tokuhisa T, Kimura H, Watanabe S, Honda I, Sakiyama S, Tagawa M, JOW Int J Cancer. 2004;109:9. doi: 10.1002/ijc.11666. [DOI] [PubMed] [Google Scholar]
- 102.Stojic L, Brun R, Jiricny J. DNA Repair (Amst) 2004;3:1091. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 103.Martomo SA, Yang WW, Gearhart PJ. J Exp Med. 2004;200:61. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cascalho M, Wong J, Steinberg C, Wabl M. Science. 1998;279:1207. doi: 10.1126/science.279.5354.1207. [DOI] [PubMed] [Google Scholar]
- 105.Kim N, Bozek G, Lo JC, Storb U. J Exp Med. 1999;190:21. doi: 10.1084/jem.190.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Phung QH, Winter DB, Alrefai R, Gearhart PJ. J Immunol. 1999;162:3121. [PubMed] [Google Scholar]
- 107.Phung QH, Winter DB, Cranston A, Tarone RE, Bohr VA, Fishel R, Gearhart PJ. J Exp Med. 1998;187:1745. doi: 10.1084/jem.187.11.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rada C, Ehrenstein MR, Neuberger MS, Milstein C. Immunity. 1998;9:135. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- 109.Bardwell PD, Woo CJ, Wei K, Li Z, Martin A, Sack SZ, Parris T, Edelmann W, Scharff MD. Nat Immunol. 2004;5:224. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- 110.Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, Yasui A, Woodgate R, Gearhart PJ. J Exp Med. 2005;201:637. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rada C, Di Noia JM, Neuberger MS. Mol Cell. 2004;16:163. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 112.Kaina B, Ochs K, Grosch S, Fritz G, Lips J, Tomicic M, Dunkern T, Christmann M. Prog Nucleic Acid Res Mol Biol. 2001;68:41. doi: 10.1016/s0079-6603(01)68088-7. [DOI] [PubMed] [Google Scholar]
- 113.Lindahl T. Nature. 1993;362:709. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 114.Loeb LA. Cancer Res. 1991;51:3075. [PubMed] [Google Scholar]
- 115.Stavnezer J, Schrader CE. Trends Genet. 2005 doi: 10.1016/j.tig.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 116.Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. J Exp Med. 2005;202:561. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Friedberg EC, Lehmann AR, Fuchs RP. Mol Cell. 2005;18:499. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 118.Prakash S, Johnson RE, Prakash L. Annu Rev Biochem. 2005;74:317. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 119.Harriman W, Volk H, Defranoux N, Wabl M. Annu Rev Immunol. 1993;11:361. doi: 10.1146/annurev.iy.11.040193.002045. [DOI] [PubMed] [Google Scholar]
- 120.Peters A, Storb U. Immunity. 1996;4:57. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 121.Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y, Shimizu A. Science. 2003;302:2137. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 122.Honjo T, Kinoshita K, Muramatsu M. Annu Rev Immunol. 2002;20:165. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 123.Bachl J, Carlson C, Gray-Schopfer V, Dessing M, Olsson C. J Immunol. 2001;166:5051. doi: 10.4049/jimmunol.166.8.5051. [DOI] [PubMed] [Google Scholar]
- 124.Yoshikawa K, Okazaki IM, Eto T, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. Science. 2002;296:2033. doi: 10.1126/science.1071556. [DOI] [PubMed] [Google Scholar]
- 125.Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. Nat Immunol. 2003;4:435. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- 126.Beletskii A, Bhagwat AS. Proc Natl Acad Sci U S A. 1996;93:13919. doi: 10.1073/pnas.93.24.13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Klapacz J, Bhagwat AS. J Bacteriol. 2002;184:6866. doi: 10.1128/JB.184.24.6866-6872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Beletskii A, Bhagwat AS. Biol Chem. 1998;379:549. [PubMed] [Google Scholar]
- 129.Beletskii A, Bhagwat AS. J Bacteriol. 2001;183:6491. doi: 10.1128/JB.183.21.6491-6493.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Beletskii A, Grigoriev A, Joyce S, Bhagwat AS. J Mol Biol. 2000;300:1057. doi: 10.1006/jmbi.2000.3944. [DOI] [PubMed] [Google Scholar]
- 131.Klapacz J, Bhagwat AS. DNA Repair (Amst) 2005;4:806. doi: 10.1016/j.dnarep.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 132.Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW. Nature. 2005;438:508. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 133.Storb U, Klotz EL, Hackett J, Jr, Kage K, Bozek G, Martin TE. J Exp Med. 1998;188:689. doi: 10.1084/jem.188.4.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Michael N, Martin TE, Nicolae D, Kim N, Padjen K, Zhan P, Nguyen H, Pinkert C, Storb U. Immunity. 2002;16:123. doi: 10.1016/s1074-7613(02)00261-3. [DOI] [PubMed] [Google Scholar]
- 135.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. Nat Immunol. 2003;4:442. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 136.Yu K, Roy D, Bayramyan M, Haworth IS, Lieber MR. Mol Cell Biol. 2005;25:1730. doi: 10.1128/MCB.25.5.1730-1736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu LF, Wang JC. Proc Natl Acad Sci U S A. 1987;84:7024. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shen HM, Storb U. Proc Natl Acad Sci U S A. 2004;101:12997. doi: 10.1073/pnas.0404974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rothenfluh HS, Taylor L, Bothwell AL, Both GW, Steele EJ. Eur J Immunol. 1993;23:2152. doi: 10.1002/eji.1830230916. [DOI] [PubMed] [Google Scholar]
- 140.Wright BE. J Bacteriol. 2000;182:2993. doi: 10.1128/jb.182.11.2993-3001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wright BE, Schmidt KH, Minnick MF. Genes Immun. 2004;5:176. doi: 10.1038/sj.gene.6364053. [DOI] [PubMed] [Google Scholar]
- 142.Kuppers R, Dalla-Favera R. Oncogene. 2001;20:5580. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- 143.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. Cell. 2004;118:431. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 144.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. J Exp Med. 2003;197:1173. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhu Y, Nonoyama S, Morio T, Muramatsu M, Honjo T, Mizutani S. J Med Dent Sci. 2003;50:41. [PubMed] [Google Scholar]
- 146.Quartier P, Bustamante J, Sanal O, Plebani A, Debre M, Deville A, Litzman J, Levy J, Fermand JP, Lane P, Horneff G, Aksu G, Yalcin I, Davies G, Tezcan I, Ersoy F, Catalan N, Imai K, Fischer A, Durandy A. Clin Immunol. 2004;110:22. doi: 10.1016/j.clim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 147.Minegishi Y, Lavoie A, Cunningham-Rundles C, Bedard PM, Hebert J, Cote L, Dan K, Sedlak D, Buckley RH, Fischer A, Durandy A, Conley ME. Clin Immunol. 2000;97:203. doi: 10.1006/clim.2000.4956. [DOI] [PubMed] [Google Scholar]
- 148.Imai K, Zhu Y, Revy P, Morio T, Mizutani S, Fischer A, Nonoyama S, Durandy A. Clin Immunol. 2005;115:277. doi: 10.1016/j.clim.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 149.Fiorini C, Jilani S, Losi CG, Silini A, Giliani S, Ferrari S, Notarangelo LD, Plebani A, Sfar T, Helal A. Eur J Pediatr. 2004;163:704. doi: 10.1007/s00431-004-1540-8. [DOI] [PubMed] [Google Scholar]