

Abstract
Most computational protein structure prediction methods are designed for either template-based or template-free (ab initio) structure prediction. The approaches that integrate the prediction capabilities of both template-based modeling and template-free modeling are needed to synergistically combine the two kinds of methods to improve protein structure prediction. In this work, we develop a new method to integrate several protein structure prediction methods including our template-based MULTICOM server, our ab initio contact-based protein structure prediction method CONFOLD, our multi-template-based model generation tool MTMG, and locally installed external Rosetta, I-TASSER and RaptorX protein structure prediction tools to improve protein structure prediction of a full-spectrum difficulty ranging from easy, to medium and to hard. Our method participated in the 11th community-wide Critical Assessment of Techniques for Protein Structure Prediction (CASP11) in 2014 as MULTICOM-NOVEL server. It was ranked among top 10 methods for protein tertiary structure prediction according to the official CASP11 assessment, which demonstrates that integrating complementary modeling methods is useful for advancing protein structure prediction.
Keywords: Model generation, model selection, protein structure prediction, sequence alignment, template-based modeling, template-free modeling
1. INTRODUCTION
The tertiary structure of a protein is important for understanding its functions. Experimental techniques such as x-ray crystallography and nucleic magnetic resonance can determine protein structure with high-resolution, however, they are too expensive to be applied to all the proteins. Computational protein structure prediction methods aim to construct the tertiary (three-dimensional) structure of a protein from its amino acid sequence [1, 2]. With a large number of known protein sequences, but a relatively small number of known protein structures, fast and accurate computational methods for protein tertiary structure prediction need to be developed.
A number of useful protein structure prediction methods that can build good structural models for a large portion of proteins have been developed [3-7]. These methods can be generally classified into two categories: template-based modeling – predicting the structure of a protein by using the known structure of its homologous proteins as template, and template-free modeling – reconstructing the structure of a protein without using the structural knowledge of any significant template protein. Even though these two kinds of methods are complementary, few methods have been developed to combine the strengths of the two kinds of methods to improve protein structure prediction. Therefore, more hybrid approaches of integrating both template-based modeling and template-free modeling are needed [8-14].
In this spirit, we developed a new approach called MULTICOM-NOVEL to integrate a variety of template-based and template-free model sampling methods to improve structure prediction. It integrates the predictions of our MULTICOM tertiary structure prediction server [15-17], our in-house multiple-template-based modeling tool MTMG, our ab initio residue-residue contact-based model generation method CONFOLD [18] built on the CNS system [19, 20], and locally installed external state-of-the-art structure prediction tools such as Rosetta [21] and I-TASSER [22, 23]. MULTICOM-NOVEL participated in the 11th Critical Assessment of Techniques for Protein Structure Prediction (CASP11) in 2014, and was ranked among top 10 methods out of 44 servers according to the official CASP11 assessment (http://www.predictioncenter.org/casp11/).
2. METHODS
2.1. Overview of the Protein Conformation Generation Process
MULTICOM-NOVEL generated an ensemble of protein models for a protein target using multiple complementary methods in each step of protein structure prediction, such as sequence/profile comparison tools (PSI-BLAST [24], HHSearch [25], and RaptorX [26]) for template identification, target-template alignment tools (PSI-BLAST, HHSearch, RaptorX, MSACompro [27], HHMsato [28], and Promals3d [29]) for target-template alignment, MULTICOM template and alignment combination protocol [15, 16] for alignment combination, our in-house multiple-template-based modelling tool MTMG and external Modeller[30] for template-based model generation, locally installed external tool RaptorX for generating template-based models, local Rosetta for ab initio modeling, local I-TASSER [22, 23] for template-based modeling, our in-house contact-based modeling CONFOLD [18] for ab initio modeling, and our MULTICOM server for template-based modeling [15-17]. The ensemble of models predicted for a target were then evaluated by two methods: a single-model absolute model quality assessment tool – ModelEvaluator [31] and a fully pairwise model comparison tool – APOLLO [32]. From the ensemble of the models, MULTICOM-NOVEL chose top five models ranked by the weighted sum of APOLLO scores and ModelEvaluator scores as final predictions.
In addition to using external I-TASSER, Rosetta, RaptorX to generate some models, MULTICOM-NOVEL primarily used our in-house protein structure prediction tools to generate conformations for a target protein in the following three ways. First, it used our previously developed MULTICOM server [15-17] to predict models for each target. MULTICOM combines multiple sources of complementary information and alternative methods at five major steps of protein structure prediction: template identification, query-template alignment and combination, model generation, model quality assessment, and model refinement. MULTICOM was ranked among the top methods in template-based modeling and template-free modeling during CASP9 and CASP10 in 2010 and 2012, respectively.
Second, for template-based modeling, we used our in-house MTMG to generate models based on all the sequence alignments between a target and templates generated by MULTICOM-NOVEL. MULTICOM-NOVEL built two template databases for identifying homologous templates for a target. It automatically downloaded new protein structures released in the Protein Data Bank (PDB) [33] every week. The ATOM file (i.e. coordinates) and FASTA sequence of each protein chain in each PDB file were extracted. All the extracted FASTA sequences after filtering identical sequences and their structural information were stored in the first template database. In order to construct the second template database, the protein sequences were filtered at 90% identity threshold by CD-HIT [34] in order to remove highly similar proteins. HHSearch was used to build a hidden Markov model (HMM) profile file for each of the remaining sequences. The HMM profile files were stored in the second template database. PSI-BLAST and HHSearch were used to search a target protein against the two template databases separately to identify homologous templates, which were used by MTMG - a novel probabilistic multi-template based model generation tool to generate structural models for the target. MTMG uses information from the multiple sequence alignment of a target and multiple templates and the structures of the templates to predict structures for the target. Based on the level of the consistency between template structures, MTMG either directly uses the weighted average coordinates of templates or samples positions (x, y, z coordinates) for residues according to the weighted point cloud.
Third, for ab initio template-free modeling, we used our in-house residue-residue contact guided modelling method (CONFOLD) that takes residue-residue contact predictions and three-class secondary structure (helix, strand, and coil) predictions as input to build three-dimensional models using distance geometry simulated annealing protocol implemented in the CNS suite. CONFOLD transforms contacts and secondary structure information into appropriate distance, angle and hydrogen bond restraints to guide conformation sampling. CONFOLD used top L/2 contacts each predicted by NNcon[35] and SVMcon[36], where L is the sequence length, and secondary structures predicted by PSPro[37] to generate restraints for model generation. The average of the distance between the residue pairs predicted to be in contact was used to select top 20 models for each target.
2.2. Integration of Model Generation Methods
Figure 1 illustrates how different methods are integrated in MULTICOM-NOVEL. It used PSI-BLAST and HHSearch to search a protein target against the two template databases separately to identify homologous templates. In each iteration of search, the identified templates were ranked by e-values from low to high, and templates with low e-value (i.e. e-value < 1) were used as candidate templates.
Figure 1.

Integration of protein structure prediction methods and components in MULTICOM-NOVEL.
A hard sequence region of a target was defined as the continuous residues of the target sequence that were not covered by any homologous templates in the previous iterations of search. A short hard uncovered region was ignored because the model generation tools such as Modeller [30] and MTMG can handle it well by constructing a loop for it. A hard region that spans >= 30 residues for a protein with >= 100 residues or >=20 residues for a protein with <100 residues was the focus of the search in the next iteration. If no homologous templates were found for the longest hard region, it was marked “final hard” and would not be searched again. Search stops if no homologous templates could be found for any hard region. If PSI-BLAST and HHSearch identified at least one homologous template for a target and no final hard region at the end of search existed, the target was marked “easy”. If there was at least one homologous template for the target and at least one hard region, the target was marked “medium”. If no homologous template was found for any regions of a target, the target was marked “hard”. Figure 2 illustrates how a sequence is searched during iterations and how the regions of the sequence are annotated as hard and easy regions by MULTICOM-NOVEL.
Figure 2.

Illustration of the template search algorithm. The target sequence is searched against the template databases by HHSearch or PSI-BLAST in three iterations. The sequence region covered by a homologous template is labeled as easy region, otherwise hard region. The hard region left in the last iteration is labeled as final hard region. The example shown here has both easy region and hard region and therefore is considered a “medium” target.
For an easy or medium target, MULTICOM-NOVEL extracted the pairwise sequence alignment between the target and each homologous template and combined them into a multiple sequence alignment for PSI-BLAST and HHSearch separately. If the homologous template with lowest e-value covered at least 90% of the target sequence, its alignment was extracted from PSI-BLAST and/or HHSearch and was specially marked as the single best sequence alignment. The templates identified by PSI-BLAST and HHSearch were also combined to generate a consensus list of templates. Three alignment generation tools (MSACompro[27], HHMsato [28], Promals3d [29]) were used to align the target with these consensus templates to generate one-target multiple-template alignment. The hard regions of a target not covered by a template in the alignments were marked for template-free modeling. Figure 3 shows the workflow of target-template sequence alignment process in MULTICOM-NOVEL.
Figure 3.

The workflow of target-template sequence alignment process in MULTICOM-NOVEL.
Modeller [30] and MTMG were used to generate template-based models for the target based on all the sequence alignments generated in the previous steps and template structures. For medium targets, Rosetta [21] was used to generate models for each of the hard regions in the target. The predicted models of the hard regions together with template-based models were used to construct the full-length models for the target. For hard targets, locally installed I-TASSER, Rosetta, and our in-house ab initio contact-based modeling method CONFOLD were used to predict models from scratch. Due to limitation of CPU, I-TASSER, Rosetta, and CONFOLD generated 5, 100, and 20 models separately for each hard target. In addition, RaptorX and MULTICOM predicted 5 and 50 models for every (easy, medium, or hard) target separately. All the models generated by these methods formed a model pool that went through model selection.
2.3. Model Selection
MULTICOM-NOVEL used APOLLO [32] to rank all the models predicted by each method except for those generated by RaptorX, I-TASSER, and Rosetta. The default ranking of those models assigned by RaptorX, I-TASSER and Rosetta were used. Top 5 models generated by each method were pooled together and evaluated by APOLLO [32] and ModelEvaluator[31], separately. The rankings generated by the two methods were complementary because APOLLO [32] is a fully pairwise model comparison tool that ranks models based on its structural similarity with other models, but ModelEvaluator is a single-model quality assessment tool that assigns an absolute quality score to a model regardless of other models. The weighted sum of the APOLLO score and ModelEvaluator score was used to rank all these models. The weight assigned to APOLLO score and ModelEvaluator score is 0.8 and 0.5, respectively.
MULTICOM-NOVEL picked up top 5 models according to the final ranking. In order to increase the diversity of the models, for easy and medium targets, the top 1 models generated by RaptorX, MULTICOM, multiple sequence alignment, and pairwise sequence alignment substituted some of the top 5 models from bottom to top if they had not already existed within top 5 models. For hard targets, the top 1 model generated byCONFOLD, MULTICOM, RaptorX, Rosetta, and I-TASSER substituted some of the top 5 models from bottom to top if they had not existed within top 5 models. After the adjustment, the final top 5 models were submitted to CASP11.
2.4. Data Set
In order to objectively evaluate the performance of MULTICOM-NOVEL, we blindly tested it in the 11th Critical Assessment of Techniques for Protein Structure Prediction (CASP11) in 2014. CASP11 released 100 protein targets whose structures were not available to the community. By the date, 85 official targets and 105 domains (from 79 targets) are available to assess the performance of the method. Among these 79 targets, 54 targets have only one domain and other 25 targets contain multiple domains. For example, T0759 is comprised of 109 residues, and contains two domains: T0759-D1 (residues 12~45) and T0759-D2 (residues 46~107). T0760 consists of 242 residues, and contains only one domain: T0760-D1 (residues 33~242). This diverse and large dataset contains protein topologies having different difficulty levels, making it ideal for objective evaluation of our method.
3. RESULTS AND DISCUSSION
MULTICOM-NOVEL participated in the 11th Critical Assessment of Techniques for Protein Structure Prediction (CASP11) in 2014. According to the CASP11 official assessment, MULTICOM-NOVEL was ranked among top 10 automated server methods for protein tertiary structure prediction out of 44 server groups. In addition to referring to the official CASP11 assessment, we conducted a detailed evaluation of the models generated by MULTICOM-NOVEL on the 85 CASP11 targets and 105 domains in order to understand when it worked and when it did not. Table 1 reports the average GDT-TS scores and TM-scores [38] of top 1 and best of 5 models predicted by MULTICOM-NOVEL. The average TM-score of the top 1 submitted models for all the targets and domains are 0.57 and 0.53 separately, which are greater than 0.5 threshold that generally indicate that the topology of a model is correct.
Table 1.
The Average GDT-TS Scores and TM-scores of Top One and Best of Five Models on 85 CASP11 Targets and 105 Domains.
| Target | Top One | Best of Five | ||
|---|---|---|---|---|
| GDT-TS | TM-score | GDT-TS | TM-score | |
| Full length targets |
0.48 | 0.57 | 0.51 | 0.60 |
| Domains | 0.47 | 0.53 | 0.51 | 0.57 |
From our analysis, the top 1 models of MULTICOM-NOVEL won other top 1 models submitted by CASP11 server predictors on three targets (T0760, T0832, T0855) and three domains (T0760-D1, T0832-D1, T0855-D1). The best submitted models of MULTICOM-NOVEL were better than other server predicted models in CASP11 on one target (T0760) and four domains (T0760-D1, T0761-D1, T0790-D1, T0855-D1). The results indicate that the target-based results and the domain-based results are largely consistent and our method can predict good protein models for both an entire protein and domains.
We compared top 5 models for each of 85 CASP11 targets and 105 domains predicted by MULTICOM-NOVEL in order to evaluate the performance of the ranking strategy. Figure 4 shows the histogram of the differences of GDT-TS scores between best of top 5 models and top 1 modelson 105 domains. The difference between GDT-TS scores of best of top 5 models and top 1 models is small (<0.05) on 72% of the domains, suggesting the model ranking strategy worked reasonably well. For 34 of these 76 domains, top 1 models were indeed the best models as well. Even though the ranking strategy worked well on average, it failed on some domains. For example, the GDT-TS score of the top 1 model of T0780-D2 was 0.14, much less than GDT-TS score 0.52 of the best model. The loss for this case was 0.38. For full length targets, the difference between GDT-TS scores of best of top 5 models and top 1 models is small (<0.05) on 69 targets, and top 1 models were the best models on 30 targets. The largest loss of GDT-TS score occurred on T0768, and the loss was 0.21.
Figure 4.

The histogram of the differences of GDT-TS scores between best of top 5 models and top 1 models generated by MULTICOM-NOVEL on 105 CASP11 domains.
We further investigated the entire MULTICOM-NOVEL model pool in order to check whether there were better models than the top 5 ranked models in the model pool. The best models in the model pool had higher GDT-TS scores than top 1 selected models of MULTICOM-NOVEL on 99 out of 105 domains and 83 out of 85 targets separately. The best models in the model pool had higher GDT-TS scores than best of top 5 selected models on 87 domains and 73 targets separately.Our analysis indicates that there were better models for most of the targets and domains that had not been selected by the ranking method, suggesting that our technique of selecting the best model out of the entire model pool can be further improved.
We also assessed the performance of different model generation methods in MULTICOM-NOVEL. Figures 5 and 6 show the GDT-TS scores of the best models generated by the methods on 75 template-based modeling (TBM) and 30 template-free (FM) CASP11 domains. A GDT-TS score 0 means that a method did not generate any model for the domain. Our in-house MULTICOM server contributed the best models for 58 TBM domains and 7 FM domains. Rosetta contributed the best models for 6 TBM domains and 18 FM domains. RaptorX generated the best models for 9 TBM domains. I-TASSER, CONFOLD and other template-based methods also predicted the best models for some domains. Our analysis shows that the modeling capabilities of the components in MULTICOM-NOVEL cover all the difficulties of proteins, from easy to medium, and to hard. Therefore, combining the models generated by these complementary methods is a good direction to improve protein structure modeling.
Figure 5.

Comparisons of GDT-TS scores of the best models generated by the individual methods in MULTICOM-NOVEL on 75 TBM CASP11 domains.
Figure 6.

Comparisons of GDT-TS scores of the best models generated by the individual methods in MULTICOM-NOVEL on 30 FM CASP11 domains.
Figure 7 shows one example that the top 1 MULTICOM-NOVEL model is the best model among all the CASP11 models submitted by all the server predictors participating in CASP11. The top 1 model was generated by our MULTICOM server [15-17] for T0760-D1 (domain 1 of target T0760) using 4J8QA and 3K0YA as templates. The model has GDT-TS score 0.75, TM-score 0.84, and RMSD 3.14 Å. Figure 8 illustrates the distributions of GDT-TS scores of all the MULTICOM-NOVEL server models (A) and all the CASP server models (B) of T0760-D1. The results show that the CASP model pool has higher GDT-TS score on average, but the MULTICOM-NOVEL model pool contains the best models.
Figure 7.
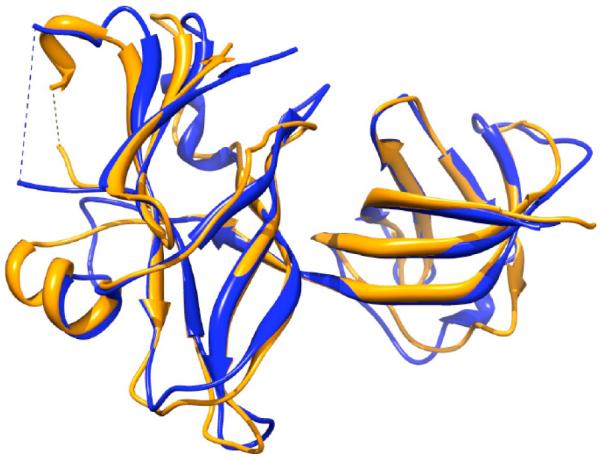
Structural superposition between the native structure of T0760-D1 (blue) and the top-one model of MULTICOM-NOVEL (gold).
Figure 8.

Distributions of GDT-TS scores of MULTICOM-NOVEL server models (A) and CASP server models (B) of T0760-D1.
CONCLUSION
We developed MULTICOM-NOVEL – a new method to combine template-based modeling and template-free modeling to improve protein structure prediction. The blind evaluation of MULTICOM-NOVEL in the CASP11 experiment shows that combination of multiple different methods can improve the quality of modeling for all kinds of targets of different difficulty. It also demonstrates that a systematic integration of existing proteins structure prediction methods, with addition of some in-house methods to supplement and complement the existing methods, can perform relatively well for template-based as well as template-free protein modeling.
ACKNOWLEDGEMENTS
JL, BA, JC designed, developed and tested the system. JL, BA, JC wrote, edited and approved the manuscript.
The work was partially supported by an NIH grant R01GM093123 to JC.
Footnotes
CONFLICTOF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
- [1].Eisenhaber F, Persson B, Argos P. Protein structure prediction: recognition of primary, secondary, and tertiary structural features from amino acid sequence. Crit. Rev. Biochem. Mol. Biol. 1995;30:1–94. doi: 10.3109/10409239509085139. [DOI] [PubMed] [Google Scholar]
- [2].Rost B. Protein structure prediction in 1D, 2D, and 3D. Encyclop.Comput. Chem. 1998;3:2242–2255. [Google Scholar]
- [3].Floudas C. Computational methods in protein structure prediction. Biotechnol.Bioeng. 2007;97:207–213. doi: 10.1002/bit.21411. [DOI] [PubMed] [Google Scholar]
- [4].Shah M, Passovets S, Kim D, Ellrott K, Wang L, Vokler I, LoCascio P, Xu D, Xu Y. A computational pipeline for protein structure prediction and analysis at genome scale. Bioinformatics. 2003;19:1985. doi: 10.1093/bioinformatics/btg262. [DOI] [PubMed] [Google Scholar]
- [5].Fox BG, Goulding C, Malkowski MG, Stewart L, Deacon A. Structural genomics: from genes to structures with valuable materials and many questions in between. Nat. Methods. 2008;5:129–132. doi: 10.1038/nmeth0208-129. [DOI] [PubMed] [Google Scholar]
- [6].Lemer CMR, Rooman MJ, Wodak SJ. Protein structure prediction by threading methods: evaluation of current techniques. Proteins: Struct.,Funct., Bioinf. 1995;23:337–355. doi: 10.1002/prot.340230308. [DOI] [PubMed] [Google Scholar]
- [7].Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins: Struct.,Funct., Bioinf. 1995;23:ii–iv. doi: 10.1002/prot.340230303. [DOI] [PubMed] [Google Scholar]
- [8].Bowie J, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- [9].Ring C, Cohen F. Modeling protein structures: construction and their applications. FASEB J. 1993;7:783–790. doi: 10.1096/fasebj.7.9.8330685. [DOI] [PubMed] [Google Scholar]
- [10].Sali A. Comparative protein modeling by satisfaction of spatial restraints. Mol. Med. Today. 1995;1:270–277. doi: 10.1016/s1357-4310(95)91170-7. [DOI] [PubMed] [Google Scholar]
- [11].Fiser A, Do RKG, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bujnicki JM. Protein-structure prediction by recombination of fragments. Chembiochem. 2006;7:19–27. doi: 10.1002/cbic.200500235. [DOI] [PubMed] [Google Scholar]
- [13].Gopal SM, Klenin K, Wenzel W. Template-free protein structure prediction and quality assessment with an all-atom free-energy model. Proteins: Struct.,Funct., Bioinf. 2009;77:330–341. doi: 10.1002/prot.22438. [DOI] [PubMed] [Google Scholar]
- [14].Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins: Struct.,Funct., Bioinf. 2012;80:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang Z, Eickholt J, Cheng J. MULTICOM: a multi-level combination approach to protein structure prediction and its assessments in CASP8. Bioinformatics. 2010;26:882–888. doi: 10.1093/bioinformatics/btq058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li J, Deng X, Eickholt J, Cheng J. Designing and benchmarking the MULTICOM protein structure prediction system. BMC Struct.Biol. 2013;13:2. doi: 10.1186/1472-6807-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheng J, Li J, Wang Z, Eickholt J, Deng X. The MULTICOM toolbox for protein structure prediction. BMC Bioinformatics. 2012;13:65. doi: 10.1186/1471-2105-13-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Adhikari B, Bhattacharya D, Cao R, Cheng J. CONFOLD: Residue-Residue Contact-guided ab initio Protein Folding. Proteins. 2015 doi: 10.1002/prot.24829. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS. Crystallography & NMR system: a new software suite for macromolecular structure determination. ActaCrystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- [20].Adhikari B, Bhattacharya D, Deng X, Li J, Cheng J. A Contact-Assisted Approach to Protein Structure Prediction and Its Assessment in CASP10. The workshop on artificial intelligence and robotics methods in computational biology of 27th AAAI Conference; Bellevue, WA, USA. 2013. [Google Scholar]
- [21].Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Soding J, Biegert A, Lupas A. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nature protocols. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Deng X, Cheng J. MSACompro: Protein Multiple Sequence Alignment Using Predicted Secondary Structure, Solvent Accessibility, and Residue-Residue Contacts. BMC Bioinformatics. 2011;12:472. doi: 10.1186/1471-2105-12-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Deng X, Cheng J. Enhancing HMM-based protein profile-profile alignment with structural features and evolutionary coupling information. BMC Bioinformatics. 2014;15:252. doi: 10.1186/1471-2105-15-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pei J, Kim B-H, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- [31].Wang Z, Tegge AN, Cheng J. Evaluating the absolute quality of a single protein model using structural features and support vector machines. Proteins: Struct.,Funct., Bioinf. 2009;75:638–647. doi: 10.1002/prot.22275. [DOI] [PubMed] [Google Scholar]
- [32].Wang Z, Eickholt J, Cheng J. APOLLO: a quality assessment service for single and multiple protein models. Bioinformatics. 2011;27:1715–1716. doi: 10.1093/bioinformatics/btr268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bernstein FC, Koetzle TF, Williams GJB, Meyer EF. The protein data bank: A computer-based archival file for macro-molecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- [34].Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- [35].Tegge AN, Wang Z, Eickholt J, Cheng J. NNcon: improved protein contact map prediction using 2D-recursive neural networks. Nucleic Acids Res. 2009;37:W515–W518. doi: 10.1093/nar/gkp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cheng J, Baldi P. Improved residue contact prediction using support vector machines and a large feature set. BMC Bioinformatics. 2007;8:113. doi: 10.1186/1471-2105-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cheng J, Randall A, Sweredoski M, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005;33:W72–W76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins: Struct.,Funct., Bioinf. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]