

Abstract
Mitochondria are a source of reactive oxygen species (ROS). Mitochondrial diseases are the result of inherited defects in mitochondrially-expressed genes. One potential pathomechanism for mitochondrial disease is oxidative stress. Oxidative stress can occur as the result of increased ROS production, or decreased ROS protection. The role of oxidative stresses in the five most common inherited mitochondrial diseases; Friedreich's ataxia (FA), LHON, MELAS, MERRF and Leigh Syndrome (LS) is discussed. Published reports for oxidative stress involvement in pathomechanism in these five mitochondrial diseases are reviewed. The strongest for oxidative stress pathomechanism among the five diseases was in Friedreich's ataxia. In addition, a meta-analysis was carried out to provide an unbiased evaluation of the role of oxidative stress in the five diseases, by searching for oxidative stress citation count frequency within each disease. Of the five most common mitochondrial diseases, the strongest support for oxidative stress is in Friedreich's ataxia (6.42%), followed by LHON (2.45%), MELAS (2.18%), MERRF (1.71%), and LS (1.03%). The increased frequency of oxidative stress citations was significant relative to the mean of the total pool of five diseases (p<0.01) and the mean of the four non-Friedreich's diseases (p<0.0001). Thus there is support for oxidative stress in all five most common mitochondrial diseases, but the strongest, significant support is for Friedreich's ataxia.
Introduction
Inheritance of a nuclear or mitochondrial DNA mutation in a mitochondrially expressed gene causes mitochondrial disease [1]. There is no explicit, detailed pathomechanism for any mitochondrial disease and no approved or effective therapy has been developed to this date. Although the most famous physiological role of mitochondria is as the producer of adenosine triphosphate (ATP), mitochondria also plays important roles in reactive oxygen species (ROS) generation and protection, Ca2+ handling, nucleotide metabolism, the urea cycle and apoptosis. Multiple mitochondrial disorders exhibit with neurological deficits or neurodegeneration, including Friedrich's Ataxia (FA), Leber's hereditary optic neuropathy(LHON), Leigh Syndrome (LS), Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes(MELAS) and Myoclonic epilepsy with ragged-red fibers(MERRF) [1, 2].
We accept that the term ‘oxidative stress’ defines an imbalance between the production of ROS, and antioxidant systems that protects from ROS. Because mitochondria are the site of the electron transport chain that transfers electrons to molecular Oxygen (O2) as part of its physiological mechanism, it is reasonable to expect that defects in this pathway could cause increased ROS. Indeed, known pharmacological agents that interact with electron transport and oxidative phosphorylation system causes increased ROS, such as antimycin A (electron shuttle) and oligomycin (ATP synthase inhibitor), which both increase measurable mitochondrial ROS production. In line with this thought, disease-causing mutations in mitochondrial disease which for example resides in the coding region of gene involved in electron transport chain or oxidative phosphorylation could cause increased ROS, and conversely, mutations that decrease the mitochondrion's protective system, including thioredoxin reductase, but also peroxiredoxins, superoxide dismutases and glutathione peroxidases could cause oxidative stress through protection deficiency [3-5].
The consequences of increased mitochondrial ROS production or decreased mitochondrial ROS protection can include damage of mitochondrial DNA and RNA, proteins and lipids. Oxidative stress can induce DNA damage by incorporating an oxidized base during DNA polymerization or oxidizing a base that is already integrated into the DNA of which the most commonly measured is 8-hydroxyguanosine [6, 7]. Proteins are oxidized in their backbone creating protein-protein cross links and fragmentations. Amino acids on the protein sidechains can also be oxidized and reduces or prevents the proteins from functioning properly [8]. Oxidation of lipids can results in the formation of lipid peroxyl radicals and hydroperoxides which propagates the further formation of lipid peroxides and can damage DNA and proteins [9].
Although research into mitochondrial disease has identified multiple potential pathomechanisms, treatment options are extremely limited and there is no cure for any mitochondrial diseases. Antioxidant therapy, for example idebenone, has not been found to be effective in any placebo-controlled, double-blind study of mitochondrial disease [10]. However, the strength of support of oxidative stress as a disease mechanism has not been critically reviewed in inherited mitochondrial disease, which is the point of this work. Thus, we critically review the potential role of oxidative stress in the five most common inherited mitochondrial diseases: FA, MELAS, MERRF, LS and LHON.
As a counterweight to the reviewer's subjective bias, we also carried out a literature search of the frequency of published papers in the five most common mitochondrial diseases for terms related to oxidative stress. Articles on the five mitochondrial diseases FA, MELAS, MERRF, LS and LHON had disease specific pools ranging from 2353 (FA) to 410 (MERRF). However, the disease-specific frequency of incidence of the term oxidative stress within each disease group ranted from a low of 1.03 (LS) to 6.42(FA) [Table 1, Fig 1]. Thus the conclusions of both types of review are consistent. There is support for oxidative stress in all five of the most common mitochondrial diseases, but the most experimental evidence, and the highest disease-specific citation frequency is for Friedreich's ataxia. These results could have impact for the design and implementation of therapeutic studies involving antioxidant protection, given that there is currently no cure for any of the 5 most common mitochondrial diseases.
Table 1.
Pubmed search of mitochondrial dysfunction diseases. The search includes total articles pertaining to the disease and subset of the searches with emphasis on oxidative stress. Search criteria included terms; “disease name” and “Oxidative Stress” but excluded review articles and “disease name” in the authorship.
| Disease names | Disease publication | Oxidative stress | Frequency (%) |
|---|---|---|---|
| Friedriech's Ataxia (FA) | 2353 | 151 | 6.42 |
| Leber's hereditary optic neuropathy (LHON) | 1019 | 25 | 2.45 |
| Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) | 1147 | 25 | 2.18 |
| Myoclonic Epilepsy with Ragged Red Fibers (MERRF) | 410 | 7 | 1.71 |
| Leigh Syndrome (LS) | 1063 | 11 | 1.03 |
Fig 1.
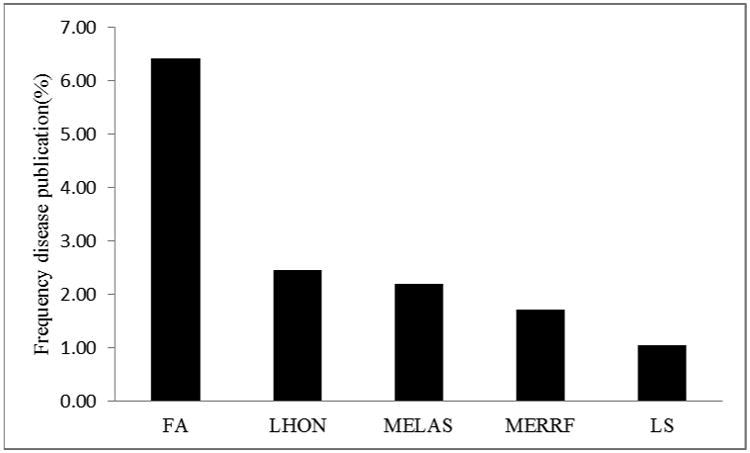
Frequency (%) of primary publication searches that support the role of oxidative stress in the pathomechanism of mitochondrial diseases.
Friedriech's ataxia-FA
Friedriech's ataxia (FA) is an autosomal recessive disease caused by trinucleotide repeat expansion of GAA in the first intron of frataxin gene or a point mutation truncating the transcript, reducing the expression of the predominantly mitochondrial frataxin gene [11-13]. The pathomechanism includes spinocerebellar dorsal root ganglion death, degeneration of the cerebellum, hypertrophic cardiomyopathy and diabetes [2, 14, 15].
Frataxin's only demonstrated physiological role is in the biogenesis of iron-sulfur clusters, and specifically at the formation of persulfide sulfur [5, 16-20]. The mechanism by which iron-sulfur cluster deficiency in this process causes increased ROS production or decreased ROS protection is not clear; however there is support for the idea that the defect in sulfur metabolism decreases thiol-dependent antioxidant protection.
It has also been proposed that frataxin is an iron-chelator, or iron chaperone, that delivers iron to the iron-sulfur cluster biogenesis machinery, suggesting that decreased frataxin causes increase ROS through increased free iron and redox-cycling, i.e. Fenton chemistry, however, this has not been definitively demonstrated. The elucidation of the functionalities of mammalian frataxin more strongly supports a mechanism which is more sulfo-centric rather than ferrocentric.
An increase in oxidative stress has been a leading hypothesis in the pathogenesis of FA since the identification of the gene in 1996 [21].
Support for Oxidative stress from FA patient cells
Deficient antioxidant protection
Early studies in this subject looked into the sensitivity of FA patient cells to extracellular stress induction. A study utilizing fibroblasts identified decreased viability of FA patient cells when exposed to free iron and hydrogen peroxide which was reversed by an additional treatment by either intracellular iron/Ca2+ chelator or apoptosis inhibitor [22]. This oxidant sensitivity was rescued by transfection of human frataxin back into the mutant cells [23]. The oxidant sensitivity was also demonstrated by other groups independently [24]. Further on, a disabling of the NRF2, a master regulator of antioxidant pathway in FA cells has been implicated in both human cells and animals models [24-26]. A study in 2010 by Marmolino et al. observed down regulation of PGC1a, a major regulator of mitochondrial biogenesis and antioxidant response in patient fibroblast cells [17]. Thus there is support from multiple groups in cell and animal model systems that antioxidant responses are disabled in FA.
The reduction of many antioxidants such as glutathione (GSH) was also observed in FA patient fibroblast and spinal cord which is thought to have produced cytoskeletal abnormalities of which the fibroblast cells were rescued by GSH treatment [27, 28]. Furthermore, patient blood and fibroblast cells has also been sampled for various antioxidant enzymes and has been shown to have reduced superoxide dismutase (SOD) and glutathione transferase (GST) activity [24, 29].
The chronic elevation of oxidative stress is also thought to arise from either the direct activity of frataxin in its regulation of antioxidative genes, a dysregulation of Fe-S cluster biogenesis affecting activities of antioxidative enzymes or silencing of a neighboring gene PIP5K1B [30, 31]. Further research is necessary to make strong assertion of this finding.
Increased ROS generation
The earliest hypothesis for FA was that frataxin was a mitochondrial iron chelator, and that its deficiency led to increased iron levels, redox cycling, and increased ROS and oxidative damage. And there is some support for this idea, including that there is increased free iron in the mitochondria. We did not observe a significant increase in iron in patient cells as compared to controls [23]. In general, patient cells indicate stronger support for a decrease in antioxidant protection rather than an increase in pro-oxidants.
FA, oxidative stress and inflammation
Oxidative stress can incite inflammation, and inflammation can cause oxidative stress. Frataxin knockdown in neural Schwann cells causes an explicit inflammation, oxidative stress and induction of inflammatory arachidonate metabolites which precedes cell death [32]. This death is inhibited by anti-inflammatories and antiapoptotics. Similarly, there is induction of prostaglandin synthase, more specifically COX2, and inflammatory activation in Friedreich's patient cells and animal models [33] This appears to be mediated by transcription factors CREB and AP1. Oxidative stress is also thought to mediate apoptosis in FA patient lymphoblast and induced pluripotent stem cell-derived neurons by Bad, DP5 and Bim, which is ameliorated by forskolin treatment [34]. The current model of disease is that reduction of frataxin leads to a decrease in antioxidant protection (such as NRF2) triggering the neuroinflammatory and neurodegenerative consequences which manifest as clinical symptoms of the disease.
Support for Oxidative stress from FA patient biomarker studies
In 2000, Schulz et al. showed that FA patient urine can be monitored for general oxidative stress by measuring 8-hydroxy-2′-deoxyguanosine (8OH2′dG), a marker of oxidative DNA damage which was on average 2.6 fold elevated in patients and reduce by treatment with an antioxidant, idebenone [35]. Similar studies observed increased nuclear and mitochondrial DNA damage in FA patient peripheral blood cells and elevation of albumin, a marker of oxidative damage in blood plasma of patients [36, 37]. Activities of antioxidative enzymes and molecules are also indicated to be reduced in frataxin deficient patients and animal models. GSH acts as a reducing agent donating an electron to a ROS and has been implicated in the pathogenesis of many neurodegenerative diseases including FA [38]. Studies have shown a reduction in glutathione concentration in patient blood plasma and lymphocytes [39-41]. Biomarker of oxidative stress such as glutathione imbalance (GSH/GSSG) in peripheral whole blood of patients may provide information on the systemic severity of the disease and has been proposed by a 2014 study by Enns et al. to be useful in monitoring efficacy of therapies [42].
Support for oxidative stress in FA S. cerevisiae, C. elegans, E. coli and Drosophila models
Frataxin-deficient S. cerevisiae C. elegans and Drosophila models exhibit increased sensitivity to pro-oxidative stress environments [43-45]. Frataxin deficient S. cerevisiae have reduced antioxidant defenses, increases in ROS, and oxidative damage to mitochondrial and nuclear DNA [46, 47]. Likewise, frataxin deficient S. cerevisiae models also show reduced activity of mitochondrial and cytosolic SOD and reduced concentration of GSH indicative of oxidative stress [41, 48, 49]. On the other hand, when frataxin is overexpressed in Drosophila, there is a systemic increase in antioxidant capability, resistance to oxidative stress insults, and longevity [50]. Furthermore, frataxin homolog in S. cerevisiae model appears to have antioxidative response but not iron chelating function [51-53]. Interestingly, a 2009 study by Seguin et al. showed a dose dependent altered function of frataxin in S. cerevisiae when taken together with findings of other studies indicates the possibility that human frataxin has the capacity to regulate multiple pathways not overlapping with S. cerevisiae homologs and that depending on the concentration of frataxin expression in various cell types, its function can be changed [54].
Lack of support for oxidative stress exists in an E. coli FA model. In the case of bacterial homolog of frataxin, cyaY in E. coli when overexpressed showed no alteration in the sensitivity to hydrogen peroxide or concentration of intracellular iron, suggesting either that cyaY is a paralog to human frataxin, or that the bacterial frataxin has function outside of oxidative stress regulation [55].
Differing support in mouse models of FA
Complete knockout of frataxin is embryonic lethal, but two kinds of mouse models of FA exist. The first kind is one in which there is a frataxin deficiency (YG8, KIKO) [56], and the second kind in which frataxin is knocked out conditionally, for example in the cardiac tissue [57].
In the frataxin deficient model of FA, i.e. YG8, there is support for decrease in multiple antioxidant gene expressions; Gclm, Hmox, Nrf2, Nqo1, Prx3, Sod1/2 and TxnRD1 in neural tissues of an FA mouse model [5], and decrease in aconitase activity [58]. Similarly, in both YG8 and KIKO mice, there is support for increased inflammatory markers; interleukins, Nfκb and Tnf, and metabolites, prostaglandins, a class of eicosanoids involved in the onset of inflammations [33].
By contrast, work with a conditional frataxin knockout in the cardiac tissue indicated that Sod expression was only reduced in frataxin deficient mouse heart tissue and not in cerebellar tissue suggesting a degree of disagreement on the generalization of the reduced antioxidant enzymatic function [59]. The same study proposed that iron accumulation and reduction of bio-available iron-sulfur clusters as a more likely pathogenic candidate rather than oxidative stress [59].
A 2012 study by Kim et al. interestingly demonstrated a frataxin dependent neuroprotection from ROS in Parkinson's mouse model which also suffers from oxidative stress[60]. The possibility that frataxin has multiple tissue and cell specific functions might exist and further research is necessary to elucidate this information.
Antioxidants and FA
Therapeutic options for FA patients remain limited and no cure has been identified. Given the support for oxidative stress in the disease, antioxidants have and continue to be tested. Idebenone was a promising drug which was initially identified to reduce oxidative stress in FA cell models, but placebo-controlled crossover studies did not find a significant effect on the ataxic phenotype of patients [35, 61, 62]. Clinical trials of EPI-743 are ongoing. Many patients take coenzyme Q but no demonstration of its effectiveness has occurred [10, 63]. Treatment with oral estrogen like compounds, polyunsaturated fatty acids diet and deferiprone has been shown to reduce oxidative stress by mitigating the reduced antioxidant response activity in aconitase, sequestering the effects of ROS or chelating free iron [64-67]. Treatment of FA patient with low dose tocotrienol supplement was identified to increase the expression of frataxin gene. While further studies are necessary in understanding how tocotrienol is regulating frataxin expression, it is thought that recovery of frataxin expression will also reduce ROS concentration in patients [68]. Trophic factors have also been tested for protective effects against oxidative stress in FA patient periodontal ligament cells and brain-derived neurotrophic factor has been identified to not only elevate frataxin expression but also reverses enzyme expression to that of healthy level, notably SODs and NADPH oxidases (NOX) [69].
Leber's hereditary Optic Neuropathy-LHON
Leber's hereditary optic neuropathy (LHON) is a maternally inherited disease characterized by the bilateral central vision loss at an early age attributed to the degeneration of the retinal ganglion cells (RGCs) with European prevalence of 15:100,000 [70]. The disease is caused by mitochondrial point mutations, most commonly in positions G11778A/ND4, G3460A/ND1, and T14484C/ND6 reducing the functional capacity of NADH:ubiquinone oxidoreductase (complex I) [71-74].
Mitochondrial respiratory chain is a major source of intercellular ROS and the dysfunction of complex I in LHON enables electrons to leak producing excess ROS. It is thought that oxidative stress as a consequence of the mutation is responsible for the cellular damage resulting in apoptosis activation of RGC [75]. A major consequence of oxidative stress is the induction of DNA damage and the levels of 8OH2′dG, a marker of DNA damage has been shown to be elevated in LHON patient leukocyte compared to healthy individuals [76]. The increase in oxidative stress is also exacerbated by the reduction of antioxidant defenses; glutathione peroxidases, glutathione reductase, CuZn superoxide dismutase (SOD) and MnSOD [3]. Human osteosarcoma cytoplasmic hybrid (cybrid) cells with LHON mutations were also observed to have reduced GSH activity [77]. It has been suggested that reduced ATP concentration resulting from complex I deficiency is involved in the pathogenesis of LHON, but in both human cell and mouse models, ATP production is unaltered while oxidative stress remains elevated indicating a greater role of ROS as a cause of RCG degeneration [75, 78].
Current research suggests that the altered redox state of the LHON mitochondria can lead to a non-inflammatory apoptotic event. The mitochondrial apoptosis in LHON is thought to be mediated by the inhibition Bcl-2 decoupling and translocation of Bax/Bak causing the release of Cyt-c leading to the activation of Apaf1 and ultimately the cleavage and activation of caspase family of proteins [79]. LHON patient peripheral blood lymphocytes were measured to be more sensitive to 2-deoxy-D-ribose induced apoptosis with reduction in mitochondrial membrane potential by flow cytometry indicating a basally elevated state of oxidative stress in LHON patients with mitochondrially driven apoptosis [80]. Concurrently, LHON cybrid cells were also more sensitive to Fas-induced apoptosis when compared to control cells alluding to a compromise in the steady state of mitochondrial stability [81]. Interestingly, a study using LHON cybrid cells showed that treatments with vitamin E, N-acetylcysteine and various quinone derived antioxidants including MitoQ10, an antioxidant which has been shown to effectively reach the mitochondria ameliorated cell death induced by tertiary-butyl hydroperoxide (t-BH) or rotenone treatment [77]. While the mechanism of how Bcl2 is inhibited in LHON remains a mystery, the complex I deficiency is likely creating excess ROS overwhelming the cell's ability to regulate the chromic oxidative stress promoting cell death by apoptosis specifically in the RGC.
MELAS and MERRF
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) and Myoclonic epilepsy with ragged-red fibers (MERRF) are two of the most common mitochondrial encephalomyopathies caused by mitochondrial point mutations m.A3243G and m.A8344G encoding mt-tRNA recognizing codons of leucine and lysine respectively [82-84]. MELAS patients are presented with recurring stroke-like episodes, epilepsy, sudden headache with vomiting and convulsions, lactic acidosis of the blood and dementia prevalence of 236:100,000 in individuals of European descent [85-87]. MERRF patients have progressive myoclonic and generalized tonic–clonic seizures, ataxia, deafness, dementia, and myopathy with prevalence of up to 1:400,000 in northern Europe [88, 89].
Although little is known about systemic effects of the tRNA mutation, MELAS and MERRF cells are characterized by the accumulation of ROS and patients suffer from oxidative stress [4, 90]. ROS was detected in patient brain tissue and skin fibroblast cells using a common DNA damage marker, 8OH2′dG, decreased GSH/GSSG ratio and elevated oxidative damage to lipids [90, 91]. The pathogenesis of both diseases are marked by deficiencies of complex I and/or IV leading to the ROS production and inducing the expression and activity of genes involved in antioxidant defense including superoxide dismutases and catalyse in patient muscle tissue [4, 92-95]. Concurrently, cybrid cell lines carrying mutant mitochondrial DNA were found to be more susceptible to oxidative insult perhaps as a result of ROS production overwhelming the antioxidative capacity of the cells [91]. Coenzyme Q10 and taurine has been suggested to alleviate disease progression of MELAS and MERRF indicating the possibility that ROS is the involved in pathomechanism [96-98].
Leigh Syndrome-LS
Leigh syndrome is an inherited mitochondrial disease arising from one of up to 35 mutations in the nuclear or mitochondrial DNA, most commonly in SURF1 and COX assembly genes [99, 100]. The disease affects 1:40,000 individuals with onset approximately 3 to 12 months of age [100, 101]. Patients have reduced capacity to synthesize ATP resulting in multifocal spongiform degeneration affecting the central nervous system [102, 103].
A clinical study in 2008 by Koopman et al. identified elevation of ROS in LS patient derived fibroblast cells. The patients had mutations in the COX assembly genes resulting in reduced complex I activity and when treated with vitamin E derivative, Trolox, the concentration of ROS in patient cells were dramatically reduced [104]. Furthermore, increase of ROS has been measured in a different LS patient fibroblast with reduction in complex V activity and decreased antioxidant defenses, SOD1 and SOD2 [105]. In a complex I deficient animal model of LS, the ndufs4 knockout mouse, there is more protein oxidative damage in the brain resulting from progressive glial activation that promotes neuronal death by both apoptotic and necrotic pathways [106]. Similarly, in the mouse embryonic fibroblasts (MEF) cell of ndufs4fky mice, there is an increased production of superoxides and higher sensitivity to oxidative stress. The same results were not observed in the astrocyte cells even though both cell types exhibit reduced capacity of complex I [107]. And a perturbation of the electron transport chain in drosophila by complex II mutation resulted in increased ROS causing degeneration of the synapses and cell bodies and a condition resembling LS [108]. Interestingly, the study ruled out energy depletion as a pathogenesis of the symptoms as ATP levels remain unchanged and treatment with antioxidant, α-tocopherol prevented synapse degeneration. Thus from these experimental data there is some evidence that perturbation of the Leigh Syndrome mutations causes an increase in ROS.
Meta-analysis of oxidative stress in mitochondrial disease
To mitigate bias in terms of author knowledge, a Pubmed search of each of the 5 most common mitochondrial diseases was carried out, and the disease-specific frequency with the terms ‘oxidative stress’ [Table 1]. The most disease specific citations were Friedreich's>LHON>MELAS>MERRF>LS. The frequencies of the term ‘oxidative stress’ in MELAS, LHON, MERRF, and LS were 2.45, 2.18, 1.71, and 1.03% respectively, and the mean value was 1.84%. By contrast, the disease-specific frequencies for the term ‘oxidative stress’ in Friedreich's ataxia publications was 6.42%, almost 3-fold higher, and the difference from the non-FA mitochondrial disease mean was significant at p<0.0005, (X2=11.8, 1 d.f.). Thus, there is physical and experimental evidence for oxidative stress involvement in all of the 5 most common mitochondrial diseases, however the unbiased test provides the strongest and significant support for Friedreich's ataxia, with LHON as second.
Reactive Nitrogen Species in Mitochondrial Genetic Diseases
Reactive Nitrogen Species (RNS) have been implicated as potential physiological and pathophysiological actors, most often in endothelial cells and airways. There is potential for RNS to be a player in mitochondrial pathophysiology, especially in MELAS and MERRF. Crossing the search terms ‘Reactive Nitrogen Species’ and the mitochondrial disease search terms (Friedreich's ataxia, MELAS, MERRF, LHON or Leigh Syndrome) only resulted in hits in the MELAS mitochondrial disease, with 9 results. Multiple groups have noted alterations in citrulline or arginine or both in MELAS subjects [109, 110]. One study has suggested that subjects with MELAS had lower NO synthesis rate due to the reduced availability of NO precursers; arginine and citrulline. MELAS patients were found to have reduced citrulline flux leading to decreased de novo arginine synthesis, and increased arginine clearance rate which is thought to reduce the plasma arginine. The reduction in the arginine availability for NO synthesis taken together with the increase in the concentration of plasma asymmetric dimethylarginine (ADMA), a regulator of NO synthesis in MELAS patients, is thought to contribute to the NO deficit in MELAS patients. [109]. However work in MELAS cybrid cell lines indicated no differences in NO levels or inducibility [111]. Thus the extent to which these differences in RNS-promoting metabolite concentrations and an overall decreased NO synthesis rate may contribute to endothelial permeability, stiffness, or inflammation and thus possibly to stroke-like episodes are unclear.
Concluding remarks
Inherited mitochondrial diseases are orphan diseases currently lacking in effective treatment. We have reviewed the support for oxidative stress as a component of the pathomechanism of the 5 most common mitochondrial diseases. There are a substantial publications supporting that oxidative stress plays some role in the pathomechanism of all 5 mitochondrial diseases, and thus antioxidant therapy could be considered in all five. Based on the number of published experiments related to oxidative stress and an unbiased meta-analysis of the primary literature, the strongest support for an oxidative stress pathomechanism occurs for Friedreich's ataxia, 6.42%. LHON is second with a score of 2.45%, but this is not significantly higher than the remaining three. These results tend to support the idea that an antioxidant therapeutic strategy is relevant in Friedreich's ataxia and perhaps LHON. As a candidate for the most involvement of oxidative stress in the pathomechanism of disease state, more studies are necessary in FA elucidating the relation between frataxin deficiency and the reduction of antioxidant responses.
Highlights.
Mitochondrial diseases are rare, orphan diseases with no known, proven, curative therapy
The involvement of oxidative stress in pathomechanism of 5 mitochondrial diseases is reviewed
The strongest support for an oxidative pathomechanism was observed in Friedreich's ataxia, however support for an oxidative pathomechanism was also identified in MELAS, MERRF, LS and LHON
Acknowledgments
This work was supported in part by grants NIH P01AG025532, R01NS077777, R01EY012245
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 2.Small JR, Thomas PK, Schapira AH. Dorsal root ganglion proteins in Friedreich's ataxia. Neuroscience Letters. 1993;163:3. doi: 10.1016/0304-3940(93)90377-w. [DOI] [PubMed] [Google Scholar]
- 3.Floreani M, Napoli E, Martinuzzi A, Pantano G, De Riva V, Trevisan R, Bisetto E, Valente L, Carelli V, Dabbeni-Sala F. Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber's hereditary optic neuropathy. The FEBS journal. 2005;272:1124–1135. doi: 10.1111/j.1742-4658.2004.04542.x. [DOI] [PubMed] [Google Scholar]
- 4.Wu SB, Ma YS, Wu YT, Chen YC, Wei YH. Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Molecular neurobiology. 2010;41:256–266. doi: 10.1007/s12035-010-8123-7. [DOI] [PubMed] [Google Scholar]
- 5.Shan Y, Schoenfeld RA, Hayashi G, Napoli E, Akiyama T, Iodi Carstens M, Carstens EE, Pook MA, Cortopassi GA. Frataxin Deficiency Leads to Defects in Expression of Antioxidants and Nrf2 Expression in Dorsal Root Ganglia of the Friedreich's Ataxia YG8R Mouse Model. Antioxidants & redox signaling. 2013 doi: 10.1089/ars.2012.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barzilai A, Yamamoto KI. DNA damage responses to oxidative stress. DNA Repair. 2004;3:1109–1115. doi: 10.1016/j.dnarep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Garner E, Costanzo V. Studying the DNA damage response using in vitro model systems. DNA Repair. 2009;8:1025–1037. doi: 10.1016/j.dnarep.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 8.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. The Journal of biological chemistry. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 9.Ayala A, Munoz MF, Arguelles S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Medicine and Cellular Longevity. 2014;2014:31. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkinson MH, Schulz JB, Giunti P. Co-enzyme Q10 and idebenone use in Friedreich's ataxia. Journal of neurochemistry. 2013;126(Suppl 1):125–141. doi: 10.1111/jnc.12322. [DOI] [PubMed] [Google Scholar]
- 11.Campuzano V, Montermini L, Molto D, Pianese L, Cossee M, Francesca Cavalcanti t, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani S, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel P, Di Donato S, Mandel J, Cocozza S, Koenig MMP. Friedreich's Ataxia: Autosomal Recessive Disease Caused by anIntronic GAA Triplet Repeat Expansion. Science. 1996;271:6. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 12.Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish S, Faucheux B, Trouillas P, Authier F, Dürr A, Mandel J, Vescovi A, Pandolfo M, Koenig M. Frataxin is reduced in Friechdreich ataxia patients and is associated with mitochondrial membranes. Human molecular genetics. 1997;6:10. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- 13.Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel J, Brice A, Koenig M. Clinical and genetic abnormalities in patients with Friedreich's ataxia. The New England journal of medicine. 1996;335:7. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- 14.Durr A, Brice A. Clinical and genetic aspects of spinocerebellar degeneration. Current opinion in Neurology. 2000;13:7. doi: 10.1097/00019052-200008000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Ristow M, Mulder H, Pomplun D, Schulz TJ, Müller-Schmehl K, Krause A, Fex M, Puccio H, Müller J, Isken F, Spranger J, Müller-Wieland D, Magnuson MA, Möhlig M, Koenig M, Pfeiffer AFH. Frataxin deficiency in pancreatic islets causes diabetes due to loss of β cell mass. Journal of Clinical Investigation. 2003;112:527–534. doi: 10.1172/JCI18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai CL, Barondeau DP. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- 17.Marmolino D, Manto M, Acquaviva F, Vergara P, Ravella A, Monticelli A, Pandolfo M. PGC-1alpha down-regulation affects the antioxidant response in Friedreich's ataxia. PLoS One. 2010;5:e10025. doi: 10.1371/journal.pone.0010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calabrese V, Lodi R, Tonon C, D'Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. Journal of the neurological sciences. 2005;233:145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Vaubel RA, Isaya G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Molecular and cellular neurosciences. 2013;55:50–61. doi: 10.1016/j.mcn.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bridwell-Rabb J, Fox NG, Tsai CL, Winn AM, Barondeau DP. Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry. 2014;53:4904–4913. doi: 10.1021/bi500532e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koenig M, Mandel JL. Deciphering the cause of Friedreich ataxia. Current opinion in neurobiology. 1997;7:689–694. doi: 10.1016/s0959-4388(97)80090-6. [DOI] [PubMed] [Google Scholar]
- 22.Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, Cortopassi G. The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Human Molecular Genetics. 1999;8:6. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 23.Tan G, Chen LS, Lonnerdal B, Gellera C, Taroni FA, Cortopassi GA. Frataxin expression rescues mitochondrial dysfunctions in FRDA cells. Human molecular genetics. 2001;10:2099–2107. doi: 10.1093/hmg/10.19.2099. [DOI] [PubMed] [Google Scholar]
- 24.Chantrel-Groussard K, Geromel V, Puccio H, Koenig M, Munnich A, Rötig A, Rustin P. Disabled early recruitment of antioxidant defense in Friedreich's ataxia. Human molecular genetics. 2001;10:8. doi: 10.1093/hmg/10.19.2061. [DOI] [PubMed] [Google Scholar]
- 25.Paupe V, Dassa E, Goncalves S, Auchere F, Lonn M, Holmgren A, Rustin P. Impaired Nuclear Nrf2 Translocation Undermines the Oxidative Stress Response in Friedreich Ataxia. PLoS One. 2009;4:11. doi: 10.1371/journal.pone.0004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Oria V, Petrini S, Travaglini L, Priori C, Piermarini E, Petrillo S, Carletti B, Bertini E, Piemonte F. Frataxin Deficiency Leads to Reduced Expression and Impaired Translocation of NF-E2-Related Factor (Nrf2) in Cultured Motor Neurons. International journal of molecular sciences. 2013;14:7853–7865. doi: 10.3390/ijms14047853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pastore A, Tozzi G, Gaeta LM, Bertini E, Serafini V, Di Cesare S, Bonetto V, Casoni F, Carrozzo R, Federici G, Piemonte F. Actin glutathionylation increases in fibroblasts of patients with Friedreich's ataxia: a potential role in the pathogenesis of the disease. The Journal of biological chemistry. 2003;278:42588–42595. doi: 10.1074/jbc.M301872200. [DOI] [PubMed] [Google Scholar]
- 28.Sparaco M, Gaeta LM, Santorelli FM, Passarelli C, Tozzi G, Bertini E, Simonati A, Scaravilli F, Taroni F, Duyckaerts C, Feleppa M, Piemonte F. Friedreich's ataxia: oxidative stress and cytoskeletal abnormalities. Journal of the neurological sciences. 2009;287:111–118. doi: 10.1016/j.jns.2009.08.052. [DOI] [PubMed] [Google Scholar]
- 29.Tozzi G, Nuccetelli M, Lo Bello M, Bernardini S, Bellincampi L, Ballerini S, Gaeta LM, Casali C, Pastore A, Federici G, Bertini E, Piemonte F. Antioxidant enzymes in blood of patients with Friedreich's ataxia. Archives of disease in childhood. 2002;86:376–379. doi: 10.1136/adc.86.5.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomes CM, Santos R. Neurodegeneration in Friedreich's ataxia: from defective frataxin to oxidative stress. Oxid Med Cell Longev. 2013;2013:487534. doi: 10.1155/2013/487534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bayot A, Rustin P. Friedreich's ataxia, frataxin, PIP5K1B: echo of a distant fracas. Oxid Med Cell Longev. 2013;2013:725635. doi: 10.1155/2013/725635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu C, Cortopassi G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Archives of biochemistry and biophysics. 2007;457:111–122. doi: 10.1016/j.abb.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi G, Shen Y, Pedersen TL, Newman JW, Pook M, Cortopassi G. Frataxin deficiency increases cyclooxygenase 2 and prostaglandins in cell and animal models of Friedreich's ataxia. Human molecular genetics. 2014 doi: 10.1093/hmg/ddu407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Igoillo-Esteve M, Gurgul-Convey E, Hu A, Romagueira Bichara Dos Santos L, Abdulkarim B, Chintawar S, Marselli L, Marchetti P, Jonas JC, Eizirik DL, Pandolfo M, Cnop M. Unveiling a common mechanism of apoptosis in beta-cells and neurons in Friedreich's ataxia. Human molecular genetics. 2014 doi: 10.1093/hmg/ddu745. [DOI] [PubMed] [Google Scholar]
- 35.Schulz J, Dehmer T, Schols L, Mende H, Vorgerd M, Burk K, Matson W, Dichgans J, Beal M, MB B. Oxidative stress in patients with Friedreich ataxia. Neurology. 2000;55:3. doi: 10.1212/wnl.55.11.1719. [DOI] [PubMed] [Google Scholar]
- 36.Haugen AC, Di Prospero NA, Parker JS, Fannin RD, Chou J, Meyer JN, Halweg C, Collins JB, Durr A, Fischbeck K, Van Houten B. Altered gene expression and DNA damage in peripheral blood cells from Friedreich's ataxia patients: cellular model of pathology. PLoS genetics. 2010;6:e1000812. doi: 10.1371/journal.pgen.1000812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swarup V, Srivastava AK, Padma MV, Rajeswari MR. Quantitative profiling and identification of differentially expressed plasma proteins in Friedreich's ataxia. Journal of neuroscience research. 2013;91:1483–1491. doi: 10.1002/jnr.23262. [DOI] [PubMed] [Google Scholar]
- 38.Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012;4:1399–1440. doi: 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. European journal of biochemistry / FEBS. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 40.Piemonte F, Pastore A, Tozzi G, Tagliacozzi D, Santorelli F, Carrozzo R, Casali C, Damiano M, Federici G, Bertini E. Glutathione in blood of patients with Friedreich's ataxia. European Journal of Clinical Investigation. 2001;31:5. doi: 10.1046/j.1365-2362.2001.00922.x. [DOI] [PubMed] [Google Scholar]
- 41.Bulteau AL, Planamente S, Jornea L, Dur A, Lesuisse E, Camadro JM, Auchere F. Changes in mitochondrial glutathione levels and protein thiol oxidation in yfh1 yeast cells and the lymphoblasts of patients with Friedreich's ataxia. Biochimica et biophysica acta. 2012;1822:212–225. doi: 10.1016/j.bbadis.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Enns GM, Moore T, Le A, Atkuri K, Shah MK, Cusmano-Ozog K, Niemi AK, Cowan TM. Degree of glutathione deficiency and redox imbalance depend on subtype of mitochondrial disease and clinical status. PLoS One. 2014;9:e100001. doi: 10.1371/journal.pone.0100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campanella A, Isaya G, O'Neill HA, Santambrogio P, Cozzi A, Arosio P, Levi S. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Human molecular genetics. 2004;13:2279–2288. doi: 10.1093/hmg/ddh232. [DOI] [PubMed] [Google Scholar]
- 44.Vazquez-Manrique RP, Gonzalez-Cabo P, Ros S, Aziz H, Baylis HA, Palau F. Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20:172–174. doi: 10.1096/fj.05-4212fje. [DOI] [PubMed] [Google Scholar]
- 45.Llorens JV, Navarro JA, Martinez-Sebastian MJ, Baylies MK, Schneuwly S, Botella JA, Molto MD. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:333–344. doi: 10.1096/fj.05-5709com. [DOI] [PubMed] [Google Scholar]
- 46.Lefevre S, Brossas C, Auchere F, Boggetto N, Camadro JM, Santos R. Apn1 AP-endonuclease is essential for the repair of oxidatively damaged DNA bases in yeast frataxin-deficient cells. Human molecular genetics. 2012;21:4060–4072. doi: 10.1093/hmg/dds230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lefevre S, Sliwa D, Rustin P, Camadro JM, Santos R. Oxidative stress induces mitochondrial fragmentation in frataxin-deficient cells. Biochemical and biophysical research communications. 2012;418:336–341. doi: 10.1016/j.bbrc.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 48.Irazusta V, Obis E, Moreno-Cermeno A, Cabiscol E, Ros J, Tamarit J. Yeast frataxin mutants display decreased superoxide dismutase activity crucial to promote protein oxidative damage. Free radical biology & medicine. 2010;48:411–420. doi: 10.1016/j.freeradbiomed.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 49.Auchere F, Santos R, Planamente S, Lesuisse E, Camadro JM. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich's ataxia. Human molecular genetics. 2008;17:2790–2802. doi: 10.1093/hmg/ddn178. [DOI] [PubMed] [Google Scholar]
- 50.Runko AP, Griswold AJ, Min KT. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS letters. 2008;582:715–719. doi: 10.1016/j.febslet.2008.01.046. [DOI] [PubMed] [Google Scholar]
- 51.Seguin A, Sutak R, Bulteau AL, Garcia-Serres R, Oddou JL, Lefevre S, Santos R, Dancis A, Camadro JM, Latour JM, Lesuisse E. Evidence that yeast frataxin is not an iron storage protein in vivo. Biochimica et biophysica acta. 2010;1802:531–538. doi: 10.1016/j.bbadis.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 52.Marobbio CM, Pisano I, Porcelli V, Lasorsa FM, Palmieri L. Rapamycin reduces oxidative stress in frataxin-deficient yeast cells. Mitochondrion. 2012;12:156–161. doi: 10.1016/j.mito.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Lefevre S, Sliwa D, Auchere F, Brossas C, Ruckenstuhl C, Boggetto N, Lesuisse E, Madeo F, Camadro JM, Santos R. The yeast metacaspase is implicated in oxidative stress response in frataxin-deficient cells. FEBS letters. 2012;586:143–148. doi: 10.1016/j.febslet.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 54.Seguin A, Bayot A, Dancis A, Rogowska-Wrzesinska A, Auchere F, Camadro JM, Bulteau AL, Lesuisse E. Overexpression of the yeast frataxin homolog (Yfh1): contrasting effects on iron-sulfur cluster assembly, heme synthesis and resistance to oxidative stress. Mitochondrion. 2009;9:130–138. doi: 10.1016/j.mito.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 55.Li DS, Ohshima K, Jiralerspong S, Bojanowski MW, Pandolfo M. Knock-out of the cyaY gene in Escherichia coli does not affect cellular iron content and sensitivity to oxidants. FEBS letters. 1999;456:13–16. doi: 10.1016/s0014-5793(99)00896-0. [DOI] [PubMed] [Google Scholar]
- 56.Miranda CJ, Santos MM, Ohshima K, Smith J, Li L, Bunting M, Cosse M, Koenig M, Sequeiros J, Kaplan J, Massimo Pandolfo M. Frataxin knockin mouse. FEBS letters. 2002;512:291–297. doi: 10.1016/s0014-5793(02)02251-2. [DOI] [PubMed] [Google Scholar]
- 57.Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 58.Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, Webster Z, Blake J, Cooper JM, King R, Pook MA. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seznec H, Simon D, Bouton C, Reutenauer L, Hertzog A, Golik P, Procaccio V, Patel M, Drapier JC, Koenig M, Puccio H. Friedreich ataxia: the oxidative stress paradox. Human molecular genetics. 2005;14:463–474. doi: 10.1093/hmg/ddi042. [DOI] [PubMed] [Google Scholar]
- 60.Kim MJ, Kim DW, Jeong HJ, Sohn EJ, Shin MJ, Ahn EH, Kwon SW, Kim YN, Kim DS, Park J, Eum WS, Hwang HS, Choi SY. Tat-Frataxin protects dopaminergic neuronal cells against MPTP-induced toxicity in a mouse model of Parkinson's disease. Biochimie. 2012;94:2448–2456. doi: 10.1016/j.biochi.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 61.Wilson RB. Therapeutic developments in Friedreich ataxia. Journal of child neurology. 2012;27:1212–1216. doi: 10.1177/0883073812449691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buyse G, Mertens L, Di Salvo G, Matthijs I, Weidemann F, Eyskens B, Goossens W, Goemans N, Sutherland GR, Van Hove JL. Idebenone treatment in Friedreich's ataxia: neurological, cardiac, and biochemical monitoring. Neurology. 2003;60:1679–1681. doi: 10.1212/01.wnl.0000068549.52812.0f. [DOI] [PubMed] [Google Scholar]
- 63.Cooper JM, Korlipara LV, Hart PE, Bradley JL, Schapira AH. Coenzyme Q10 and vitamin E deficiency in Friedreich's ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2008;15:1371–1379. doi: 10.1111/j.1468-1331.2008.02318.x. [DOI] [PubMed] [Google Scholar]
- 64.Richardson TE, Yu AE, Wen Y, Yang SH, Simpkins JW. Estrogen prevents oxidative damage to the mitochondria in Friedreich's ataxia skin fibroblasts. PLoS One. 2012;7:e34600. doi: 10.1371/journal.pone.0034600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cotticelli MG, Crabbe AM, Wilson RB, Shchepinov MS. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox biology. 2013;1:398–404. doi: 10.1016/j.redox.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kontoghiorghe CN, Kolnagou A, Kontoghiorghes GJ. Antioxidant targeting by deferiprone in diseases related to oxidative damage. Frontiers in bioscience (Landmark edition) 2014;19:862–885. doi: 10.2741/4253. [DOI] [PubMed] [Google Scholar]
- 67.Richardson TE, Yang SH, Wen Y, Simpkins JW. Estrogen protection in Friedreich's ataxia skin fibroblasts. Endocrinology. 2011;152:2742–2749. doi: 10.1210/en.2011-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abruzzo PM, Marini M, Bolotta A, Malisardi G, Manfredini S, Ghezzo A, Pini A, Tasco G, Casadio R. Frataxin mRNA isoforms in FRDA patients and normal subjects: effect of tocotrienol supplementation. BioMed research international. 2013;2013:276808. doi: 10.1155/2013/276808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones J, Estirado A, Redondo C, Bueno C, Martinez S. Human adipose stem cell-conditioned medium increases survival of Friedreich's ataxia cells submitted to oxidative stress. Stem cells and development. 2012;21:2817–2826. doi: 10.1089/scd.2012.0029. [DOI] [PubMed] [Google Scholar]
- 70.Sadun AA, La Morgia C, Carelli V. Leber's Hereditary Optic Neuropathy. Current treatment options in neurology. 2011;13:109–117. doi: 10.1007/s11940-010-0100-y. [DOI] [PubMed] [Google Scholar]
- 71.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, 2nd, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 72.Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet. 1991;48:1147–1153. [PMC free article] [PubMed] [Google Scholar]
- 73.Mackey D, Howell N. A variant of Leber hereditary optic neuropathy characterized by recovery of vision and by an unusual mitochondrial genetic etiology. Am J Hum Genet. 1992;51:1218–1228. [PMC free article] [PubMed] [Google Scholar]
- 74.Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochemical and biophysical research communications. 1992;187:1551–1557. doi: 10.1016/0006-291x(92)90479-5. [DOI] [PubMed] [Google Scholar]
- 75.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Progress in retinal and eye research. 2004;23:53–89. doi: 10.1016/j.preteyeres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 76.Yen MY, Kao SH, Wang AG, Wei YH. Increased 8-Hydroxy-2′-Deoxyguanosine in Leukocyte DNA in Leber's Hereditary Optic Neuropathy. Investigative Ophthalmology & Visual Science. 2004;45:1688–1691. doi: 10.1167/iovs.03-0568. [DOI] [PubMed] [Google Scholar]
- 77.Ghelli A, Porcelli AM, Zanna C, Martinuzzi A, Carelli V, Rugolo M. Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids. Invest Ophthalmol Vis Sci. 2008;49:671–676. doi: 10.1167/iovs.07-0880. [DOI] [PubMed] [Google Scholar]
- 78.Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, Carelli V, Ross-Cisneros FN, Baciu P, Sung E, McManus MJ, Pan BX, Gil DW, MacGregor GR, Wallace DC. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proceedings of the National Academy of Sciences. 2012;109:20065–20070. doi: 10.1073/pnas.1217113109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annual review of genetics. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Battisti C, Formichi P, Cardaioli E, Bianchi S, Mangiavacchi P, Tripodi SA, Tosi P, Federico A. Cell response to oxidative stress induced apoptosis in patients with Leber's hereditary optic neuropathy. Journal of neurology, neurosurgery, and psychiatry. 2004;75:1731–1736. doi: 10.1136/jnnp.2003.024372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghelli A, Zanna C, Porcelli AM, Schapira AH, Martinuzzi A, Carelli V, Rugolo M. Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. The Journal of biological chemistry. 2003;278:4145–4150. doi: 10.1074/jbc.M210285200. [DOI] [PubMed] [Google Scholar]
- 82.Kobayashi Y, Momoi MY, Tominaga K, Momoi T, Nihei K, Yanagisawa M, Kagawa Y, Ohta S. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes) Biochemical and biophysical research communications. 1990;173:816–822. doi: 10.1016/s0006-291x(05)80860-5. [DOI] [PubMed] [Google Scholar]
- 83.Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 84.Enriquez JA, Chomyn A, Attardi G. MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nat Genet. 1995;10:47–55. doi: 10.1038/ng0595-47. [DOI] [PubMed] [Google Scholar]
- 85.Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M, Nonaka I. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42:545–550. doi: 10.1212/wnl.42.3.545. [DOI] [PubMed] [Google Scholar]
- 86.Manwaring N, Jones MM, Wang JJ, Rochtchina E, Howard C, Mitchell P, Sue CM. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007;7:230–233. doi: 10.1016/j.mito.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 87.Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Annals of neurology. 1984;16:481–488. doi: 10.1002/ana.410160409. [DOI] [PubMed] [Google Scholar]
- 88.Berkovic SF, Carpenter S, Evans A, Karpati G, Shoubridge EA, Andermann F, Meyer E, Tyler JL, Diksic M, Arnold D, et al. Myoclonus epilepsy and ragged-red fibres (MERRF). 1. A clinical, pathological, biochemical, magnetic resonance spectrographic and positron emission tomographic study. Brain. 1989;112(Pt 5):1231–1260. doi: 10.1093/brain/112.5.1231. [DOI] [PubMed] [Google Scholar]
- 89.Chinnery PF, Johnson MA, Wardell TM, Singh-Kler R, Hayes C, Brown DT, Taylor RW, Bindoff LA, Turnbull DM. The epidemiology of pathogenic mitochondrial DNA mutations. Annals of neurology. 2000;48:188–193. [PubMed] [Google Scholar]
- 90.Katayama Y, Maeda K, Iizuka T, Hayashi M, Hashizume Y, Sanada M, Kawai H, Kashiwagi A. Accumulation of oxidative stress around the stroke-like lesions of MELAS patients. Mitochondrion. 2009;9:306–313. doi: 10.1016/j.mito.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 91.Pang CY, Lee HC, Wei YH. Enhanced oxidative damage in human cells harboring A3243G mutation of mitochondrial DNA: implication of oxidative stress in the pathogenesis of mitochondrial diabetes. Diabetes research and clinical practice. 2001;54(Suppl 2):S45–56. doi: 10.1016/s0168-8227(01)00335-7. [DOI] [PubMed] [Google Scholar]
- 92.Ohkoshi N, Mizusawa H, Shiraiwa N, Shoji S, Harada K, Yoshizawa K. Superoxide dismutases of muscle in mitochondrial encephalomyopathies. Muscle & nerve. 1995;18:1265–1271. doi: 10.1002/mus.880181108. [DOI] [PubMed] [Google Scholar]
- 93.Filosto M, Tonin P, Vattemi G, Spagnolo M, Rizzuto N, Tomelleri G. Antioxidant agents have a different expression pattern in muscle fibers of patients with mitochondrial diseases. Acta neuropathologica. 2002;103:215–220. doi: 10.1007/s004010100455. [DOI] [PubMed] [Google Scholar]
- 94.James AM, Wei YH, Pang CY, Murphy MP. Altered mitochondrial function in fibroblasts containing MELAS or MERRF mitochondrial DNA mutations. Biochem J. 1996;318(Pt 2):401–407. doi: 10.1042/bj3180401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rusanen H, Majamaa K, Hassinen IE. Increased activities of antioxidant enzymes and decreased ATP concentration in cultured myoblasts with the 3243A-->G mutation in mitochondrial DNA. Biochimica et biophysica acta. 2000;1500:10–16. doi: 10.1016/s0925-4439(99)00081-2. [DOI] [PubMed] [Google Scholar]
- 96.De la Mata M, Garrido-Maraver J, Cotan D, Cordero MD, Oropesa-Avila M, Izquierdo LG, De Miguel M, Lorite JB, Infante ER, Ybot P, Jackson S, Sanchez-Alcazar JA. Recovery of MERRF fibroblasts and cybrids pathophysiology by coenzyme Q10. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2012;9:446–463. doi: 10.1007/s13311-012-0103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rikimaru M, Ohsawa Y, Wolf AM, Nishimaki K, Ichimiya H, Kamimura N, Nishimatsu S, Ohta S, Sunada Y. Taurine ameliorates impaired the mitochondrial function and prevents stroke-like episodes in patients with MELAS. Internal medicine (Tokyo, Japan) 2012;51:3351–3357. doi: 10.2169/internalmedicine.51.7529. [DOI] [PubMed] [Google Scholar]
- 98.Cotan D, Cordero MD, Garrido-Maraver J, Oropesa-Avila M, Rodriguez-Hernandez A, Gomez Izquierdo L, De la Mata M, De Miguel M, Lorite JB, Infante ER, Jackson S, Navas P, Sanchez-Alcazar JA. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:2669–2687. doi: 10.1096/fj.10-165340. [DOI] [PubMed] [Google Scholar]
- 99.Gerards M, Kamps R, van Oevelen J, Boesten I, Jongen E, de Koning B, Scholte HR, de Angst I, Schoonderwoerd K, Sefiani A, Ratbi I, Coppieters W, Karim L, de Coo R, van den Bosch B, Smeets H. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain. 2013;136:882–890. doi: 10.1093/brain/awt013. [DOI] [PubMed] [Google Scholar]
- 100.Genetics Home Reference: Leigh syndrome. 2011 http://ghr.nlm.nih.gov/condition/leigh-syndrome.
- 101.Sofou K, De Coo IF, Isohanni P, Ostergaard E, Naess K, De Meirleir L, Tzoulis C, Uusimaa J, De Angst IB, Lonnqvist T, Pihko H, Mankinen K, Bindoff LA, Tulinius M, Darin N. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet journal of rare diseases. 2014;9:52. doi: 10.1186/1750-1172-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wong LJ. Mitochondrial syndromes with leukoencephalopathies. Seminars in neurology. 2012;32:55–61. doi: 10.1055/s-0032-1306387. [DOI] [PubMed] [Google Scholar]
- 103.Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Annals of neurology. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 104.Koopman WJ, Verkaart S, van Emst-de Vries SE, Grefte S, Smeitink JA, Nijtmans LG, Willems PH. Mitigation of NADH: ubiquinone oxidoreductase deficiency by chronic Trolox treatment. Biochimica et biophysica acta. 2008;1777:853–859. doi: 10.1016/j.bbabio.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 105.Karkucinska-Wieckowska A, Lebiedzinska M, Jurkiewicz E, Pajdowska M, Trubicka J, Szymanska-Debinska T, Suski J, Pinton P, Duszynski J, Pronicki M, Wieckowski MR, Pronicka E. Increased reactive oxygen species (ROS) production and low catalase level in fibroblasts of a girl with MEGDEL association (Leigh syndrome, deafness, 3-methylglutaconic aciduria) Folia neuropathologica / Association of Polish Neuropathologists and Medical Research Centre, Polish Academy of Sciences. 2011;49:56–63. [PubMed] [Google Scholar]
- 106.Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10996–11001. doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bird MJ, Wijeyeratne XW, Komen JC, Laskowski A, Ryan MT, Thorburn DR, Frazier AE. Neuronal and astrocyte dysfunction diverges from embryonic fibroblasts in the Ndufs4fky/fky mouse. Bioscience reports. 2014;34 doi: 10.1042/BSR20140151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mast JD, Tomalty KM, Vogel H, Clandinin TR. Reactive oxygen species act remotely to cause synapse loss in a Drosophila model of developmental mitochondrial encephalopathy. Development (Cambridge, England) 2008;135:2669–2679. doi: 10.1242/dev.020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.El-Hattab AW, Emrick LT, Chanprasert S, Craigen WJ, Scaglia F. Mitochondria: role of citrulline and arginine supplementation in MELAS syndrome. The international journal of biochemistry & cell biology. 2014;48:85–91. doi: 10.1016/j.biocel.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 110.Naini A, Kaufmann P, Shanske S, Engelstad K, De Vivo DC, Schon EA. Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox”. Journal of the neurological sciences. 2005;229-230:187–193. doi: 10.1016/j.jns.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 111.Sandhu JK, Sodja C, McRae K, Li Y, Rippstein P, Wei YH, Lach B, Lee F, Bucurescu S, Harper ME, Sikorska M. Effects of nitric oxide donors on cybrids harbouring the mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) A3243G mitochondrial DNA mutation. Biochem J. 2005;391:191–202. doi: 10.1042/BJ20050272. [DOI] [PMC free article] [PubMed] [Google Scholar]