

Abstract
Calcium signaling regulates synaptic plasticity and many other functions in striatal medium spiny neurons to modulate basal ganglia function. Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a major calcium-dependent signaling protein that couples calcium entry to diverse cellular changes. CaMKII activation results in autophosphorylation at Thr286 and sustained calcium-independent CaMKII activity after calcium signals dissipate. However, little is known about the mechanisms regulating striatal CaMKII. To address this, mouse brain slices were treated with pharmacological modulators of calcium channels and punches of dorsal striatum were immunoblotted for CaMKII Thr286 autophosphorylation as an index of CaMKII activation. KCl depolarization increased levels of CaMKII autophosphorylation ∼2-fold; this increase was blocked by an LTCC antagonist and was mimicked by treatment with pharmacological LTCC activators. The chelation of extracellular calcium robustly decreased basal CaMKII autophosphorylation within 5 min and increased levels of total CaMKII in cytosolic fractions, in addition to decreasing the phosphorylation of CaMKII sites in the GluN2B subunit of NMDA receptors and the GluA1 subunit of AMPA receptors. We also found that the maintenance of basal levels of CaMKII autophosphorylation requires low-voltage gated T-type calcium channels, but not LTCCs or R-type calcium channels. Our findings indicate that CaMKII activity is dynamically regulated by multiple calcium channels in the striatum thus coupling calcium entry to key downstream substrates.
Keywords: synaptic plasticity, L-type calcium channels, T-type calcium channels, medium spiny neuron, subcellular fractionation
Introduction
Refined motor control and cognitive functions such as decision-making and habit learning depend on proper function of the striatum, the major input to the basal ganglia system. Over 90% of striatal neurons are medium spiny neurons (MSNs) which integrate and process converging glutamatergic and dopaminergic inputs via modifications of synaptic efficacy to regulate striatal output. Moreover, alterations of the activity or morphology of striatal MSNs are associated with dysregulation of striatal function in many psychiatric and neurological disorders (Glenn & Yang 2012, Ehrlich 2012, Looi & Walterfang 2013, Roberts et al. 1996, Stephens et al. 2005, Zaja-Milatovic et al. 2005, Plotkin & Surmeier 2015, Day et al. 2006). However, signaling mechanisms underlying the regulation of striatal function and plasticity are poorly understood.
The multifunctional serine-threonine kinase, calcium/calmodulin-dependent protein kinase-II (CaMKII), can respond to diverse calcium signals to initiate many responses. Calcium/ calmodulin binding to CaMKII activates autophosphorylation at Thr286, inducing prolonged, calcium-independent activation, which allows CaMKII to execute various forms of neuroplasticity following transient increases of postsynaptic calcium. In other brain regions, Thr286 autophosphorylation promotes interactions with and/or phosphorylation of many downstream targets, including NMDA receptor (NMDAR) subunits (Bayer et al. 2006, Strack & Colbran 1998, Barria & Malinow 2005) and AMPA receptor (AMPAR) subunits (Barria et al. 1997a, Barria et al. 1997b, Hayashi et al. 2000). CaMKII autophosphorylation at Thr286, and CaMKII binding to GluN2B subunits of the NMDAR are essential for normal hippocampal long-term potentiation and various forms of learning and memory (reviewed in Sanhueza & Lisman 2013, Hell 2014). However, CaMKII also plays an important role in some forms of synaptic depression (Mockett et al. 2011, Mayford et al. 1995, Shonesy et al. 2013, Coultrap et al. 2014). Moreover, CaMKII activation is important for the initiation of excitation-transcription coupling (Ma et al. 2014) and for the modulation of neuronal excitability (Nelson et al. 2005, Klug et al. 2012b, Sametsky et al. 2009), morphology (Okamoto et al. 2009, Jourdain et al. 2003, Fink et al. 2003, Wu & Cline 1998), and toxicity (Ashpole et al. 2012, Vest et al. 2010, Hajimohammadreza et al. 1995).
CaMKII is critical for normal striatal function. Under basal conditions, Thr286 autophosphorylation of CaMKII is significantly higher in the striatum relative to other brain regions (Baucum et al. 2013), though mechanisms behind this are not understood. Striatal CaMKII Thr286 autophosphorylation is further increased following the lesion of dopamine inputs in rodent models of Parkinson Disease, and this increase can be reversed by repeated injections of levodopa, the primary dopamine replacement therapy used to treat patients with Parkinson Disease (Brown et al. 2005, Picconi et al. 2004). Moreover, CaMKII inhibition reverses the disruption of corticostriatal synaptic plasticity and the deficit in spontaneous motor behavior caused by dopamine depletion (Picconi et al. 2004). While the links between CaMKII activity and striatal dysfunction are not completely understood, CaMKII is very abundant in striatal MSNs (Fukunaga et al. 1988, Erondu & Kennedy 1985, Ouimet et al. 1984). Moreover, striatal CaMKII interacts strongly with other proteins (Baucum et al. 2013), and has been found to regulate excitatory transmission and intrinsic excitability (Klug et al. 2012a) and control endocannabinoid-dependent plasticity (Shonesy et al. 2013).
Calcium signaling in striatal MSNs is complex. Relative contributions of calcium influx into dendritic spines via multiple ligand- and voltage-gated calcium channels depends on the resting membrane potential, which fluctuates between a downstate membrane potential near -80mV and a depolarized ‘upstate’ membrane potential near -50mV (Carter & Sabatini 2004). For example, T-type voltage-gated calcium channels (TTCCs) play a substantial role in calcium influx in the downstate, but not in the upstate. In contrast, L-type voltage-gated calcium channels (LTCCs) play little role in the downstate, but are responsible for much of calcium influx from the upstate. R-type voltage-gated calcium channels appear to make significant contributions in both the downstate and upstate (Carter & Sabatini 2004). Notably, calcium influx via specific channels can be linked to distinct downstream responses in striatal MSNs. For example, the CaV1.3 subtype of LTCC is specifically linked to the induction of endocannabinoid-dependent synaptic plasticity (Adermark & Lovinger 2007), as well as to the loss of dendritic spines following dopamine depletion in a parkinsonian mouse model (Day et al. 2006). However, the specific signaling mechanisms that are engaged downstream of calcium entry via these different channels are poorly understood.
Here we utilize in vitro slice pharmacology and biochemistry to investigate the link between calcium entry and CaMKII activation in the dorsal striatum. We report that Thr286 autophosphorylation of CaMKII and its synaptic localization under basal conditions is maintained by continual influx of extracellular calcium. Whereas TTCCs significantly contribute to the maintenance of this basal CaMKII autophosphorylation, the activation of LTCCs further increases CaMKII autophosphorylation. Overall, our data begin to define the roles of different VGCCs in regulating striatal CaMKII.
Methods
Animal care
Adult, male mice at postnatal day 35-49 on a C57Bl6/J background (Jackson Laboratories) were used for all experiments and were housed on a 12-h light-dark cycle with food and water ad libitum. All experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee and in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH.
Acute slice preparation and treatment
Mice were anesthetized using isofluorane and then decapitated. Brains were removed, cut into left and right hemispheres, and then 300 μm coronal slices were made at 1-4°C in oxygenated (95% v/v O2, 5% v/v CO2) dissecting solution (208 mM sucrose, 2.5 mM KCl, 1 mM CaCl2, 4 mM MgCl2, 4 mM MgSO4, 1.6 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, and 3 mM Na-pyruvate) using a Vibratome 3000 (The Vibratome Company). Typically, a total of 5-7 striatal hemi-slices (left/right hemisphere combined) were obtained from each mouse brain (1.1 to 0.14 mm from Bregma). Slices were allowed to recover on a nylon mesh for 1 h at 30°C in oxygenated ACSF (113 mM NaCl, 2.5-5 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1 mM NaH2PO4, 26 mM NaHCO3, 20 mM glucose, and 3 mM Na-pyruvate) followed by addition of picrotoxin (50 μM) for 30 min. Slices were then transferred to oxygenated 30°C ACSF solutions supplemented with vehicle or drug for 1-30 min, as defined in the figure legends. The following drugs were used and dissolved in water or DMSO based on manufacturer's instructions: BAPTA (Sigma), (S)-BayK8644 (Tocris), FPL64176 (Tocris), isradipine (National Institute of Mental Health Chemical Synthesis and Drug Supply Program), KCl (Sigma), mibefradil (Tocris), Nickel Chloride (Sigma), nimodipine (MP Biomedicals), SNX-482 (Peptides International), and TTX (Tocris).
Homogenization of striatal tissue punches
Punches (2 mm diameter) of dorsal striatum, containing both lateral and medial regions, were collected from slices on ice after incubation. Total lysates were prepared by immediately homogenizing striatal tissue punches in lysis buffer (2% SDS, 2 mM EGTA, 0.2 mM PMSF, 1 mM benzamidine, 10 μg/ml leupeptin, 10 μM pepstatin, and 1 μM microcystin). Protein concentrations in total striatal lysates were determined by BCA assay (Thermo Scientific), using a bovine serum albumin standard.
Subcellular fractions of freshly dissected striatal tissue punches were prepared as previously described (Gustin et al. 2010). Briefly, two striatal punches were pooled and homogenized in 100 μl Isotonic Buffer (IB: 150 mM KCl, 50 mM Tris-HCl pH 7.5, 1 mM DTT, 0.2 mM PMSF, 1 mM Benzamidine, 1 μM Pepstatin, 10 mg/l Leupeptin, 1 μM microcystin) and incubated at 4°C with rocking for 30 min prior to centrifugation at 100,000 × g for 1 h. The supernatant (S1 fraction, “cytosolic”) was saved, and the pellet was resuspended in 100 μl IB containing 1% (v/v) Triton X-100 and then incubated at 4°C with rocking for 30 min. This lysate was then centrifuged at 18,403 × g, and the resulting supernatant (S2 fraction, “Extrasynaptic membrane”) was saved. The pellet was resuspended in 100 μl isotonic buffer containing 1% Triton X-100 and 1% deoxycholate and sonicated (S3 fraction, “Synaptic”).
Immunoblotting
Striatal lysate protein (10 μg) was mixed with 4× SDS sample buffer, heated at 70ᵒC for 10 min, and then separated by electrophoresis in a 10% SDS polyacrylamide gel. For striatal subcellular fractions, equal volumes were mixed with 4× sample buffer prior to immunoblotting. Gels were transferred in a 0.01 M CAPS/10% methanol buffer to nitrocellulose membranes, which were stained with Ponceau S to detect all proteins and then scanned using a desktop scanner. After blocking for 0.5 hr in 5% milk in Tris-Buffered Solution with 0.01% Tween-20 (TBST), membranes were probed overnight with antibodies to total CaMKIIa (1:1000-1:4000; Thermo, MA1-0147), pThr286 CaMKIIα (1:1000-1:4000; Santa Cruz Biotechnologies, SC-12886-R), pSer1303-GluN2B (1:500; Millipore, 07-398), Total GluN2B (1:1000; BD Transduction, 610417), pSer831-GluA1 (1:500; PhosphoSolutions, P1160-831), GluA1 (1:1000; Santa Cruz Biotechnologies, SC-55509), GAPDH (1:5000; Millipore, MAB374), or PSD95 (1:1000; Neuromab; 75-028). Membranes were then washed, incubated with the appropriate infra-red fluorescent secondary antibodies (1:10,000; 1 h) and imaged using an Odyssey system (LiCor Biosciences).
In vitro autophosphorylation assay
Purified CaMKIIα (1 μM) was incubated on ice for 2.5 min with 50 mM HEPES, 2 mM Mg(Ac)2, 1.5 mM CaCl2, 2 mM DTT, 2 μM CaM, and various concentrations of ATP (0.2-5 μM) to control the level of Thr286 autophosphorylation. Reactions were stopped by adding 4X SDS sample buffer, and approximately 200 ng of CaMKIIα was then loaded on a SDS-PAGE gel.
Quantification of blots and statistical analysis
For analysis of total lysates, Image J (http://imagej.nih.gov/ij/) was used to quantify total Ponceau staining in each gel lane on the nitrocellulose membrane as well as immunoblot signal intensities. Immunoblot signals for total protein levels were normalized to the Ponceau signal in the corresponding lane to correct for variations in gel loading. Phospho-site specific immunoblot signals were normalized to the immunoblot signal for the corresponding total protein in each lane. For analysis of striatal subcellular fractions, individual values were normalized to the average of the values from the same subcellular fraction of control slices analyzed in parallel.
The raw normalized ratios within each experimental condition on each experimental day (5-8 slices) were first subjected to a Grubbs outlier test, resulting in the exclusion of less than 2% of samples analyzed across all conditions. The mean signal from all vehicle control slices was then used for normalization of the individual values for all of the individual slices analyzed on that day (i.e., as a % of the mean control value on each day). The results are plotted as the mean ± SEM of the % control for the indicated total number of slices per group, collected from 4-11 mice over 1-3 days. In analysis of levels of PSD95, GAPDH, and CaMKIIα in subcellular fractions (Fig. 4F), raw normalized ratios of protein/ponceau from BAPTA treated slices were compared to protein/ponceau raw normalized ratios from control slices from the same fraction of the same animal. For autophosphorylation in subcellular fractions, raw normalized ratios of P-T286/total CaMKIIα from BAPTA treated slices were compared to P-T286/total CaMKIIα raw normalized rations from control slices from the same fraction of the same animal. Data were then analyzed using an unpaired Student's t -test, paired t -test, or one way ANOVA followed by a Tukey's post-hoc test as appropriate.
Figure 4.
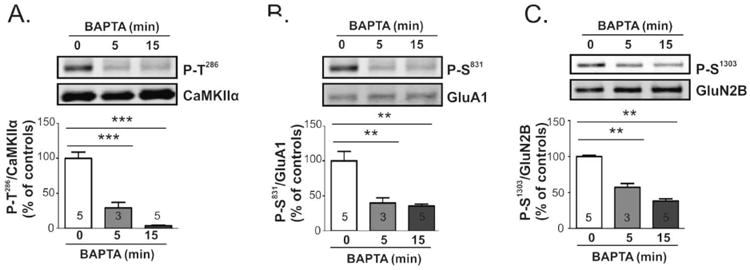
Extracellular calcium is required to sustain basal CaMKII activity and phosphorylation of CaMKII substrates. Mouse brain slices were incubated with BAPTA (5 mM) or vehicle (white bars) for 0, 5, or 15 min. Lysates of dorsal striatum were immunoblotted for P-Thr286 and total CaMKIIα (A), P-Ser831 and total GluA1 (B), or P-Ser1303 and total GluN2B (C). Representative blots are shown above bar graphs reporting P-Thr286/CaMKIIα, P-Ser831/total GluA1, and P-Ser1303/total GluN2B ratios as the mean ± SEM for the indicated number of replicates. Significance determined by one-way ANOVA (**p<0.001) followed by Tukey's post-hoc analysis (**p<0.001).
Results
Ca2+/calmodulin-dependent Thr286 autophosphorylation of CaMKIIα results in generation of an autonomously active form of the kinase that is critical for normal neuronal function. Therefore, we assessed CaMKIIα activation by comparing the levels of Thr286 autophosphorylation by immunoblotting extracts of striatal slices incubated under a variety of conditions using phospho-Thr286 site-specific and total CaMKII antibodies and measuring the ratio of signals detected. In initial pilot studies, slices were immediately flash frozen on dry ice prior to collecting punches of dorsal striatum, followed by immediate homogenization. However, when processing tissue in this manner we could not reliably detect increased levels of CaMKII autophosphorylation after incubating slices in a variety of pharmacological conditions to increase intracellular calcium (data not shown). In order to investigate whether this may reflect a ceiling effect dues to high basal levels of autophosphorylation, we compared samples collected in this manner from control (untreated) slices with samples of purified CaMKIIα that were autophosphorylated with different limiting concentrations of ATP in vitro (Fig 1A). Surprisingly, levels of Thr286 autophosphorylation in basal striatal samples were similar to the maximal levels attained in vitro, suggesting that CaMKII is near maximally phosphorylated in extracts prepared in this manner. Therefore, we optimized the collection of dorsal striatal punches after chilling slices to ∼0°C on wet ice, followed by immediate homogenization. Notably, in tissue punches collected on ice the levels of CaMKIIα autophosphorylation at Thr286 were ∼15% of the levels observed in samples collected after freezing (Fig. 1B). We interpret these observations to indicate that freezing and thawing during the isolation of striatal tissue punches can induce rapid increases in intracellular calcium concentrations, which are sensed by calmodulin to near-maximally activate CaMKIIα. Consequently, all the remaining data reported in this paper were collected from analyses of striatal tissue punches collected near 0°C on ice.
Figure 1.
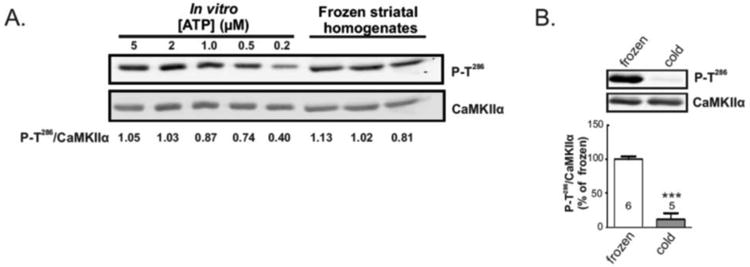
A) Comparison of Thr286 phosphorylation in purified CaMKIIα autophosphorylated in vitro and CaMKIIα in whole lysates of dorsal striatum collected after freezing slices. Numbers below each lane of the blot indicate the P-T286/total signal ratio. Thr286 autophosphorylation of purified CaMKIIα (1 μM) saturates with ATP concentration higher than 2 μM. Notably, CaMKIIα in these striatal samples appears to be Thr286 phosphorylated to similar saturating levels based on the P-T286/total ratio. B) Representative immunoblots of whole lysates of dorsal striatal punches collected either from flash frozen slices on dry ice (frozen) or from slices chilled on regular ice (cold). Quantitation of P-T286 and total CaMKIIα signals from 6 and 5 slices, respectively, revealed that Thr286 phosphorylation in slices dissected on ice is approximately 15% of the levels detected from frozen slices (below). Significance determined by unpaired Student's ttest (***p<0.001).
CaMKII autophosphorylation is increased by LTCC activation
We initially used a pharmacological approach to promote calcium influx. Depolarization of striatal slices using 40 mM KCl for 1 min resulted in a significant ∼2-fold increase in CaMKIIα Thr286 autophosphorylation in whole tissue lysates (Fig. 2A). Since CaMKII and LTCCs are both enriched in dendritic spines, (Fukunaga et al. 1988, Hell et al. 1993, Ouimet et al. 1984), we pretreated slices with the LTCC blocker nimodipine (10 μM, >25 min) and found that this prevented the KCl-induced increase of CaMKIIα Thr286 autophosphorylation. Moreover, treatment of slices with structurally distinct direct pharmacological LTCC activators (0.5 μM FPL64176 or 5 μM BayK8644) for 2 min significantly increased striatal CaMKIIα autophosphorylation in whole tissue lysates by ∼25% and ∼17%, respectively as compared to vehicle treated slices (Fig. 2B, 2C). However, CaMKII activation by FPL64176 or BayK8644 was transient, with no significant increases in levels of Thr286 autophosphorylation after 10 min (data not shown). Taken together, these results indicate that calcium influx via LTCCs transiently activates striatal CaMKIIα.
Figure 2.
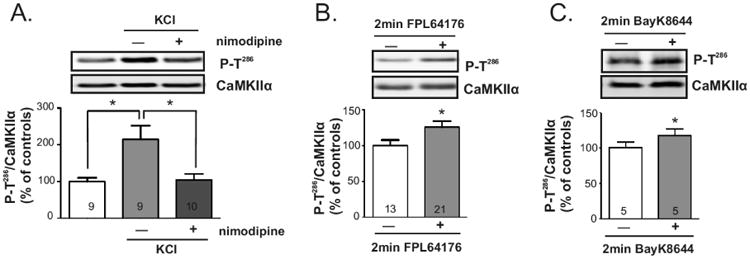
Striatal CaMKII is regulated by membrane depolarization or by selective activation of LTCCs. Brain slices were incubated with (light/dark gray bars) or without (white bars): A) 40 mM KCl for 1 min (light gray) or nimodipine (10 μM, >25 min) followed by 40 mM KCl for 1 min (dark gray). B) 500 nM FPL64176 for 2 min. C) 5 μM BayK8644 for 2 min. Whole lysates of dorsal striatum were immunoblotted for P-Thr286 and total CaMKIIα. Representative blots are shown above bar graphs reporting P-T286/total CaMKIIα ratios as the mean ± SEM for the indicated number of replicates. Significance determined by ANOVA followed by Tukey post-test (A) or by unpaired Student's -test (B,C) (*p<0.05).
Blocking spontaneous activity promotes CaMKII autophosphorylation
In order to explore the role of intrinsic neuronal activity in determining the set point of striatal CaMKIIα autophosphorylation under basal conditions, slices were incubated for 30 min with 1 μM tetrodotoxin (TTX), a sodium channel blocker. Interestingly, TTX treatment increased induced levels of CaMKIIα Thr286 autophosphorylation by ∼18% in whole tissue lysates compared to vehicle treated controls (Fig 3). Thus, it appears that spontaneous firing of action potentials reduces CaMKIIα autophosphorylation under basal conditions.
Figure 3.
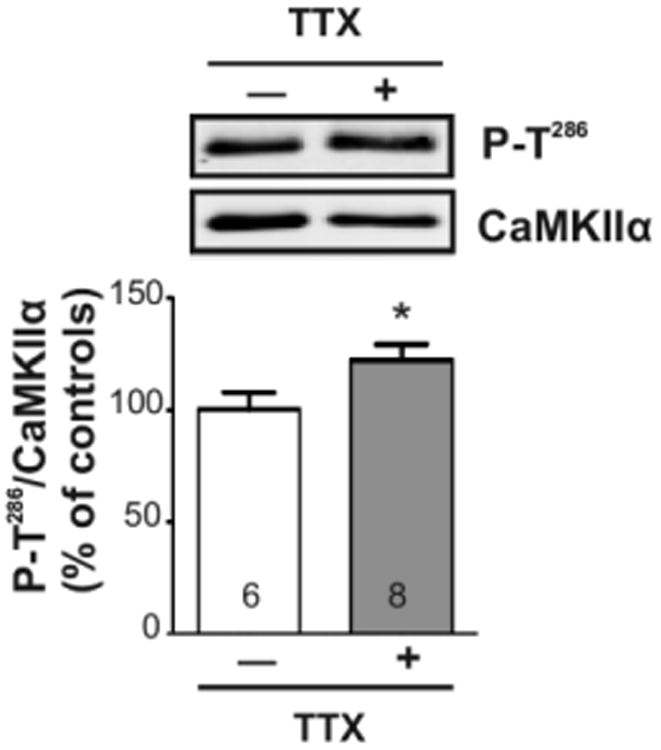
Blocking intrinsic activity increases striatal CaMKII activity. Mouse brain slices were incubated for 30 min with tetrodotoxin (TTX; 1 μM; gray bars) or vehicle (white bars). Lysates of dorsal striatal punches were immunoblotted for P-Thr286 and total CaMKIIα. Representative blots are shown above the bar graph reporting the P-Thr286/total CaMKIIα ratio as the mean ± SEM for the indicated number of replicates. Significance determined by unpaired Student's t-test (*p<0.05).
Calcium influx is necessary to maintain basal phosphorylation of CaMKII, GluA 1, and GluN2B
Since intrinsic neuronal activity was not responsible for the maintenance of significant CaMKIIα autophosphorylation in dorsal striatum under basal conditions, we investigated whether extracellular calcium was required to maintain basal CaMKIIα autophosphorylation. BAPTA (5 mM final) was added to the slice incubation media (ACSF) to rapidly chelate extracellular calcium and prevent basal calcium influx (Fig 4A). Notably, Thr286 autophosphorylation of CaMKIIα in whole tissue lysates was decreased by ∼70% after 5 min and by ∼95% after 15 min (Fig 4A), indicating that CaMKIIα was rapidly and progressively dephosphorylated following the prevention of calcium influx. Thus, continual calcium influx under basal conditions is important in driving CaMKIIα activation to maintain basal levels of Thr286 autophosphorylation.
We also examined the impact of removing extracellular calcium on the phosphorylation of two physiologically relevant synaptic substrates of CaMKII, Ser831 in the AMPAR GluA1 subunit (Mammen et al. 1997, Barria et al. 1997a) and Ser1303 in the NMDAR GluN2B subunit (Omkumar et al. 1996). Phosphorylation of both substrates was readily detected under basal conditions. The addition of BAPTA significantly reduced GluA1 phosphorylation at Ser831 (Fig. 4B) to 40% and 35% of control after 5 and 15 min, respectively. Similarly, GluN2B phosphorylation at Ser1303 (Fig 4C) was significantly reduced to 57% and 39% of control, respectively. These data indicate that ongoing calcium influx under basal conditions drives not only CaMKIIα activation, but also the phosphorylation of two key synaptic targets of CaMKII that are important for synaptic plasticity.
Calcium influx is required for normal CaMKII localization
CaMKII activation drives its translocation to dendritic spines and the postsynaptic density in the hippocampus (Shen & Meyer 1999, Strack et al. 1997c). To determine if striatal CaMKII localization is also sensitive to its activation state, we prepared subcellular fractions from striatal slices that had been treated for 15 min with 5 mM BAPTA or vehicle. Under basal conditions, CaMKIIα was detected in all three fractions, but was most abundant (normalized to volume loaded) in the S3 fraction, and least abundant in the S1 fraction (see representative blots in Fig. 5A), consistent with prior studies of dissected tissues (Baucum et al. 2013, Gustin et al. 2011). Moreover, the relative enrichment (normalized to protein) of CaMKIIα in S1, S2, and S3 fractions from control dorsal striatum was 0.54 ± 0.13, 0.91 ± 0.15, and 1.42 ± 0.18, respectively, demonstrating significant enrichment in the synaptic fraction (n=8-9. P=0.0024 by ANOVA). Thr286 phosphorylation of CaMKIIα also was readily detected in all three fractions from control slices, and the P-T286/total CaMKIIα ratio was highest in synaptic fractions (S1, 1.28 ± 0.17; S2, 0.62 ± 0.09; S3, 1.80 ± 0.23; N=9. ANOVA P=0.0003). Extracellular calcium chelation using BAPTA decreased the levels of Thr286 autophosphorylation by >85% in all three fractions, when compared to the corresponding fraction from untreated control slices (Fig. 5A, B). Interestingly, BAPTA treatment also resulted in a significant translocation of total CaMKIIα to the cytosolic fraction when quantified as the S1/S2 ratio (Con: 0.59 ± 0.10, BAPTA: 0.78 ± 0.07; paired t -test, p=0.02) or as the S1/S3 ratio (Con: 0.35 ± 0.10, BAPTA: 0.47 ± 0.08; paired ttest, p=0.05) (Fig. 5A, C). Alternatively, when total levels of CaMKIIα in S1 fractions (normalized to Ponceau) were quantified as a BAPTA/control ratio, there is a significant 68 ± 12% increase in CaMKIIα in the cytosolic S1 fraction (p = 0.0017, by Student's t-test). However, there was no significant effect of BAPTA treatment on the levels of total CaMKIIα in the S2 or S3 fractions, or in the S2/S3 ratio (Fig. 5C). To verify the effective fractionation of control and BAPTA treated tissue, samples were also blotted for PSD95 (Fig. 5A, D) and GAPDH (Fig. 5A, E). Treatment with BAPTA did not alter the levels of PSD95 in the S2 (Triton-soluble) membrane fraction or the (higher) levels in the S3 (Triton-insoluble) synaptic fraction. Moreover, levels of GAPDH in the S1 (cytosolic) fraction and S2 membrane fraction were unaffected by BAPTA. Taken together, these data indicate that chelation of extracellular calcium and dephosphorylation of Thr286 resulted in a significant translocation of total CaMKIIα to the cytosolic S1 fraction.
Figure 5.
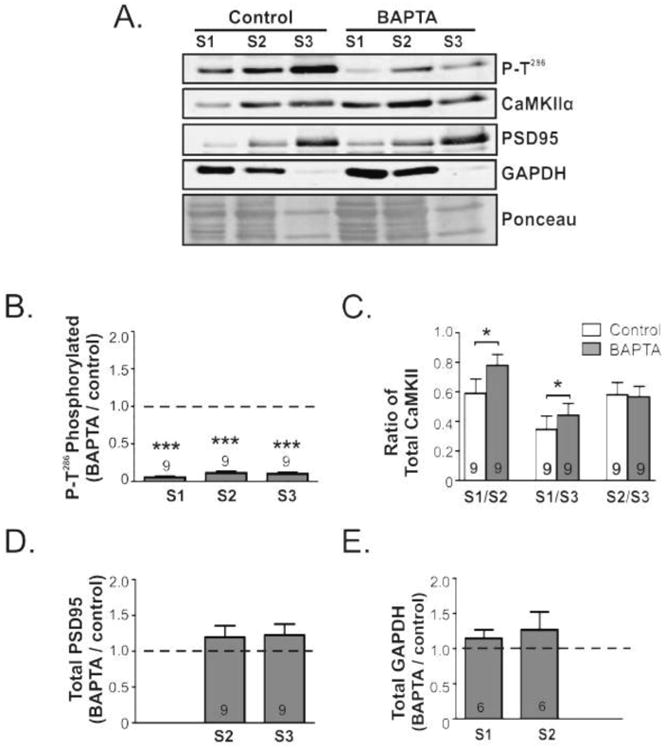
Depletion of extracellular calcium induces CaMKII translocation to the cytosolic fraction. Mouse brain slices were incubated for 15 min with BAPTA (5 mM) or vehicle. Equal volumes of cytosolic (S1), membrane (S2, Triton-Soluble), and synaptic (S3, Triton-Insoluble) fractions isolated from dorsal striatum were immunoblotted for: P-Thr286, total CaMKIIα, and also GAPDH and PSD-95 as cytosolic and synaptic markers, respectively. Representative images of each blot are shown, as well as a segment of the Ponceau stained membrane prior to immunoblotting. The relative enrichment of the P-Thr286/total CaMKIIα ratio (B) as well as the total CaMKIIα (C) PSD95 (D) GAPDH (E) normalized to the Ponceau stain in each fraction from control and BAPTA treated slices were then quantified. All bar graphs report the mean ± SEM from the indicated number of replicates. The dashed line indicates a value of 1.0 (F); the theoretical value if BAPTA had no effect. Significance determined by one sample -test (*p<0.05, ***p<0.001).
T-type voltage gated calcium channels (TTCCs) contribute to basal CaMKII autophosphorylation
Since calcium influx is essential for maintenance of basal CaMKIIα autophosphorylation, we tested for the involvement of specific calcium channels under basal condition. Striatal MSNs express both CaV1.2 and CaV1.3 LTCCs, and a stronger membrane depolarization is required to activate CaV1.2 in comparison to CaV1.3 channels. Therefore, we tested the effects of two different dihydropyridines, nimodipine and isradipine, which are reported to differentially block CaV1.2 and CaV1.3 LTCCs (Bonci et al. 1998, Fitton & Benfield 1990, Sinnegger-Brauns et al. 2009, Koschak et al. 2001). However, basal CaMKIIα Thr286 autophosphorylation was unaffected by a 30 min incubation with either 10 μM nimodipine (Fig. 6A) or 5 μM isradipine (Fig. 6B) as compared with vehicle treated controls. RTCCs make a significant contribution to calcium influx into MSN dendritic spines at both down- and up-state membrane potentials (Carter & Sabatini 2004), but Thr286 phosphorylation of CaMKIIα was unchanged by treatment with the RTCC blocker SNX-482 (400 nM; 30 min) (Fig. 6C). Finally, we examined the role of TTCCs, which appear to be active at downstate membrane potentials in the striatum (Carter & Sabatini 2004). TTCCs can be selectively inhibited using mibefradil or NiCl2 compared to other VGCCs (Martin et al. 2000, Todorovic & Lingle 1998, Fox et al. 1987). Interestingly, incubation of striatal slices for 30 min with mibefradil (5 μM) or NiCl2 (100 μM) significantly decreased Thr286 autophosphorylation of CaMKIIα by 37% (Fig. 6D) and 35% (Fig. 6E), respectively. In combination, these data indicate that basal CaMKIIα activation/autophosphorylation in the striatum is at least partially supported by calcium influx through TTCCs.
Figure 6.
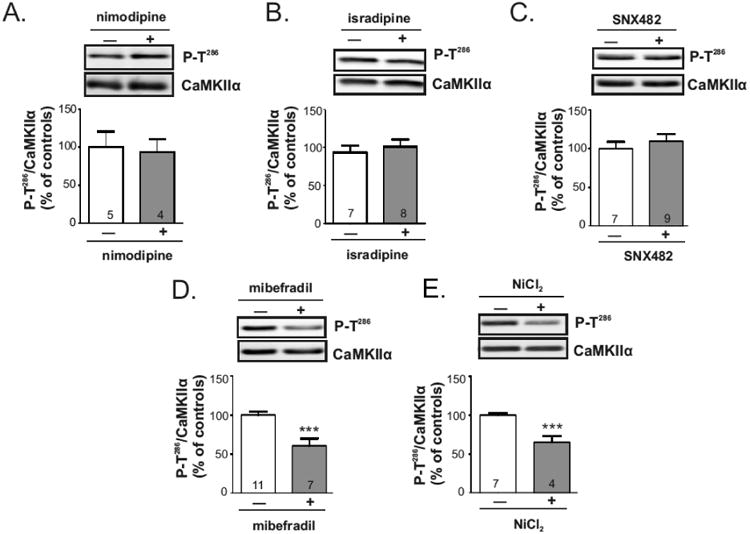
Maintenance of basal CaMKII autophosphorylation requires TTCC activity. Mouse brain slices were incubated for 30 min with A) nimodipine (10 μM), B) isradipine (5 μM), C) SNX482 (400 nM), D) mibefradil (5 μM), E) NiCl2 (100 μM), or vehicle. Whole lysates of dorsal striatum were immunoblotted for P-Thr286 and total CaMKIIα. Representative blots are shown above bar graphs reporting the P-Thr286/total CaMKIIα ratios as the mean ± SEM for the indicated number of replicates. White bars are vehicle controls and gray bars are drug-treated samples. Significance determined by unpaired Student's ttest (***p<0.001).
Discussion
Calcium influx via numerous channels dynamically regulates multiple MSN functions by recruiting diverse signaling proteins. While extensive work has identified several channels underlying striatal plasticity, our understanding of the proteins that couple calcium influx to synaptic function remains incomplete. We began to explore this by focusing on the calcium-dependent activation of CaMKII, which modulates synaptic function in multiple brain regions, including dorsal striatum (Klug et al. 2012a, Shonesy et al. 2013). We found that basal influx of extracellular calcium promotes Thr286 autophosphorylation and synaptic localization of CaMKIIα as well as the phosphorylation of physiologically relevant targets, NMDAR GluN2B and AMPAR GluA1 subunits, at CaMKII sites. We also identified a novel role for striatal TTCCs in maintaining basal striatal CaMKIIα activity, with calcium influx via LTCCs inducing a further activation of CaMKIIα following depolarization. These findings demonstrate a dynamic, calcium-dependent CaMKIIα regulation that presumably connects striatal calcium channel activity with key downstream signaling pathways.
Our studies utilize the calcium-dependent Thr286 autophosphorylation site as a proxy for CaMKIIα activation. The calcium sensitivity of CaMKIIα introduced unusual technical difficulties in that we found that freezing brain slices prior to isolating dorsal striatal tissue induced a near maximal autophosphorylation at Thr286. Previous work also found that flash freezing of rat hippocampal slices causes similar artificially elevated CaMKIIα activity as measured by increases in Thr286 autophosphorylation and CaMKII substrate peptide phosphorylation (Lengyel et al. 2001). Furthermore, this freezing-induced elevation of CaMKII activity masked experimental treatment effects and was not reversed by exclusion of phosphatase inhibitors, changes in buffer components, or by freezing on dry ice versus liquid nitrogen (Lengyel et al. 2001). Our data emphasize the importance of avoiding freeze/thaw of striatal samples prior to homogenization to avoid artificial activation of CaMKII in these types of experiments.
The sensitivity and rapid response of CaMKII autophosphorylation to increases of calcium are well known, enabling CaMKII to function as an effective integrator of repetitive calcium spikes that occur during action potential firing. Our studies examined if intrinsic spontaneous activity in striatal slices modulates CaMKII activity. Previous work using neurons cultured from other brain regions has shown that spontaneous neuronal activity induces glutamate release to promote CaMKII activation (Murphy et al. 1994, Padmanabhan et al. 2008, Eshete & Fields 2001). Thus, it was somewhat surprising that TTX treatment slightly increased CaMKIIα autophosphorylation in the striatum. The mechanism of this activation is likely complex, perhaps involving components at the molecular and system levels. At the molecular level, TTX treatments have been shown to increase the phosphorylation of striatal DARPP-32 at Thr34 (Nishi et al. 1997). Enhanced phosphorylation of DARPP-32 would be expected to inhibit protein phosphatase 1, which is known to dephosphorylate CaMKII at Thr286 (Shields et al. 1985, Strack et al. 1997a). However, the response to TTX treatment might also reflect the complex neuromodulatory environment of the striatum. As a central mediator of the basal ganglia circuit, striatal MSNs receive excitatory inputs from cortical and thalamic pathways. However, dopaminergic, cholinergic, and GABA-ergic transmission also exert potent inhibitory control of the glutamatergic inputs to MSNs (Pakhotin & Bracci 2007, Kawaguchi et al. 1995, Gerfen & Surmeier 2011, Do et al. 2013). Furthermore, these excitatory and inhibitory inputs converge on both major subtypes of striatal MSNs, which either express the Dopamine 1 Receptors (D1R-MSNs) or Dopamine 2 Receptors (D2R-MSNs). Striatal D1R-MSNs and D2R-MSNs have quite distinct properties, particularly with respect to excitability (Kreitzer & Malenka 2007b, Gertler et al. 2008, Planert et al. 2013), with D1R signaling promoting LTP (Centonze et al. 2003, Kerr & Wickens 2001) and D2R signaling promoting LTD (Kreitzer & Malenka 2007a, Calabresi et al. 1997). Dopamine enhances calcium influx via LTCCs in D1R-MSNs (Hernandez-Lopez et al. 1997), but inhibits LTCCs in D2R-MSNs (Hernandez-Lopez et al. 2000). Additionally, D1R activation can inhibit sodium channels, whereas D2R activation can activate sodium channels (Surmeier et al. 1992, Surmeier et al. 1993). Future studies would need to differentiate the contributions of these D1R- and D2R- MSN subtypes to better elucidate the regulation of CaMKII activity in the striatum.
Next, we examined the role of calcium entry in regulating striatal CaMKII. We found that membrane depolarization increases CaMKIIα autophosphorylation, indicating that striatal CaMKII can be further activated from basal conditions. We also identified an important role for calcium influx in sustaining basal levels of CaMKII autophosphorylation by adding BAPTA to the ACSF. The rapid chelation of extracellular calcium by BAPTA presumably decreases the amount of calcium in specific intracellular pools that continually drive CaMKII autophosphorylation in the face of significant basal phosphatase activity(ies) that can dephosphorylate Thr286 (Strack et al. 1997b, Fukunaga et al. 1993, Ishida et al. 1998). These findings are consistent with prior studies in cultured neurons showing that removal of extracellular calcium profoundly decreases CaMKII autophosphorylation (Scholz & Palfrey 1998, Cohen & Fields 2006). We also found that extracellular calcium chelation decreases the phosphorylation of GluN2B and GluA1, key synaptic substrates of CaMKII (Hunt & Castillo 2012, Santos et al. 2009), suggesting that basal CaMKII activity is driving the phosphorylation of physiological CaMKII substrates. However, our data cannot exclude possible contributions from broader effects of BAPTA addition on other signaling pathways or on the kinetics of calmodulin dissociation. Since BAPTA reduced Thr286 autophosphorylation of CaMKIIα in whole lysates by ∼90% within 15 min, it was not surprising that pools of CaMKIIα holoenzymes in the cytosolic (S1), membrane (S2), and synaptic (S3) fractions were similarly dephosphorylated. However, BAPTA treatments increased total levels of CaMKIIα in the cytosolic fraction as compared with control treatments, which we interpret to reflect a translocation of CaMKII out of the synaptic fraction because it is well established that calcium/calmodulin-dependent activation and Thr286 autophosphorylation drives CaMKII translocation to synapses (Strack et al. 1997c, Shen et al. 2000, Shen & Meyer 1999). While total levels of CaMKIIα in membrane (S2) or synaptic (S3) fractions were not significantly reduced by BAPTA treatment, this may reflect the higher levels of CaMKIIα in these fractions. Thus, dissociation of a statistically insignificant fraction of CaMKIIα from the synaptic or membrane fraction could significantly increase levels of CaMKIIα in the cytosolic S1 fraction. Furthermore, we recently identified over 100 proteins associated with synaptic CaMKII holoenzymes (Baucum et al. 2015), some of which could limit CaMKII translocation to other fractions following decreases in Thr286 autophosphorylation. However, we cannot exclude other contributions to changes in cytosolic CaMKII levels such as degradation or synthesis. Taken together these findings suggest that the ongoing influx of extracellular calcium under steady-state basal conditions promotes basal CaMKIIα autophosphorylation and localization to intracellular compartments in striatal MSNs where it could act on substrates necessary for synaptic plasticity.
We also examined the roles of specific VGCCs thought to mediate calcium entry in CaMKII regulation. Prior calcium imaging studies have shown that LTCCs, RTCCs, and TTCCs can make significant contributions to total calcium influx into striatal MSN spines and dendrites, depending on the resting membrane potential (Carter & Sabatini 2004). Although we have no direct control of the resting membrane potential in our in vitro slice experiments, our data indicate that LTCCs and TTCCs, but not RTCCs, make distinct contributions to CaMKIIα activation under different conditions.
First, we found that pretreatment of slices with nimodipine blocked depolarization-induced increases in CaMKIIα autophosphorylation, suggesting that LTCCs are key regulators of CaMKII during depolarization. Furthermore, CaMKII autophosphorylation was also transiently increased by the selective activation of LTCCs using FPL64176 and BayK8644. Notably, FPL64176 and BayK8644 only modestly activated CaMKII in comparison with the response to global depolarization using KCl, likely due to their mechanism of action. These compounds enhance calcium influx by prolonging LTCC opening (Kunze & Rampe 1992, Bechem & Hoffmann 1993, Rampe et al. 1993), rather than directly opening the channel, so it seems likely that FPL64176 and BayK8644 will induce more modest levels of calcium influx in comparison to 40 mM KCl. Although calcium influx via activated LTCCs can recruit CaMKII autophosphorylation, LTCCs did not play a significant role in driving CaMKIIα autophosphorylation under basal conditions because neither nimodipine, a generic LTCC blocker, nor isradipine a somewhat CaV1.3-selective LTCC blocker, had a significant effect. This is not entirely surprising becasue LTCCs would be expected to be largely closed under basal conditions. Thus, the role of LTCCs in CaMKIIα regulation is dependent on the upstate (depolarized) or downstate (basal) membrane potentials, which could have implications for understanding the essential roles of LTCCs in spine loss following dopamine depletion (Day et al. 2006), endocannabinoid dependent synaptic depression (Adermark & Lovinger 2007), and depolarization-induced spine loss (Tian et al. 2010). Indeed, CaMKII has an important role in regulating the synthesis of the major striatal endocannabinoid, 2-arachidonylglycerol (Shonesy et al. 2013) and spine morphology (Xie et al. 2007, Zheng et al. 2010). Furthermore, CaMKII could also function as a feedback regulator of striatal LTCCs, as CaMKII binds to and phosphorylates multiple subunits of the CaV1.2 and CaV1.3 LTCCs to facilitate calcium entry (Hudmon et al. 2005, Koval et al. 2010, Erxleben et al. 2006, Lee et al. 2006, Wang et al. 2009, Gao et al. 2006, Jenkins et al. 2010). This positions CaMKII to regulate LTCCs through phosphorylation or binding, in addition to acting on downstream synaptic targets that mediate striatal LTCC functions.
Second, our data indicate that TTCCs, but not RTCCs, make a significant contribution to calcium entry that is important for driving activation and Thr286 autophosphorylation of CaMKIIα under basal conditions. RTCCs and TTCCs are activated at much more hyper-polarized membrane potentials than LTCCs. Our data using SNX-482, which selectively inhibits RTCCs over other VGCCs (Newcomb et al. 1998, Bourinet et al. 2001), do not support a significant role for RTCCs in basal CaMKIIα activation, although potential off target effects on potassium channels may confound this interpretation (Kimm & Bean 2014). However, basal CaMKIIα autophosphorylation was partially blocked by mibefradil, a selective TTCC blocker. Mibefradil has been reported to also block LTCCs at somewhat higher concentrations (Martin et al. 2000, Jiménez et al. 2000), but the lack of effect of dihydropyridines (see above) strongly suggests that LTCCs are not involved under basal conditions. Moreover, CaMKIIα autophosphorylation was also decreased by incubating slices with NiCl2, which at the dosage used preferentially blocks TTCC currents over other VGCC subtypes (Todorovic & Lingle 1998, Fox et al. 1987). While NiCl2 can also inhibit RTCCs (Zamponi et al. 1996), the lack of effect of SNX-482 strongly supports a role for TTCCs in mediating the effect of NiCl2. TTCCs are less well characterized than other VGCCs, though they have been linked to spontaneous neuronal burst firing (Cain & Snutch 2010, Isope & Murphy 2005, Contreras 2006, Broicher et al. 2008) and synaptic depression (Bender et al. 2006, Birtoli & Ulrich 2004, Oliet et al. 1997, Nevian & Sakmann 2006), and were recently shown to increase CaMKII autophosphorylation in hippocampus (Moriguchi et al. 2012). The coupling of CaMKIIs to TTCC activation could mediate specific downstream signals or also be important in feedback facilitation of the CaV3.2 TTCC subtype (Yao et al. 2006, Welsby et al. 2003, Wolfe et al. 2002). Future studies further examining the coupling of TTCC and CaMKII activity could provide key insights into how these proteins contribute to striatal plasticity.
One limitation of our biochemical studies is that we are measuring the overall average CaMKIIα activation across all striatal cell types. CaMKII is highly expressed in striatal MSNs (Erondu & Kennedy 1985, Fukunaga et al. 1988, Ouimet et al. 1984), which constitute ≥90% of striatal neurons, likely reflecting the source of most of the CaMKII we assayed here. However, as mentioned previously, there are two major MSN subtypes, the D1R-MSNs or the D2R-MSNs (Gerfen et al. 1990, Surmeier et al. 1996). These dopamine receptors couple to distinct biochemical signaling pathways and are generally considered to play opposing roles in coordinating basal ganglia functions (Gerfen & Surmeier 2011, Lovinger 2010, Kreitzer 2009, Calabresi et al. 2014). Moreover, MSNs expressing D1Rs or D2Rs differ from each other in dendritic morphology (Gertler et al. 2008). Preliminary immunostaining studies have not indicated substantial differences in CaMKII expression in D1R-MSNs and D2R-MSNs (data not shown), but our current data likely reflect the combined effects of these calcium channels in both MSN subtypes. It is possible that these calcium channels selectively modulate CaMKII in one MSN subtype, but not the other. In this regard, it is interesting to note that CaV1.3 LTCCs are selectively implicated in spine loss from D2R-MSNs following dopamine depletion (Day et al. 2006) and that CaMKII can modulate both spine morphology (Xie et al. 2007, Zheng et al. 2010) and the activity of CaV1.3 LTCCs (Jenkins et al. 2010). Future studies should investigate CaMKII regulation using immunohistochemical approaches in mice expressing DrD2-eGFP or Drd1a-tdTomato transgenes to differentiate striatal MSN subtypes, and examine dendritic pools of CaMKII. Such an approach would also provide insights into CaMKII regulation in lateral and medial portions of the dorsal striatum, as well as the ventral striatum (Nucleus Accumbens), which have distinct behavioral roles.
Overall, our results highlight the complexity of CaMKII regulation by multiple calcium channels in the striatum. This could implicate CaMKII in a number of calcium mediated striatal functions, such as regulating membrane potential, integrating dopaminergic signaling, influencing gene expression, and inducing synaptic plasticity. Dysregulation between CaMKII activation and calcium entry via select VGCCs could also be important in understanding neurological diseases (e.g., Parkinson disease, Huntington disease) and psychiatric disorders involving changes in striatal function. Future studies determining how these upstream calcium entry points couple with downstream calcium-dependent signaling components could be important in the development of new therapeutic targets to neurological and psychiatric disorders.
Highlights.
We explored CaMKII regulation by pharmacologically treating striatal slices.
Depolarization recruits L-type calcium channels to increase CaMKII activation.
Calcium influx partly by T-type channels sustains CaMKII activity and location.
Acknowledgments
The authors wish to thank members of the Colbran lab for helpful comments and suggestions to this project. This work was supported by NIH R01 grants MH063232 and NS078291 to RJC. JGP was supported by NIH Training Program in Ion Channel and Transporter Biology (T32NS00749) and National Institute of Mental Health (F32MH100747). XW was supported by an award from the American Heart Association. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- CaMKII
calcium/calmodulin-dependent protein kinase II
- LTCC
L-type calcium channel
- MSN
medium spiny neuron
- RTCC
R-type calcium channel
- TTCC
T-type calcium channel
- VGCC
Voltage-gated calcium channel
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Adermark L, Lovinger DM. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:6781–6787. doi: 10.1523/JNEUROSCI.0280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, Hudmon A. Calcium/Calmodulin-dependent Protein Kinase II (CaMKII) Inhibition Induces Neurotoxicity via Dysregulation of Glutamate/Calcium Signaling and Hyperexcitability. The Journal of biological chemistry. 2012;287:8495–8506. doi: 10.1074/jbc.M111.323915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. The Journal of biological chemistry. 1997a;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997b;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Baucum AJ, 2nd, Brown AM, Colbran RJ. Differential association of postsynaptic signaling protein complexes in striatum and hippocampus. Journal of neurochemistry. 2013;124:490–501. doi: 10.1111/jnc.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucum AJ, 2nd, Shonesy BC, Rose KL, Colbran RJ. Quantitative Proteomics Analysis of CaMKII Phosphorylation and the CaMKII Interactome in the Mouse Forebrain. ACS chemical neuroscience. 2015 doi: 10.1021/cn500337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O'Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechem M, Hoffmann H. The molecular mode of action of the Ca agonist (-) BAY K 8644 on the cardiac Ca channel. Pflugers Arch. 1993;424:343–353. doi: 10.1007/BF00384362. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two Coincidence Detectors for Spike Timing-Dependent Plasticity in Somatosensory Cortex. The Journal of Neuroscience. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtoli B, Ulrich D. Firing Mode-Dependent Synaptic Plasticity in Rat Neocortical Pyramidal Neurons. The Journal of Neuroscience. 2004;24:4935–4940. doi: 10.1523/JNEUROSCI.0795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Grillner P, Mercuri NB, Bernardi G. L-Type calcium channels mediate a slow excitatory synaptic transmission in rat midbrain dopaminergic neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18:6693–6703. doi: 10.1523/JNEUROSCI.18-17-06693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Stotz SC, Spaetgens RL, Dayanithi G, Lemos J, Nargeot J, Zamponi GW. Interaction of SNX482 with domains III and IV inhibits activation gating of alpha(1E) (Ca(V)2.3) calcium channels. Biophysical Journal. 2001;81:79–88. doi: 10.1016/S0006-3495(01)75681-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broicher T, Kanyshkova T, Meuth P, Pape HC, Budde T. Correlation of T-channel coding gene expression, IT, and the low threshold Ca2+ spike in the thalamus of a rat model of absence epilepsy. Mol Cell Neurosci. 2008;39:384–399. doi: 10.1016/j.mcn.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Brown AM, Deutch AY, Colbran RJ. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. The European journal of neuroscience. 2005;22:247–256. doi: 10.1111/j.1460-9568.2005.04190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain SM, Snutch TP. Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 2010;4:475–482. doi: 10.4161/chan.4.6.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nature neuroscience. 2014;17:1022–1030. doi: 10.1038/nn.3743. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Saiardi A, Pisani A, Baik JH, Centonze D, Mercuri NB, Bernardi G, Borrelli E. Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:4536–4544. doi: 10.1523/JNEUROSCI.17-12-04536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Centonze D, Grande C, Saulle E, et al. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:8506–8512. doi: 10.1523/JNEUROSCI.23-24-08506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JE, Fields RD. CaMKII inactivation by extracellular Ca2+ depletion in dorsal root ganglion neurons. Cell Calcium. 2006;39:445–454. doi: 10.1016/j.ceca.2006.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D. The role of T-channels in the generation of thalamocortical rhythms. CNS Neurol Disord Drug Targets. 2006;5:571–585. doi: 10.2174/187152706779025526. [DOI] [PubMed] [Google Scholar]
- Coultrap SJ, Freund RK, O'Leary H, Sanderson JL, Roche KW, Dell'Acqua ML, Bayer KU. Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep. 2014;6:431–437. doi: 10.1016/j.celrep.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nature neuroscience. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Do J, Kim JI, Bakes J, Lee K, Kaang BK. Functional roles of neurotransmitters and neuromodulators in the dorsal striatum. Learning & Memory. 2013;20:21–28. doi: 10.1101/lm.025015.111. [DOI] [PubMed] [Google Scholar]
- Ehrlich M. Huntington's Disease and the Striatal Medium Spiny Neuron: Cell- Autonomous and Non-Cell-Autonomous Mechanisms of Disease. Neurotherapeutics. 2012;9:270–284. doi: 10.1007/s13311-012-0112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erondu NE, Kennedy MB. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1985;5:3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erxleben C, Liao Y, Gentile S, Chin D, Gomez-Alegria C, Mori Y, Birnbaumer L, Armstrong DL. Cyclosporin and Timothy syndrome increase mode 2 gating of CaV1.2 calcium channels through aberrant phosphorylation of S6 helices. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3932–3937. doi: 10.1073/pnas.0511322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshete F, Fields RD. Spike Frequency Decoding and Autonomous Activation of Ca2+–Calmodulin-Dependent Protein Kinase II in Dorsal Root Ganglion Neurons. The Journal of Neuroscience. 2001;21:6694–6705. doi: 10.1523/JNEUROSCI.21-17-06694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective Regulation of Neurite Extension and Synapse Formation by the β but not the α Isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Fitton A, Benfield P. Isradipine: A Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Use in Cardiovascular Disease. Drugs. 1990;40:31–74. doi: 10.2165/00003495-199040010-00004. [DOI] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. The Journal of physiology. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Goto S, Miyamoto E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J Neurochem. 1988;51:1070–1078. doi: 10.1111/j.1471-4159.1988.tb03070.x. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Kobayashi T, Tamura S, Miyamoto E. Dephosphorylation of autophosphorylated Ca2+/calmodulin-dependent protein kinase II by protein phosphatase 2C. The Journal of biological chemistry. 1993;268:133–137. [PubMed] [Google Scholar]
- Gao L, Blair LA, Salinas GD, Needleman LA, Marshall J. Insulin-like growth factor-1 modulation of CaV1.3 calcium channels depends on Ca2+ release from IP3-sensitive stores and calcium/calmodulin kinase II phosphorylation of the alpha1 subunit EF hand. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:6259–6268. doi: 10.1523/JNEUROSCI.0481-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ. Dichotomous anatomical properties of adult striatal medium spiny neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn AL, Yang Y. The potential role of the striatum in antisocial behavior and psychopathy. Biol Psychiatry. 2012;72:817–822. doi: 10.1016/j.biopsych.2012.04.027. [DOI] [PubMed] [Google Scholar]
- Gustin RM, Shonesy BC, Robinson SL, Rentz TJ, Baucum AJ, 2nd, Jalan-Sakrikar N, Winder DG, Stanwood GD, Colbran RJ. Loss of Thr286 phosphorylation disrupts synaptic CaMKIIalpha targeting, NMDAR activity and behavior in pre-adolescent mice. Molecular and cellular neurosciences. 2011;47:286–292. doi: 10.1016/j.mcn.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajimohammadreza I, Probert AW, Coughenour LL, Borosky SA, Marcoux FW, Boxer PA, Wang KK. A specific inhibitor of calcium/calmodulin-dependent protein kinase-II provides neuroprotection against NMDA- andhypoxia/hypoglycemia-induced cell death. The Journal of neuroscience: the officialjournal of the Society for Neuroscience. 1995;15:4093–4101. doi: 10.1523/JNEUROSCI.15-05-04093.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hell Johannes W. CaMKII: Claiming Center Stage in Postsynaptic Function and Organization. Neuron. 2014;81:249–265. doi: 10.1016/j.neuron.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. The Journal of Cell Biology. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Bargas J, Surmeier DJ, Reyes A, Galarraga E. D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:3334–3342. doi: 10.1523/JNEUROSCI.17-09-03334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. The Journal of Cell Biology. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Current Opinion in Neurobiology. 2012;22:496–508. doi: 10.1016/j.conb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Fujisawa H. A Novel Protein Phosphatase That Dephosphorylates and Regulates Ca2+/Calmodulin-dependent Protein Kinase II. Journal of Biological Chemistry. 1998;273:1904–1910. doi: 10.1074/jbc.273.4.1904. [DOI] [PubMed] [Google Scholar]
- Isope P, Murphy TH. Low threshold calcium currents in rat cerebellar Purkinje cell dendritic spines are mediated by T-type calcium channels. The Journal of physiology. 2005;562:257–269. doi: 10.1113/jphysiol.2004.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ, Lee A. Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:5125–5135. doi: 10.1523/JNEUROSCI.4367-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez C, Bourinet E, Leuranguer V, Sylvain R, Snutch TP, Nargeot J. Determinants of voltage-dependent inactivation affect Mibefradil block of calcium channels. Neuropharmacology. 2000;39:1–10. doi: 10.1016/s0028-3908(99)00153-7. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Fukunaga K, Muller D. Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:10645–10649. doi: 10.1523/JNEUROSCI.23-33-10645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends in Neurosciences. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kerr JND, Wickens JR. Dopamine D-1/D-5 Receptor Activation Is Required for Long-Term Potentiation in the Rat Neostriatum In Vitro. 2001;1 doi: 10.1152/jn.2001.85.1.117. [DOI] [PubMed] [Google Scholar]
- Kimm T, Bean BP. Inhibition of A-Type Potassium Current by the Peptide Toxin SNX-482. The Journal of Neuroscience. 2014;34:9182–9189. doi: 10.1523/JNEUROSCI.0339-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug JR, Mathur BN, Kash TL, et al. Genetic Inhibition of CaMKII in Dorsal Striatal Medium Spiny Neurons Reduces Functional Excitatory Synapses and Enhances Intrinsic Excitability. PloS one. 2012a;7:e45323. doi: 10.1371/journal.pone.0045323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug JR, Mathur BN, Kash TL, et al. Genetic inhibition of CaMKII in dorsal striatal medium spiny neurons reduces functional excitatory synapses and enhances intrinsic excitability. PloS one. 2012b;7:e45323. doi: 10.1371/journal.pone.0045323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. The Journal of biological chemistry. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- Koval OM, Guan X, Wu Y, et al. CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Physiology and pharmacology of striatal neurons. Annu Rev Neurosci. 2009;32:127–147. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007a;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007b;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Kunze DL, Rampe D. Characterization of the effects of a new Ca2+ channel activator, FPL 64176, in GH3 cells. Molecular pharmacology. 1992;42:666–670. [PubMed] [Google Scholar]
- Lee TS, Karl R, Moosmang S, Lenhardt P, Klugbauer N, Hofmann F, Kleppisch T, Welling A. Calmodulin kinase II is involved in voltage-dependent facilitation of the L-type Cav1.2 calcium channel: Identification of the phosphorylation sites. The Journal of biological chemistry. 2006;281:25560–25567. doi: 10.1074/jbc.M508661200. [DOI] [PubMed] [Google Scholar]
- Lengyel I, Cammarota M, Brent VA, Rostas JA. Autonomous activity and autophosphorylation of CAMPK-II in rat hippocampal slices: effects of tissue preparation. Journal of neurochemistry. 2001;76:149–154. doi: 10.1046/j.1471-4159.2001.00058.x. [DOI] [PubMed] [Google Scholar]
- Looi JCL, Walterfang M. Striatal morphology as a biomarker in neurodegenerative disease. Mol Psychiatry. 2013;18:417–424. doi: 10.1038/mp.2012.54. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Groth Rachel D, Cohen Samuel M, Emery John F, Li B, Hoedt E, Zhang G, Neubert Thomas A, Tsien Richard W. γCaMKII Shuttles Ca2+/CaM to the Nucleus to Trigger CREB Phosphorylation and Gene Expression. Cell. 2014;159:281–294. doi: 10.1016/j.cell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. The Journal of biological chemistry. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- Martin RL, Lee JH, Cribbs LL, Perez-Reyes E, Hanck DA. Mibefradil Block of Cloned T-Type Calcium Channels. Journal of Pharmacology and Experimental Therapeutics. 2000;295:302–308. [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O'Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- Mockett BG, Guevremont D, Wutte M, Hulme SR, Williams JM, Abraham WC. Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:7380–7391. doi: 10.1523/JNEUROSCI.6656-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi S, Shioda N, Yamamoto Y, Tagashira H, Fukunaga K. The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J Neurochem. 2012;121:44–53. doi: 10.1111/j.1471-4159.2012.07667.x. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Blatter LA, Bhat RV, Fiore RS, Wier WG, Baraban JM. Differential regulation of calcium/calmodulin-dependent protein kinase II and p42 MAP kinase activity by synaptic transmission. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1994;14:1320–1331. doi: 10.1523/JNEUROSCI.14-03-01320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Gittis AH, du Lac S. Decreases in CaMKII Activity Trigger Persistent Potentiation of Intrinsic Excitability in Spontaneously Firing Vestibular Nucleus Neurons. Neuron. 2005;46:623–631. doi: 10.1016/j.neuron.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb R, Szoke B, Palma A, et al. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Bosch M, Hayashi Y. The Roles of CaMKII and F-Actin in the Structural Plasticity of Dendritic Spines: A Potential Molecular Identity of a Synaptic Tag? 2009;6 doi: 10.1152/physiol.00029.2009. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. The Journal of biological chemistry. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Ouimet CC, McGuinness TL, Greengard P. Immunocytochemical localization of calcium/calmodulin-dependent protein kinase II in rat brain. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:5604–5608. doi: 10.1073/pnas.81.17.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Lambert NA, Prasad BM. Activity-dependent regulation of the dopamine transporter is mediated by Ca2+/calmodulin-dependent protein kinase signaling. European Journal of Neuroscience. 2008;28:2017–2027. doi: 10.1111/j.1460-9568.2008.06496.x. [DOI] [PubMed] [Google Scholar]
- Pakhotin P, Bracci E. Cholinergic Interneurons Control the Excitatory Input to the Striatum. The Journal of Neuroscience. 2007;27:391–400. doi: 10.1523/JNEUROSCI.3709-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Gardoni F, Centonze D, Mauceri D, Cenci MA, Bernardi G, Calabresi P, Di Luca M. Abnormal Ca2+-Calmodulin-Dependent Protein Kinase II Function Mediates Synaptic and Motor Deficits in Experimental Parkinsonism. The Journal of Neuroscience. 2004;24:5283–5291. doi: 10.1523/JNEUROSCI.1224-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planert H, Berger TK, Silberberg G. Membrane properties of striatal direct and indirect pathway neurons in mouse and rat slices and their modulation by dopamine. PloS one. 2013;8:e57054. doi: 10.1371/journal.pone.0057054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Surmeier DJ. Corticostriatal synaptic adaptations in Huntington's disease. Current opinion in neurobiology. 2015;33:53–62. doi: 10.1016/j.conb.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampe D, Anderson B, Rapien-Pryor V, Li T, Dage RC. Comparison of the in vitro and in vivo cardiovascular effects of two structurally distinct Ca++ channel activators, BAY K 8644 and FPL 64176. Journal of Pharmacology and Experimental Therapeutics. 1993;265:1125–1130. [PubMed] [Google Scholar]
- Roberts RC, Conley R, Kung L, Peretti FJ, Chute DJ. Reduced striatal spine size in schizophrenia: a postmortem ultrastructural study. Neuroreport. 1996;7:1214–1218. doi: 10.1097/00001756-199604260-00024. [DOI] [PubMed] [Google Scholar]
- Sametsky EA, Disterhoft JF, Ohno M. Autophosphorylation of αCaMKII downregulates excitability of CA1 pyramidal neurons following synaptic stimulation. Neurobiology of Learning and Memory. 2009;92:120–123. doi: 10.1016/j.nlm.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Lisman J. The CaMKII/NMDAR complex as a molecular memory. Molecular Brain. 2013;6:10. doi: 10.1186/1756-6606-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Carvalho AL, Caldeira MV, Duarte CB. Regulation of AM PA receptors and synaptic plasticity. Neuroscience. 2009;158:105–125. doi: 10.1016/j.neuroscience.2008.02.037. [DOI] [PubMed] [Google Scholar]
- Scholz WK, Palfrey HC. Activation of Ca2+/calmodulin-dependent protein kinase II by extracellular calcium in cultured hippocampal pyramidal neurons. Journal of neurochemistry. 1998;71:580–591. doi: 10.1046/j.1471-4159.1998.71020580.x. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nature neuroscience. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- Shields SM, Ingebritsen TS, Kelly PT. Identification of protein phosphatase 1 in synaptic junctions: dephosphorylation of endogenous calmodulin-dependent kinase II and synapse-enriched phosphoproteins. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1985;5:3414–3422. doi: 10.1523/JNEUROSCI.05-12-03414.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Wang X, Rose KL, et al. CaMKII regulates diacylglycerol lipase-alpha and striatal endocannabinoid signaling. Nature neuroscience. 2013 doi: 10.1038/nn.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine- binding properties of brain L-type calcium channel isoforms. Molecular pharmacology. 2009;75:407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- Stephens B, Mueller AJ, Shering AF, et al. Evidence of a breakdown of corticostriatal connections in Parkinson's disease. Neuroscience. 2005;132:741–754. doi: 10.1016/j.neuroscience.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. Journal of neurochemistry. 1997a;68:2119–2128. doi: 10.1046/j.1471-4159.1997.68052119.x. [DOI] [PubMed] [Google Scholar]
- Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential Inactivation of Postsynaptic Density-Associated and Soluble Ca2+/Calmodulin-Dependent Protein Kinase II by Protein Phosphatases 1 and 2A. Journal of Neurochemistry. 1997b;68:2119–2128. doi: 10.1046/j.1471-4159.1997.68052119.x. [DOI] [PubMed] [Google Scholar]
- Strack S, Choi S, Lovinger DM, Colbran RJ. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. The Journal of biological chemistry. 1997c;272:13467–13470. doi: 10.1074/jbc.272.21.13467. [DOI] [PubMed] [Google Scholar]
- Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. The Journal of biological chemistry. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proceedings of the National Academy of Sciences. 1992;89:10178–10182. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Reiner A, Levine MS, Ariano MA. Are neostriatal dopamine receptors co-localized? Trends in Neurosciences. 1993;16:299–305. doi: 10.1016/0166-2236(93)90103-s. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Kai L, Hockberger PE, Wokosin DL, Surmeier DJ. MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Molecular and Cellular Neuroscience. 2010;44:94–108. doi: 10.1016/j.mcn.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic SM, Lingle CJ. Pharmacological Properties of T-Type Ca2+ Current in Adult Rat Sensory Neurons: Effects of Anticonvulsant and Anesthetic Agents. 1998;1 doi: 10.1152/jn.1998.79.1.240. [DOI] [PubMed] [Google Scholar]
- Vest RS, O'Leary H, Coultrap SJ, Kindy MS, Bayer KU. Effective Post-insult Neuroprotection by a Novel Ca(2+)/ Calmodulin-dependent Protein Kinase II (CaMKII) Inhibitor. The Journal of biological chemistry. 2010;285:20675–20682. doi: 10.1074/jbc.M109.088617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Hao LY, Minobe E, Saud ZA, Han DY, Kameyama M. CaMKII phosphorylates a threonine residue in the C-terminal tail of Cav1.2 Ca(2+) channel and modulates the interaction of the channel with calmodulin. J Physiol Sci. 2009;59:283–290. doi: 10.1007/s12576-009-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ. A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:10116–10121. doi: 10.1523/JNEUROSCI.23-31-10116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Perez-Reyes E, Barrett PQ. Stimulation of recombinant Ca(v)3.2, T-type, Ca(2+) channel currents by CaMKIIgamma(C) The Journal of physiology. 2002;538:343–355. doi: 10.1113/jphysiol.2001.012839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Cline HT. Stabilization of Dendritic Arbor Structure in Vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 Controls Activity-Dependent Structural and Functional Plasticity of Dendritic Spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Davies LA, Howard JD, Adney SK, Welsby PJ, Howell N, Carey RM, Colbran RJ, Barrett PQ. Molecular basis for the modulation of native T-type Ca2+ channels in vivo by Ca2+/calmodulin-dependent protein kinase II. The Journal of clinical investigation. 2006;116:2403–2412. doi: 10.1172/JCI27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, Deutch AY, Montine TJ. Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology. 2005;64:545–547. doi: 10.1212/01.WNL.0000150591.33787.A4. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Snutch TP. Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. The Journal of membrane biology. 1996;151:77–90. doi: 10.1007/s002329900059. [DOI] [PubMed] [Google Scholar]
- Zheng H, Zeng Y, Chu J, Kam AY, Loh HH, Law PY. Modulations of NeuroD Activity Contribute to the Differential Effects of Morphine and Fentanyl on Dendritic Spine Stability. The Journal of Neuroscience. 2010;30:8102–8110. doi: 10.1523/JNEUROSCI.6069-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]