

Abstract
We have previously shown that vasa recta pericytes are known to dilate vasa recta capillaries in the presence of PGE2 and contract vasa recta capillaries when endogenous production of PGE2 is inhibited by the nonselective nonsteroidal anti-inflammatory drug (NSAID) indomethacin. In the present study, we used a live rat kidney slice model to build on these initial observations and provide novel data that demonstrate that nonselective, cyclooxygenase-1-selective, and cyclooxygenase -2-selective NSAIDs act via medullary pericytes to elicit a reduction of vasa recta diameter. Real-time images of in situ vasa recta were recorded, and vasa recta diameters at pericyte and nonpericyte sites were measured offline. PGE2 and epoprostenol (a prostacyclin analog) evoked dilation of vasa recta specifically at pericyte sites, and PGE2 significantly attenuated pericyte-mediated constriction of vasa recta evoked by both endothelin-1 and ANG II. NSAIDs (indomethacin > SC-560 > celecoxib > meloxicam) evoked significantly greater constriction of vasa recta capillaries at pericyte sites than at nonpericyte sites, and indomethacin significantly attenuated the pericyte-mediated vasodilation of vasa recta evoked by PGE2, epoprostenol, bradykinin, and S-nitroso-N-acetyl-l-penicillamine. Moreover, a reduction in PGE2 was measured using an enzyme immune assay after superfusion of kidney slices with indomethacin. In addition, immunohistochemical techiques were used to demonstrate the population of EP receptors in the medulla. Collectively, these data demonstrate that pericytes are sensitive to changes in PGE2 concentration and may serve as the primary mechanism underlying NSAID-associated renal injury and/or further compound-associated tubular damage.
Keywords: pericyte, vasa recta, nonsteroidal anti-inflammatory drugs, medulla
the kidney is the major organ responsible for the elimination of clinically prescribed drugs; hence, nephrotoxicity is a major complication of many commonly prescribed/administered medications and diagnostic agents. The clinical manifestations of nephrotoxicity are varied and differ between drugs and drug classes, with different compartments of the kidney being differentially affected (38, 48, 49). Examples of these drugs include antibiotics (7, 14, 17), antiretrovirals (33), anticancer agents (29, 59), contrast media (36, 62), immunosuppressants (4, 13, 32), and angiotensin-converting enzyme inhibitors as well as nonsteroidal anti-inflammatory drugs (NSAIDs) (64, 66).
NSAIDs are extensively prescribed for the treatment of acute and chronic pain and inflammatory diseases; this, in combination with the over-the-counter availability of these agents, means that nephrotoxic side effects are a significant concern when considering the number of patients at risk of developing renal injury as a consequence (63). NSAID-mediated nephrotoxicity is particularly prevalent in aged elderly patients and patients with renal and cardiovascular comorbidities such as hypertension, nephrotic syndrome, diabetes, congestive heart failure, and sepsis due to the existing strain on renal function (56). Current strategies to reduce NSAID-mediated nephrotoxicity are limited to a reduction in dose and identification of an appropriate therapeutic window while managing identifiable risk factors (20).
NSAID-mediated renal injury covers a broad spectrum of clinical manifestations, which include acute tubular necrosis, acute renal injury, tubulointerstitial nephritis, papillary necrosis due to reduced medullary blood flow (MBF) (1), hypertension, and salt and water retention, with the ultimate progression to chronic renal failure (60). The underlying pathophysiology of renal injury hinges on the inhibition of renal cyclooxygenase (COX) enzymes and the production of renal prostanoids, which play critical roles in many regulatory mechanisms within the kidney. Prostaglandins (PGs; PGI2, PGE2, and PGD2) elicit vasodilation of renal vessels to facilitate increased perfusion and redistribution of blood from the renal cortex to the renal medulla (40). PGE2 and PGI2 act at the glomerulus to regulate glomerular filtration rate (63), and, in the medulla, PGE2 acts to regulate transport of NaCl in the thick ascending limb of the loop of Henle and collecting duct to cause diuresis and naturesis (57, 58) while regulating vasa recta diameter via action at contractile pericytes (41, 54).
Numerous in vivo studies have previously described an NSAID-mediated reduction in MBF (1, 16, 19, 50); however, a cellular mechanism for this reduction in MBF has yet to be determined. The medullary capillary network is devoid of smooth muscle cells, and this has previously raised the question as to how this microcirculatory bed might be regulated independently of cortical blood flow (28). A combination of in vivo, in vitro, and ex vivo studies has since extensively identified the presence of contractile pericytes along vasa recta capillaries that serve the medulla and demonstrated their ability to regulate vasa recta diameter and thus MBF (8, 15, 39, 43, 44, 67). Moreover, our previous studies have focused on pericyte vasoactivity, demonstrating the ability of these cells to react to endogenous vasoactive substances (9–11).
Regulation of MBF is critical for the maintenance of corticomedullary gradients of NaCl and urea essential for urine concentration while ensuring appropriate O2 delivery and metabolic waste clearance. The consequence of these demanding local processes is a relatively hostile environment in a highly metabolic region of the kidney. Dysregulation of MBF leads to a reduction in the capacity of the kidney to concentrate urine with additional localized ischemia, leading to papillary necrosis and acute kidney injury, with long-term implications for the onset of interstitial fibrosis and chronic kidney disease (42, 47), most of which coincide with the clinical manifestations of NSAID-mediated nephrotoxicity. Pericytes themselves have a pivotal role in a variety of pathophysiological settings, which includes hypertension, fibrosis, diabetic nephropathy, and renal tumours (47), and have also recently featured as novel drug targets in other organ systems (6, 31). Furthermore, the vasoactivity of pericytes has been reported in other tissues (5, 22, 26, 27, 46, 51). Given that the kidney is a major site for drug elimination, we postulated that dysregulation/dysfunction of medullary pericytes contributes to the nephrotoxic side effects of many commonly prescribed drugs and, indeed, novel therapies in development. To this end, we used a live kidney slice model, in which the tubulovascular architecture remains intact, to investigate how a panel of NSAIDs affects the ability of pericytes to regulate vasa recta capillary diameter. Furthermore, we investigated whether the presence of other endogenous medullary vasodilators [nitric oxide (NO) and bradykinin] could attenuate the established NSAID-mediated constriction of vasa recta in situ.
MATERIALS AND METHODS
Animal experiments were conducted in accordance with national and institutional ethical and welfare standards and in compliance with the United Kingdom Home Office Scientific Procedures Act (1986). Adult male Sprague-Dawley rats (250–300 g) were euthanized by cervical dislocation, after which both kidneys were removed and decapsulated. Kidney slices were prepared as previously described (9, 10).
Functional experiments.
For functional experiments, live kidney slices were superfused with PGE2, the PGI2 analog epoprostenol, or the following panel of NSAIDs: indomethacin, SC-560, meloxicam, or celecoxib. Other agents superfused included ANG II, endothelin (ET)-1, bradykinin and the NO donor S-nitroso-N-acetyl-l-penicillamine (SNAP) as previously described (9, 10). Differential interference contrast images of pericytes on subsurface vasa recta capillaries were captured through a ×63 water-immersion objective. Pericytes were identified by their distinctive “bump on a log” morphology, as previously described (9, 46). Real-time video images of changes in vasa recta diameter were collected every 1 s by an attached Rolera XR charge-couple device camera and recorded using Image ProSoftware (Media Cybernetics, Maidenhead, UK). Video recordings were analyzed as previously described using public domain ImageJ software (National Institutes of Health; http://rsb.info.nih.gov).
Enzyme immunoassay.
The perfusate was collected from functional experiments (samples snap frozen in liquid nitrogen and stored at −80°C) where the nonselective NSAID indomethacin was superfused onto live kidney slices. The PGE2 content of these samples was assessed using commercial enzyme immunoassay kits (Cayman Chemicals, Cambridge Bioscience) as per the manufacturer's instructions.
Statistical analysis.
In all experiments, statistically significant differences between pericyte and nonpericyte sites were determined using a Student's t-test; P < 0.05 was considered significant. Similarly, in experiments in which kidney tissue was sequentially superfused with more than one compound, the pericyte-mediated change in vessel diameter evoked in response to exposure to one compound was compared with the pericyte-mediated change in vessel diameter evoked by exposure of tissue to the subsequent compounds. In these experiments, statistical significance was also determined using a Student's t-test, as described above. All values are expressed as means ± SE, n values represent numbers of pericytes (1 pericyte and 1 nonpericyte site per kidney slice), as variation observed in previous studies (10, 46) was between pericytes and not animals. All experiments were performed in at least three different animals.
RESULTS
We have previously used a live kidney slice model, in which endogenous tubulovascular architecture remains intact, to demonstrate that PGE2 acts at vasa recta pericytes to cause vasodilation and that indomethacin evoked a pericyte-mediated constriction of vasa recta (9). The data presented here elaborate on these initial observations.
Effect of prostanoids on in situ vasa recta diameter.
Superfusion of live kidney slices with PGE2 or the PGI2 analog epoprostenol (both at 1 and 10 μM) resulted in a measurable dilation of vasa recta capillaries at sites at which contractile pericytes reside, termed “pericyte sites.” The constriction evoked by both agents was significantly greater at pericyte sites (6.83 ± 1.01% with 1 μM PGE2, 8.60 ± 1.58% with 10 μM PGE2, 9.8 ± 0.83% with 1 μM epoprostenol, and 11.49 ± 1.79% with 10 μM epoprostenol, n ≤ 107 slices and n ≥ 4 animals, P < 0.01; Fig. 1) compared with nonpericyte sites along the capillary (1.44 ± 0.73% with 1 μM PGE2, 1.43 ± 0.45% with 10 μM PGE2, 1.0 ± 0.83% with 1 μM epoprostenol, and 1.23 ± 0.78% with 10 μM epoprostenol; Fig. 1). Increasing concentrations of PGE2 and epoprostenol did not significantly increase the pericyte-mediated dilation of vasa recta; lower concentrations of both agents failed to elicit an effect on vessel diameter. Furthermore, no measureable changes in tubule diameter or tubule cell diameter were measured in response to either compound.
Fig. 1.
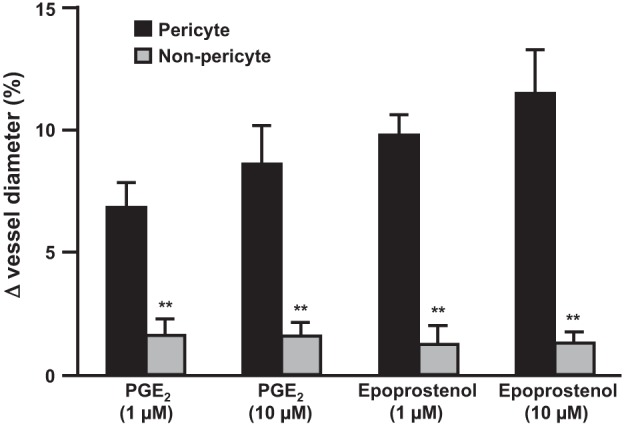
PGE2 and epoprostenol act specifically at pericytes to regulate in situ vasa recta diameter. The bar graph shows percent changes in vasa recta diameter at pericyte and nonpericyte sites. PGE2 (1 and 10 μM) and epoprostenol (1 and 10 μM) evoked a significantly greater dilation of vasa recta at pericyte sites than at nonpericyte sites. Values are means ± SE; n ≤ 7 slices and n ≥ 4 animals. **P < 0.01.
In an attempt to recapitulate an aspect of in vivo renal hemodynamics, vasa recta capillaries were preconstricted with either ET-1 or ANG II and then superfused with ET-1 or ANG II in the presence of PGE2. As previously demonstrated (9), both ET-1 and ANG II evoked a pericyte-mediated constriction of vasa recta capillaries [7.51 ± 0.84% (Fig. 2A) and 12.28 ± 1.19% (Fig. 2B), respectively, n ≤ 6 slices and n = 4 animals]. When preconstricted vasa recta were superfused with ET-1 in the presence of PGE2, the pericyte-mediated constriction of vasa recta was attenuated, resulting in total vasodilation (∼10%; Fig. 2A). Subsequent removal of PGE2 from the perfusate resulted in a second ET-1-evoked constriction of vasa recta at pericyte sites (∼10%; Fig. 2A). As for ET-1, the pericyte-mediated constriction of vasa recta evoked by ANG II was attenuated when PGE2 was included in the perfusate (∼14% total change in vessel diameter; Fig. 2B), and removal of PGE2 resulted in a second ANG II-evoked constriction (9.7 ± 1.26%; Fig. 2B). Thus, PGE2 significantly attenuated the ET-1- and ANG II-mediated constriction of vasa recta by pericytes (P < 0.05).
Fig. 2.
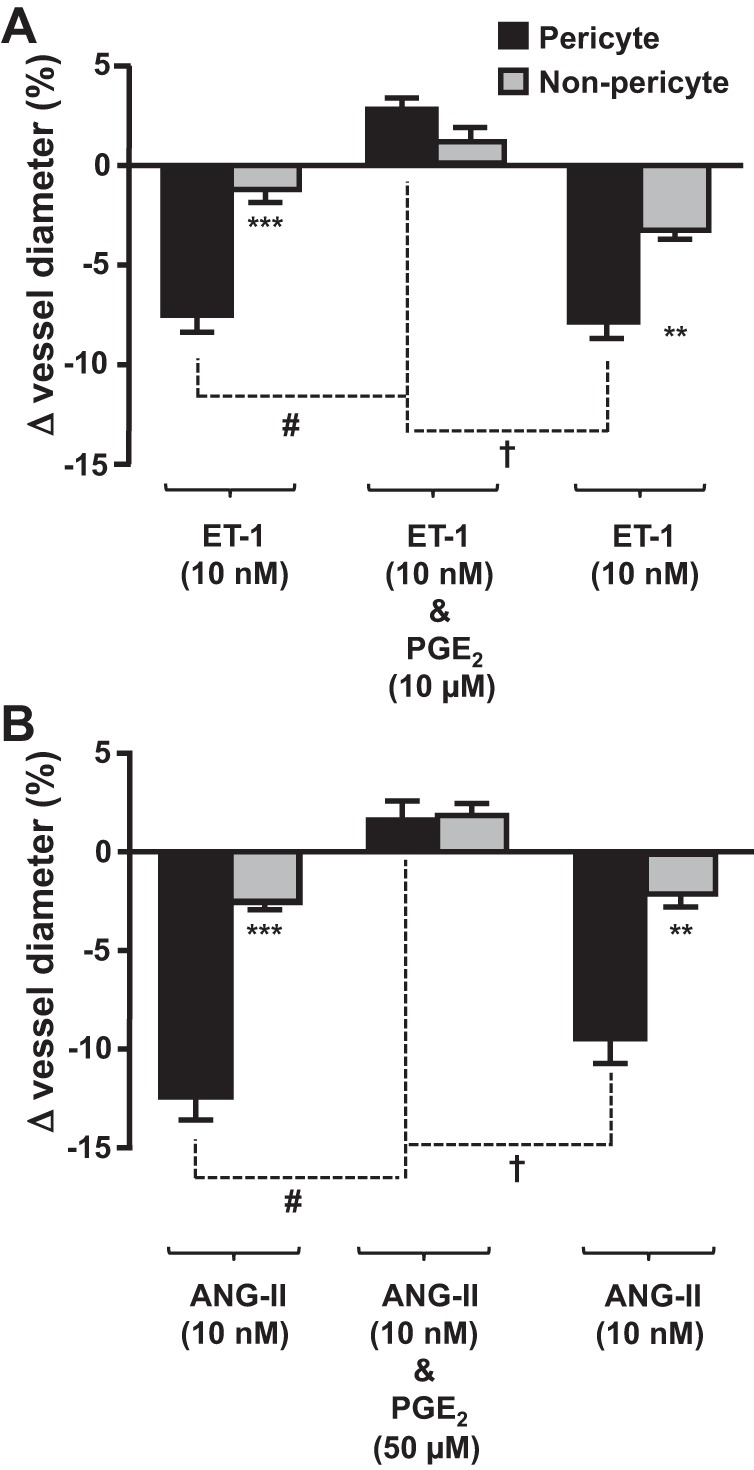
PGE2 attenuates the endothelin (ET)-1- and ANG II-evoked vasoconstriction of in situ vasa recta by pericytes. Bar graphs show percent changes in vasa recta diameter at pericyte and nonpericyte sites. ET-1 (10 nM; A) and ANG II (10 nM; B) evoked significantly (***) greater constriction at pericytes sites than at nonpericyte sites. The addition of PGE2 [10 μM (A) and 50 μM (B)] significantly (#) attenuated the pericyte-mediated vasoconstriction evoked by ET-1and ANG II, respectively. After removal of PGE2, ET-1 and ANG II alone evoked a second vasoconstriction, indicating that the effect of PGE2 was reversible. No significant change in vessel diameter was observed at nonpericyte sites. Values are means ± SE; n ≥ 6 slices and n ≥ 4 animals. **P < 0.01; ***P < 0.001; #P < 0.05; †P < 0.05.
Effects of NSAIDs on vasa recta diameter in situ.
Expression of COX-1 and COX-2 enzymes varies throughout the kidney in both healthy and inflamed tissue, and, as such, kidney slices were exposed to a range of nonselective, COX-1-selective, and COX-2-selective NSAIDs. Superfusion of live kidney slices with indomethacin (nonselective), SC-560 (COX-1 selective), meloxicam (COX-2 preferential), or celecoxib (COX-2 selective) resulted in a significantly greater vasoconstriction of vasa recta at pericyte sites (30 μM indomethacin: 9.86 ± 0.55%, n = 13 slices and n = 5 animals, P < 0.001), SC 560 (100 μM, 10.19 ± 0.31%, n = 7 slices and n = 5 animals, P < 0.001), meloxicam (1 mM, 9.19 ± 0.59%, n = 9 slices and n = 5 animals, P < 0.001), celecoxib (300 μM, 11.71 ± 0.97%, n = 5 slices and n = 4 animals, P < 0.001) compared with nonpericyte sites (Fig. 3). The concentration of each NSAID tested above was the minimum concentration found to elicit a measurable change in vessel diameter in the live kidney slice preparation. The relationship among the concentrations used in the present study, the therapeutic dose, and the amount the kidney is likely to be exposed to are shown in Table 1 and considered in the discussion below.
Fig. 3.

Superfusion of live kidney tissue with nonselective nonsteroidal anti-inflammatory drugs (NSAIDs) caused pericyte-mediated constriction of vasa recta. Representative traces (A,i–D,i) of percent changes in vessel diameter recorded over time at pericyte sites (black lines) and nonpericyte sites (shaded lines) when kidney slices were exposed to either indomethacin (30 μM; A,i), SC-560 (100 μM; B,i), meloxicam (1 mM; C,i), or celecoxib (300 μM; D,i) are shown. Corresponding differential interference contrast images of pericyte (P) and nonpericyte sites before drug exposure (ii), during superfusion of the drug (iii), and after exposure, during washout of the drug (iv), for each NSAID used [indomethacin (A), SC-560 (B), meloxicam (C), and celecoxib (D)] are also shown. Pericytes are denoted by black dotted circles; red dotted lines and yellow dotted lines indicate where changes in vessel diameter were measured at pericyte sites and nonpericyte sites, respectively. E: superfusion of either indomethacin (30 μM), SC-560 (100 μM), meloxicam (1 mM), or celecoxib (300 μM) onto live kidney slices evoked a significantly greater constriction of vasa recta at pericyte sites compared with nonpericyte sites. NS, nonselective; COX-1, cyclooxygenase-1 specific; COX-2 S, cyclooxygenase-2 selective; COX-2, cyclooxygenase-2 specific. Values are means ± SE; n ≥ 5 slices and n ≥ 4 animals. ***P < 0.001.
Table 1.
Comparison of clinical nonselective nonsteroidal anti-inflammatory drug doses with doses used in functional experiments
| Compound | Tablet/Capsule Concentration (Active), mg | 5% of Tablet/Capsule Concentration (Potential Renal Exposure During Excretion), mg | Concentration Used in Functional Experiments |
|---|---|---|---|
| Meloxicam | 7.5 | 0.38 | 2.45 mg (1 mM) |
| 15 | 0.75 | ||
| Indomethacin | 25 | 1.25 | 0.075 mg (30 μM) |
| 50 | 2.5 | ||
| Celecoxib | 50 | 2.5 | 0.81 mg (300 μM) |
| 100 | 5 | ||
| 50 | 10 | ||
| 400 | 20 |
Renal exposure to clinical nonselective nonsteroidal anti-inflammatory drug doses was extrapolated and compared with concentrations used in functional experiments.
Both COX-1-selective and COX-2 selective NSAIDs evoked pericyte-mediated vasoconstriction in the absence of an inflammatory stimulus. Moreover, there was no significant difference in the periciyte-mediated constriction measured in response to all agents tested despite the literature suggesting COX-2-selective NSAIDs to be less nephrotoxic. Inhibition of the production of vasodilatory prostanoids via both COX-1 and COX-2 elicited a comparable pericyte-mediated constriction of vasa recta capillaries, which was the same order of magnitude as that measured in response to bath application of other renal vasoconstrictor agents (ET-1, ATP, and N-nitro-l-arginine methyl ester) (9, 10). Given that both COX-1-selective and COX-2-selective NSAIDs elicited a comparable constriction of vasa recta via pericytes, the nonselective NSAID indomethacin was used in subsequent experiments to inhibit both subtypes of the COX enzyme. To further corroborate the nonselective action of indomethacin, using immunohistochemical techniques, we identified both subtypes of COX enzymes in the medulla in close proximity to the vasculature. Furthermore, enzyme immunoassay experiments confirmed that the indomethacin (nonselective NSAID)-induced vasoconstriction observed in functional experiments was due to a significant decrease in measurable PGE2 in the perfusate (10.61 ± 2.24%, n = 7, P < 0.01; Fig. 4).
Fig. 4.
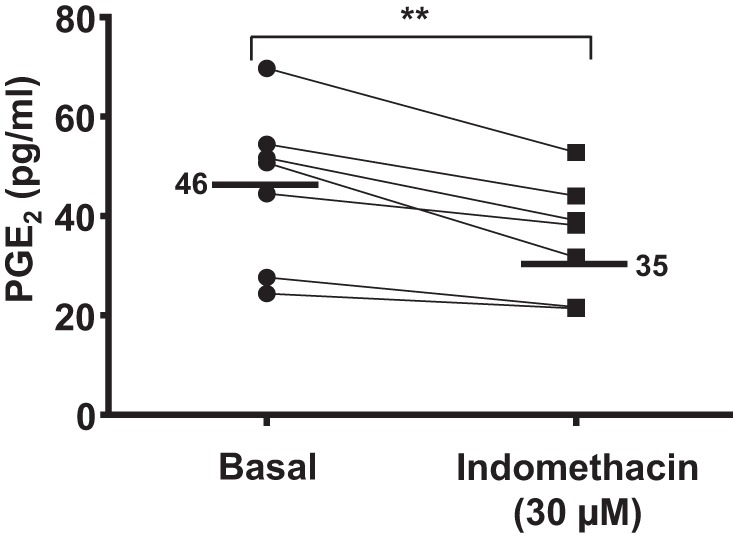
PGE2 concentration in the experimental perfusate before and during exposure to the nonselective NSAID indomethacin. The line graph shows the basal concentration of PGE2 and the PGE2 concentration in the perfusate during exposure to indomethacin (30 μM). Mean basal and indomethacin-exposed values are denoted by black lines. In the presence of indomethacin, there was a significant reduction in PGE2 compared with basal levels. n = 7 slices and n = 4 animals. **P < 0.01.
Determination of the potency of NSAID-mediated vasoconstriction.
Bradykinin and NO both stimulate COX activity and contribute to PGE2-mediated vasodilation (45, 52, 53). To examine the potential interactions between endogenous vasodilators and NSAIDs, live kidney slices were exposed to indomethacin (nonselective NSAID) in the presence of bradykinin or SNAP to stimulate NO production. Pericyte-mediated vasodilation in response to bradykinin and SNAP [9.3 ± 2.05% (Fig. 5A) and 8.64 ± 1.66% (Fig. 5B), respectively, n ≤ 8 slices and n ≤ 6 animals, P < 0.001] was significantly attenuated when indomethacin was included in the perfusate (total change in vessel diameter: ∼10% and ∼10%, respectively), resulting in pericyte-mediated constriction (Fig. 5B). This constriction was reversed upon washout of indomethacin, resulting in subsequent pericyte-mediated vasodilation in the presence of bradykinin and SNAP [8.3 ± 1.6% (Fig. 5A) and 10.0 ± 1.0% (Fig. 5B), respectively]. The time taken for indomethacin to significantly attenuate the pericyte-mediated bradykinin- and SNAP-evoked dilation was significantly greater (51.53 ± 6.23 s, P < 0.01) than the time taken for indomethacin to elicit a pericyte-mediated dilation of naïve vasa recta.
Fig. 5.
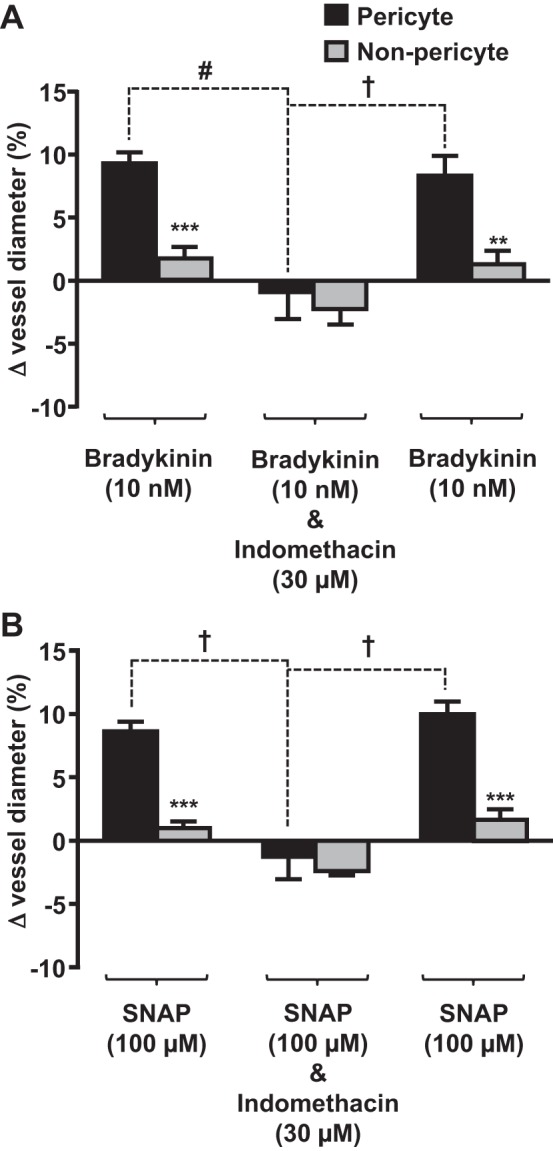
Indomethacin acts at pericyte sites to attenuate bradykinin- and S-nitroso-N-acetyl-l-penicillamine (SNAP)-evoked vasodilation of in situ vasa recta. Bar graphs show percent changes in vasa recta diameter at pericyte and nonpericyte sites. Bradykinin (10 nM; A) and SNAP (10 μM; B) evoked significantly (***) greater dilation at pericyte sites than at nonpericyte sites. The addition of indomethacin (30 μM; A and B) significantly (#) attenuated the pericyte-mediated vasodilation evoked by bradykinin and SNAP. After removal of indomethacin, vasodilation was observed in the presence of bradykinin and SNAP alone, indicating that the effect of indomethacin was reversible. No significant change in vessel diameter was observed at nonpericyte sites. Values are means ± SE; n ≤ 9 slices and n ≤ 8 animals. **P < 0.01; ***P < 0.001; #P < 0.01; †P < 0.001.
To investigate what might occur if it were possible to stimulate in vivo synthesis of PGE2 or PGI2, in the presence of indomethacin, live kidney slices were superfused with indomethacin in the presence of PGE2 (Fig. 6A) or epoprostenol (Fig. 6B). In parallel experiments, vasa recta capillaries were initially constricted by pericytes when live tissue was superfused with indomethacin (6.25 ± 0.43 and 7.87 ± 0.49; Fig. 6, A and B). In the presence of PGE2 or epoprostenol, the indomethacin-mediated constriction was attenuated, resulting in a pericyte-mediated dilation of vasa recta (3.41 ± 1.21% and 1.02 ± 0.73% from baseline, respectively; Fig. 6), revealing a total change in vessel diameter of ∼10% and ∼9%, respectively. Removal of exogenous PGE2 and epoprostenol resulted in an indomethacin-mediated restoration of pericyte-evoked constriction of vasa recta [6.97 ± 0.72%, n = 6 slices and n = 5 animals (Fig. 6A), and 8.07 ± 0.38%, n = 4 slices and n = 3 animals (Fig. 6B)]. Little to no change in vasa recta diameter was measured at nonpericyte sites (Fig. 6).
Fig. 6.
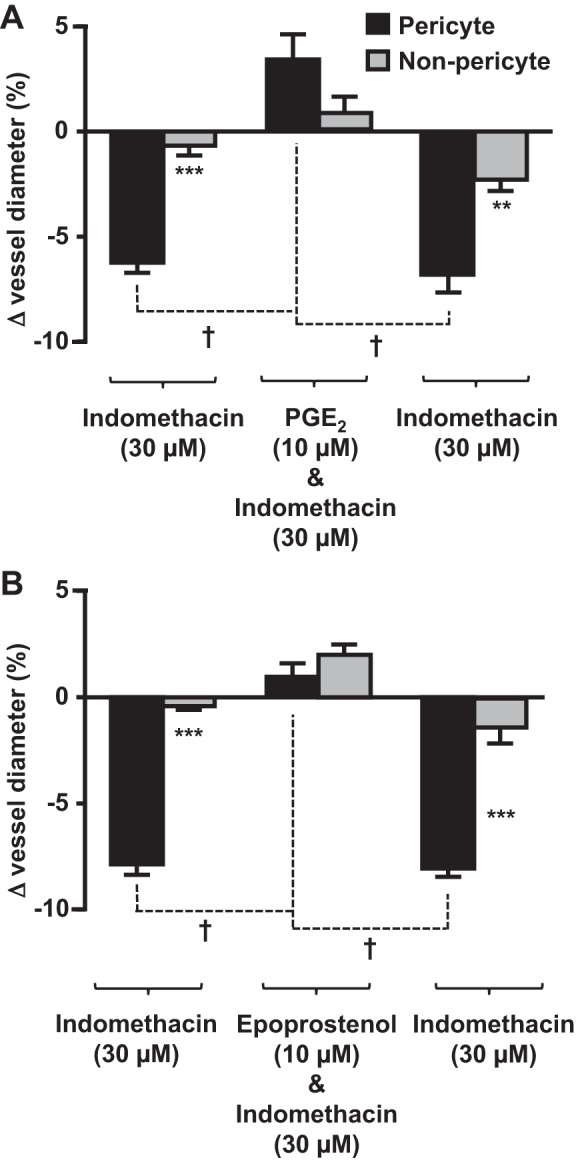
PGE2 and epoprostenol act at pericyte sites to attenuate indomethacin-evoked vasoconstriction of in situ vasa recta. Bar graphs show percent changes in vasa recta diameter at pericyte and nonpericyte sites. Indomethacin (30 μM) evoked a significantly (***) greater constriction at pericyte sites than at nonpericyte sites (A and B). The addition of PGE2 (10 μM; A) and epoprostenol (10 μM; B) significantly (#) attenuated the pericyte-mediated vasoconstriction evoked by indomethacin. After removal of PGE2 (A) and epoprosentol (B), vasoconstriction (†) was observed in the presence of indomethacin alone, indicating that the effects of PGE2 and epoprostenol were reversible. No significant change in vessel diameter was observed at nonpericyte sites. Values are means ± SE; n ≤ 6 slices and n ≤ 5 animals. **P < 0.01; ***P < 0.001; †P < 0.001.
DISCUSSION
NSAIDs have long been associated with renal injury, although the precise mechanisms underlying this associated renal injury have yet to be fully delineated. NSAIDs themselves are not vasoactive agents but act to inhibit COXs and the synthesis and bioavailability of vasodilatory PGs. Clinically, this has the beneficial effect of reducing pain and inflammation and the detrimental effect of dysregulating MBF due to the reduced bioavailability of PGE2 and PGI2 (23).
The data presented here identify contractile pericytes as the key cell type responsible for 1) increasing vasa recta diameter when live tissue slices were exposed to exogenous PGE2 and PGI2 (epoprostanol) and 2) decreasing vasa recta diameter when live tissue was exposed to a panel of nonselective, COX-1-selective, and COX-2- selective NSAIDs (Figs. 1 and 3). PGE2 is a potent vasodilator in the medulla and, as such, can attenuate ET-1 and ANG II-mediated vasoconstriction (Fig. 2) of vasa recta by pericytes. This may be important in vivo as a reduction in bioavailable PGE2, due to NSAID-mediated inhibition of COXs, would result in a pericyte-mediated decrease in vasa recta diameter and an associated reduction in MBF. We have previously described how pericyte-mediated regulation of vasa recta diameter is thought to underlie the changes in MBF that can occur in both physiological and pathophysiological settings (9, 10). Given that PGE2 is the most prominent prostanoid expressed in the medulla and its key role in both regulation of MBF and NaCl transport in the loop of Henle, the consequences of reduced production of PGE2 are significant for kidney function. The NSAID-mediated reduction in vasa recta diameter observed here may indeed be due to a reduction in the bioavailability of vasodilatory PGE2 but may also involve the shunting of arachidonic acid (AA) metabolism via alternative enzymes such as lipooxygenases or cytochrome P-450s, resulting in the production of vasoconstrictors such as leukotrienes and hydroxyeicosatetraenoic acid (see Fig. 7) (30, 55). Additionally, AA can be nonenzymatically metabolized, leading to the production of PGE2α, which also acts as a vasoconstrictor (35). While measuring the production of multiple AA metabolites was beyond the remit of this study, the reduced production of PGE2 in the perfusate collected during functional experiments, during which live kidney slices were superfused with indomethacin, confirms that the reduced bioavailability of vasodilatory PGE2 is a key component of the vasoconstriction observed.
Fig. 7.

Schematic of the proposed mechanism for the NSAID-induced reduction in vasa recta diameter. Treatment with NSAIDs shunts the metabolism of arachidonic acid (AA) from the COX pathway to other pathways, which results in the production of alternative vasoconstrictor agents. Increased availability of agonists favoring vasoconstriction leads to a reduction in mean blood blow (MBF). Whether these agonists act directly on pericytes or indirectly via endothelial signaling has yet to be established; however, the data presented here indicate that pericytes are the key players in mediating the reduction of vasa recta diameter. LOX, lipooxgenase; Cyt P450, cytochrome P-450; LB4, leukotriene B4; 5-HETE, 5-hydroxyeicosatetraenoic acid; PDG2α, PGE2α.
The data presented here demonstrate that prostanoids act specifically at pericytes to increase capillary diameter and that NSAID-mediated inhibition of endogenous prostanoids can reduce vasa recta capillary diameter via renal pericytes. In addition, these data demonstrate that the effect of inhibiting the production of PGE2 in the renal medulla cannot be overcome by exogenous application of endogenous vasodilators (bradykinin and SNAP) regardless of whether they act specifically at pericytes or mediate secondary production of PGE2. Given the location of COX-1 and COX-2 in the medulla (12) (see Fig. 8) and the endothelial expression of EP receptors (25) (see Fig. 9), we hypothesize that epithelial cell-derived PGE2 acts at endothelial EP receptors and that endothelial-pericyte cell communication [via previously defined mechanisms such as increased K+ channel activity (5) or Ca2+ signaling between cell types (68)] culminates in pericyte-mediated vasodilation of vasa recta. Further work is undoubtedly required to validate this hypothesis, but given the lack of contractile machinery expression in endothelial cells and the spatially localized constriction of vasa recta at pericyte sites, this hypothesis certainly seems plausible. The ability of indomethacin to mediate pericyte-specific vasoconstriction beyond baseline diameter in the presence of SNAP and bradykinin suggests alternative metabolites of AA might additionally act at pericytes to further constrict vasa recta. This is, of course, merely speculation on our part, as measuring the production of a vast array of potential metabolites was beyond the scope of this study. Moreover, it should be noted that in vivo many other factors, such as capillary and tubular flow, pH, and spatial changes in O2 tension and osmolality, may also influence the ability of SNAP and bradykinin to evoke vasodilation in the presence of a COX inhibitor. However, the effect these parameters may confer on pericyte-mediated changes in vessel diameter cannot be assessed in the slice model.
Fig. 8.

Confocal images of COX-1 and COX-2 expression in the renal medulla. Before fixation with paraformaldehyde, vasa recta capillaries of live kidney slices were labeled with Alexa 488-conjugated IB4 (green; A,i and B,i); outlines of the vessels are indicated by arrows, the potential location of pericytes, identified by their bump-on-a-log morphology, on vessels are indicated by arrowheads. A,ii: postfixation, COX-1 expression was labeled with anti-COX-1 primary antibody and probed with Alexa 555-conjugated secondary antibody (red). B,ii: COX-2 expression was labeled with anti-COX-2 primary antibody and probed with Alexa 555-conjugated secondary antibody (red). Both COX-1 and COX-2 were located on the apical membrane of tubular cells in the renal medulla (A,ii and B,ii, respectively). *Location of tubule cells. A,iii: overlay of images of A,i and ii, showing the localization of COX-1 (red) and vasa recta. B,iii: overlay of images of B,i and ii, showing the localization of COX-2 (red) and vasa recta (green). n = 6 slices and n = 3 animals.
Fig. 9.

Confocal images of EP2 and EP4 receptor expression in the renal medulla. Before fixation with paraformaldehyde, vasa recta capillaries of live kidney slices were labeled with Alexa 488-conjugated IB4 antibody (green; A,i and B,i). A,ii: EP2 receptor expression was labeled with anti-EP2 primary antibody and probed with Alexa 555-conjugated secondary antibody (red). A,iii: overlay of images of A,i and ii, showing the close proximity of vasa recta (green) and EP2 receptor expression (red). B,ii: EP4 receptor expression was labeled with anti-EP4 primary antibody and probed with Alexa 555-conjugated secondary antibody (red). B,iii: overlay of images of B,i and ii, suggesting colocalization of vasa recta (green) and EP4 receptor expression (red). n = 6 slices and n = 3 animals.
While the concentration of PGE2 used in this study seems high (1–10 μM) compared with the physiological concentration of PGE2 in the kidney (∼10 nM), the vessels imaged in this study reside ∼50 μm below the surface of the tissue, and determination of the concentration of the vasoactive agent at that depth is not possible. Furthermore, endogenous PGE2 is susceptible to oxidation by 15-hydroxyprostaglandin dehydrogenase and is readily absorbed by epithelial cells in the cortical and medullary thick ascending limb of the loop of Henle and proximal tubule (24), thus reducing the bioavailability of PGE2 as a result. The concentration of NSAIDs used in our experiments was selected primarily with the aim of inducing a measurable function effect while working within the therapeutic dose window. Meloxicam, for example, is prescribed as 7.5- or 15-mg tablets (3); taking into account metabolism of the drug in the liver, the kidneys would be exposed to ∼5% of the original dose, which equates to 0.38 and 0.75 mg, respectively (see Table 1). In the present study, bath application (as opposed to intraluminal) of 1 mM meloxicam, which equates to 2.45 mg, evoked a 9.2% vasoconstriction of vasa recta at pericyte sites. Taking into account the reduced concentration of meloxicam at subsurface vessels, the concentration ranges used for meloxicam and other NSAIDS (see Table 1 for concentration and therapeutic dose comparisons) used in this study seem appropriate.
To further contextualize the implication for our findings in a clinical setting, we considered the following: kidneys remove 50–90% of conjugated metabolites and between trace and 50% of the active compound depending on the extent of liver metabolism; thus, in vivo, it is likely that the kidneys could be exposed to toxic levels of active NSAID compounds (37). The resultant chronic suppression of PGE2 synthesis and ensuing pericyte-mediated constriction of vasa recta can ultimately lead to localized ischemia. Pericytes constrict irreversibly under ischemic conditions (21, 46), and, as such, long-term irreversible damage will likely ensue, resulting in chronic disease manifestation. Reciprocally, renal excretion of NSAIDs and their metabolites relies fundamentally on efficient filtration and tubular secretion, and the chronic reduction in renal blood flow and filtration would therefore further hinder appropriate elimination of NSAIDs, leading to their accumulation in the blood (18, 61, 65). The metabolism and excretion of NSAIDs are highly dependent on organ systems operating optimally, which in a clinical setting might seldom be a reality due to preexisting health complications and/or comorbidity. In addition to poor elimination, inactive NSAID metabolites can become reactivated in the kidney, thus exposing the kidney to even higher concentrations of active compounds (34).
Collectively, the data presented here identify pericyte-mediated constriction of vasa recta as the cellular mechanism underlying the previously reported in vivo reduction in MBF (2). The pericyte-mediated decrease in vasa recta diameter evoked by both selective and nonselective NSAIDs described here provides a novel mechanism for NSAID-mediated nephrotoxicity, and we conclude that the pericyte-mediated changes in vasa recta diameter are likely to significantly influence renal function and the onset of long-term disease (42, 47). Given the borderline hypoxic environment of the medulla, a decrease in vessel diameter would invariably have a severe impact on blood flow to this region. Although measurement of MBF was beyond the scope of this study, the observed reductions in vessel diameter strongly suggest that there would be a concomitant reduction in blood flow in an in vivo setting. Furthermore, our data demonstrate the sensitivity of pericytes in the medulla to changes in PG production and robustly show the vascular response to fluxes in PG production. Whether this novel mechanism represents the primary nephrotoxic mechanisms or is secondary to tubular toxicity now requires clarification, and, as such, this will be the focus of future studies.
GRANTS
This work was supported by the Medical Research Council.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: T.M.K.-L. and C.C. performed experiments; T.M.K.-L., C.C., and C.M.P.-W. analyzed data; T.M.K.-L. and C.M.P.-W. interpreted results of experiments; T.M.K.-L. and C.M.P.-W. prepared figures; T.M.K.-L. drafted manuscript; T.M.K.-L., S.S.W., and C.M.P.-W. approved final version of manuscript; S.S.W. and C.M.P.-W. conception and design of research; S.S.W. and C.M.P.-W. edited and revised manuscript.
ACKNOWLEDGMENTS
Present address of T. Kennedy-Lydon: National Heart and Lung Institute, Imperial College London, London, UK.
REFERENCES
- 1.Agmon Y, Brezis M. Effects of nonsteroidal anti-inflammatory drugs upon intrarenal blood flow: selective medullary hypoperfusion. Exp Nephrol 1: 357–363, 1993. [PubMed] [Google Scholar]
- 2.Agmon Y, Dinour D, Brezis M. Disparate effects of adenosine A1- and A2-receptor agonists on intrarenal blood flow. Am J Physiol Renal Fluid Electrolyte Physiol 265: F802–F806, 1993. [DOI] [PubMed] [Google Scholar]
- 3.British National Formulary. British National Formulary (online). http://www.bnf.org/bnf/index.htm [3 August 2015]. [Google Scholar]
- 4.Burdmann EA, Andoh TF, Yu L, Bennett WM. Cyclosporine nephrotoxicity. Semin Nephrol 23: 465–476, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Burnette JO, White RE. PGI2 opens potassium channels in retinal pericytes by cyclic AMP-stimulated, cross-activation of PKG. Exp Eye Res 83: 1359–1365, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, Dunner K, Pati S, Bankson JA, Pasqualini R, Arap W, Bryan NS, Taegtmeyer H, Langley RR, Yao H, Kupferman ME, Entman ML, Dickinson ME, Khakoo AY. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med 5: 187ra169, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa S, Nucci M. Can we decrease amphotericin nephrotoxicity? Curr Opin Crit Care 7: 379–383, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Cowley AW, Mori T, Mattson D, Zou AP. Role of renal NO production in the regulation of medullary blood flow. Am J Physiol Regul Integr Comp Physiol 284: R1355–R1369, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Crawford C, Kennedy-Lydon T, Sprott C, Desai T, Sawbridge L, Munday J, Unwin RJ, Wildman SS, Peppiatt-Wildman CM. An intact kidney slice model to investigate vasa recta properties and function in situ. Nephron Physiol 120: p17–p31, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crawford C, Kennedy-Lydon TM, Callaghan H, Sprott C, Simmons RL, Sawbridge L, Syme HM, Unwin RJ, Wildman SS, Peppiatt-Wildman CM. Extracellular nucleotides affect pericyte-mediated regulation of rat in situ vasa recta diameter. Acta Physiol (Oxf) 202: 241–251, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Crawford C, Wildman SS, Kelly MC, Kennedy-Lydon TM, Peppiatt-Wildman CM. Sympathetic nerve-derived ATP regulates renal medullary vasa recta diameter via pericyte cells: a role for regulating medullary blood flow? Front Physiol 4: 307, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Câmpean V, Theilig F, Paliege A, Breyer M, Bachmann S. Key enzymes for renal prostaglandin synthesis: site-specific expression in rodent kidney (rat, mouse). Am J Physiol Renal Physiol 285: F19–F32, 2003. [DOI] [PubMed] [Google Scholar]
- 13.de Mattos AM, Olyaei AJ, Bennett WM. Nephrotoxicity of immunosuppressive drugs: long-term consequences and challenges for the future. Am J Kidney Dis 35: 333–346, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Deray G. Amphotericin B nephrotoxicity. J Antimicrob Chemother 49, Suppl 1: 37–41, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Dickhout JG, Mori T, Cowley AW. Tubulovascular nitric oxide crosstalk: buffering of angiotensin II-induced medullary vasoconstriction. Circ Res 91: 487–493, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Dunham EW, Zimmerman BG. Release of prostaglandin-like material from dog kidney during nerve stimulation. Am J Physiol 219: 1279–1285, 1970. [DOI] [PubMed] [Google Scholar]
- 17.Fanos V, Cataldi L. Amphotericin B-induced nephrotoxicity: a review. J Chemother 12: 463–470, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Gambertoglio JG, Aweeka FT, Blythe WB. Use of Drugs in Patients With Renal Failure. Boston, MA: Little Brown, 1993. [Google Scholar]
- 19.Green T, Gonzalez AA, Mitchell KD, Navar LG. The complex interplay between cyclooxygenase-2 and angiotensin II in regulating kidney function. Curr Opin Nephrol Hypertens 21: 7–14, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo X, Nzerue C. How to prevent, recognize, and treat drug-induced nephrotoxicity. Cleve Clin J Med 69: 284–296, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508: 55–60, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton N, Vayro S, Wigley R, Butt AM. Axons and astrocytes release ATP and glutamate to evoke calcium signals in NG2-glia. Glia 58: 66–79, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol 70: 357–377, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Hatano R, Onoe K, Obara M, Matsubara M, Kanai Y, Muto S, Asano S. Sex hormones induce a gender-related difference in renal expression of a novel prostaglandin transporter, OAT-PG, influencing basal PGE2 concentration. Am J Physiol Renal Physiol 302: F342–F349, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Jensen BL, Stubbe J, Hansen PB, Andreasen D, Skøtt O. Localization of prostaglandin E2 EP2 and EP4 receptors in the rat kidney. Am J Physiol Renal Physiol 280: F1001–F1009, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Kawamura H, Kobayashi M, Li Q, Yamanishi S, Katsumura K, Minami M, Wu DM, Puro DG. Effects of angiotensin II on the pericyte-containing microvasculature of the rat retina. J Physiol 561: 671–683, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawamura H, Sugiyama T, Wu DM, Kobayashi M, Yamanishi S, Katsumura K, Puro DG. ATP: a vasoactive signal in the pericyte-containing microvasculature of the rat retina. J Physiol 551: 787–799, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy-Lydon T, Crawford C, Wildman S, Peppiatt-Wildman C. Renal pericytes: regulators of medullary blood flow. Acta Physiol (Oxf) 207: 212–225, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kintzel PE. Anticancer drug-induced kidney disorders. Drug Saf 24: 19–38, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Kroetz DL, Zeldin DC. Cytochrome P450 pathways of arachidonic acid metabolism. Curr Opin Lipidol 13: 273–283, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Lebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C, Bréant C, Mathivet T, Larrivée B, Thomas JL, Arthur HM, Westermann CJ, Disch F, Mager JJ, Snijder RJ, Eichmann A, Mummery CL. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med 16: 420–428, 2010. [DOI] [PubMed] [Google Scholar]
- 32.Liptak P, Ivanyi B. Primer: histopathology of calcineurin-inhibitor toxicity in renal allografts. Nat Clin Pract Nephrol 2: 398– and quiz after 404, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Maurice-Estepa L, Daudon M, Katlama C, Jouanneau C, Sazdovitch V, Lacour B, Beaufils H. Identification of crystals in kidneys of AIDS patients treated with foscarnet. Am J Kidney Dis 32: 392–400, 1998. [DOI] [PubMed] [Google Scholar]
- 34.Miller MJ, Bednar MM, McGiff JC. Renal metabolism of sulindac: functional implications. J Pharmacol Exp Ther 231: 449–456, 1984. [PubMed] [Google Scholar]
- 35.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci USA 87: 9383–9387, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy SW, Barrett BJ, Parfrey PS. Contrast nephropathy. J Am Soc Nephrol 11: 177–182, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Murray MD, Brater DC. Renal toxicity of the nonsteroidal anti-inflammatory drugs. Annu Rev Pharmacol Toxicol 33: 435–465, 1993. [DOI] [PubMed] [Google Scholar]
- 38.Nolin T, Himmelfarb J. Mechanisms of drug-induced nephrotoxicity. Hand Exp Pharmacol 196: 111–130, 2010. [DOI] [PubMed] [Google Scholar]
- 39.O'Connor PM, Cowley AW. Medullary thick ascending limb buffer vasoconstriction of renal outer-medullary vasa recta in salt-resistant but not salt-sensitive rats. Hypertension 60: 965–972, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oates JA, FitzGerald GA, Branch RA, Jackson EK, Knapp HR, Roberts LJ. Clinical implications of prostaglandin and thromboxane A2 formation (1). N Engl J Med 319: 689–698, 1988. [DOI] [PubMed] [Google Scholar]
- 41.Pallone TL. Vasoconstriction of outer medullary vasa recta by angiotensin II is modulated by prostaglandin E2. Am J Physiol Renal Fluid Electrolyte Physiol 266: F850–F857, 1994. [DOI] [PubMed] [Google Scholar]
- 42.Pallone TL, Robertson CR, Jamison RL. Renal medullary microcirculation. Physiol Rev 70: 885–920, 1990. [DOI] [PubMed] [Google Scholar]
- 43.Pallone TL, Silldorff EP. Pericyte regulation of renal medullary blood flow. Exp Nephrol 9: 165–170, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Pallone TL, Zhang Z, Rhinehart K. Physiology of the renal medullary microcirculation. Am J Physiol Renal Physiol 284: F253–F266, 2003. [DOI] [PubMed] [Google Scholar]
- 45.Pang L, Knox A. PGE2 release by bradykinin in human airway smooth muscle cells: involvement of cyclooxygenase-2 induction. Am J Physiol Lung Cell Mol Physiol 273: L1132–L1140, 1997. [DOI] [PubMed] [Google Scholar]
- 46.Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature 443: 700–704, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peppiatt-Wildman C. The evolving role of renal pericytes. Curr Opin Nephrol Hypertens 22: 10–16, 2013. [DOI] [PubMed] [Google Scholar]
- 48.Perazella MA. Drug-induced nephropathy: an update. Expert Opin Drug Saf 4: 689–706, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Perazella MA. Drug-induced renal failure: update on new medications and unique mechanisms of nephrotoxicity. Am J Med Sci 325: 349–362, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest 110: 61–69, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reynaert H, Thompson MG, Thomas T, Geerts A. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut 50: 571–581, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salvemini D, Masferrer JL. Interactions of nitric oxide with cyclooxygenase: in vitro, ex vivo, and in vivo studies. Methods Enzymol 269: 7240–7244, 1996. [DOI] [PubMed] [Google Scholar]
- 53.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA 90: 7240–7244, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silldorff EP, Yang S, Pallone TL. Prostaglandin E2 abrogates endothelin-induced vasoconstriction in renal outer medullary descending vasa recta of the rat. J Clin Invest 95: 2734–2740, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith WL. The eicosanoids and their biochemical mechanisms of action. Biochem J 259: 315–324, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stillman MT, Schlesinger PA. Nonsteroidal anti-inflammatory drug nephrotoxicity. Should we be concerned? Arch Intern Med 150: 268–270, 1990. [PubMed] [Google Scholar]
- 57.Stokes JB. Effect of prostaglandin E2 on chloride transport across the rabbit thick ascending limb of Henle. Selective inhibitions of the medullary portion. J Clin Invest 64: 495–502, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stokes JB, Kokko JP. Inhibition of sodium transport by prostaglandin E2 across the isolated, perfused rabbit collecting tubule. J Clin Invest 59: 1099–1104, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taguchi T, Nazneen A, Abid MR, Razzaque MS. Cisplatin-associated nephrotoxicity and pathological events. Contrib Nephrol 148: 107–121, 2005. [DOI] [PubMed] [Google Scholar]
- 60.Tse W, Adu D. Non-steroidal anti-inflammatory drugs and the kidney. In: Oxford Textbook of Nephrology (2nd ed), edited by Davison A, Cameron S, Grunfeld J, Kerr D, Ritz E, Winerals C. Oxford: Oxford Publication, 1998, p. 1145–1156. [Google Scholar]
- 61.Verbeeck RK, Blackburn JL, Loewen GR. Clinical pharmacokinetics of non-steroidal anti-inflammatory drugs. Clin Pharmacokinet 8: 297–331, 1983. [DOI] [PubMed] [Google Scholar]
- 62.Waybill MM, Waybill PN. Contrast media-induced nephrotoxicity: identification of patients at risk and algorithms for prevention. J Vasc Interv Radiol 12: 3–9, 2001. [DOI] [PubMed] [Google Scholar]
- 63.Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med 106: 13S–24S, 1999. [DOI] [PubMed] [Google Scholar]
- 64.Whelton A, Stout RL, Spilman PS, Klassen DK. Renal effects of ibuprofen, piroxicam, and sulindac in patients with asymptomatic renal failure. A prospective, randomized, crossover comparison. Ann Intern Med 112: 568–576, 1990. [DOI] [PubMed] [Google Scholar]
- 65.Woodhouse KW, Wynne H. The pharmacokinetics of non-steroidal anti-inflammatory drugs in the elderly. Clin Pharmacokinet 12: 111–122, 1987. [DOI] [PubMed] [Google Scholar]
- 66.Wynckel A, Ebikili B, Melin JP, Randoux C, Lavaud S, Chanard J. Long-term follow-up of acute renal failure caused by angiotensin converting enzyme inhibitors. Am J Hypertens 11: 1080–1086, 1998. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Z, Pallone TL. Response of descending vasa recta to luminal pressure. Am J Physiol Renal Physiol 287: F535–F542, 2004. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Z, Payne K, Cao C, Pallone TL. Mural propagation of descending vasa recta responses to mechanical stimulation. Am J Physiol Renal Physiol 305: F286–F294, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]